
Last Updated: April 1, 2025
Introduction to the Steroid Hormones
The steroid hormones are all derived from cholesterol. Moreover, with the exception of vitamin D, they all contain the same cyclopentanophenanthrene ring and atomic numbering system as cholesterol. The conversion of C27 cholesterol to the 18-, 19-, and 21-carbon steroid hormones (designated by the nomenclature C with a subscript number indicating the number of carbon atoms, e.g. C19 for androstanes) involves the rate-limiting, irreversible cleavage of a 6-carbon residue from cholesterol, producing pregnenolone (C21) plus isocaproaldehyde.
Common names of the steroid hormones are widely recognized, but systematic nomenclature is gaining acceptance and familiarity with both nomenclatures is increasingly important. Steroids with 21 carbon atoms are known systematically as pregnanes, whereas those containing 19 and 18 carbon atoms are known as androstanes and estranes, respectively. The important mammalian steroid hormones are shown below along with the structure of the precursor, pregnenolone. Retinoic acid and vitamin D are not derived from pregnenolone, but from vitamin A and cholesterol respectively.
Table of the Major Steroid Hormones
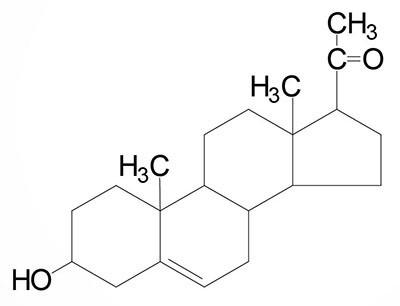 | Pregnenolone: produced directly from cholesterol, the precursor molecule for all C18, C19, and C21 steroids |
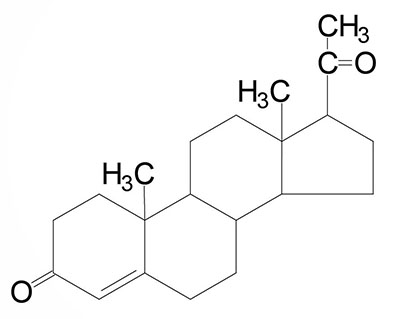 | Progesterone: a progestogen, produced directly from pregnenolone and secreted from the corpus luteum, responsible for changes associated with luteal phase of the menstrual cycle, differentiation factor for mammary glands |
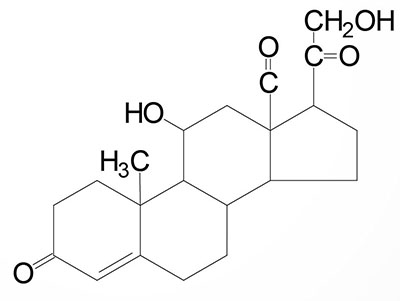 | Aldosterone: the principal mineralocorticoid, produced from progesterone in the zona glomerulosa of adrenal cortex, raises blood pressure and fluid volume, increases Na+ uptake via the kidney |
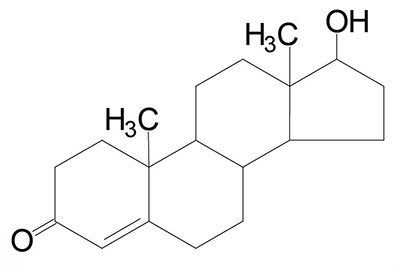 | Testosterone: an androgen, male sex hormone synthesized in the testes, responsible for secondary male sex characteristics, produced from progesterone |
 | Estradiol: an estrogen, principal female sex hormone, produced in the ovary, responsible for secondary female sex characteristics |
 | Cortisol: dominant glucocorticoid in humans, synthesized from progesterone in the zona fasciculata of the adrenal cortex, involved in stress adaptation, increases gluconeogenesis in liver, elevates blood pressure, increases Na+ uptake by the kidney, numerous effects on the immune system |
All the steroid hormones exert their action by passing through the plasma membrane and binding to intracellular receptors (also referred to as nuclear receptors, NR). The mechanism of action of the thyroid hormones is similar in that they also interact with intracellular nuclear receptors. Both the steroid and thyroid hormone-receptor complexes exert their actions by binding to specific nucleotide sequences in the DNA of responsive genes. These DNA sequences are identified as hormone response elements, HRE. The interaction of steroid-receptor complexes with DNA leads to altered rates of transcription of the associated genes.
Introduction to the Thyroid Hormones
The thyroid hormones (called thyronines) are all derived from the amino acid tyrosine within specific follicle cells of the thyroid gland. The primary hormone secreted from the thyroid gland is thyroxine (T4: 3,5,3’5′-tetraiodothyronine). Another hormone secreted from the thyroid, but at much lower levels, is triiodothyronine (T3).
The primary site of T3 generation is in peripheral tissues via the deiodination of thyroxine. T3 is the most biologically active form of thyroid hormone and exerts its effects by binding to the thyroid hormone receptor (TR). TR is a member of the steroid hormone/thyroid hormone superfamily of nuclear receptors. There are two known TR genes, one encoding TRα and the other encoding TRβ.
Synthesis of the thyroid hormones is controlled by the actions of the anterior pituitary hormone, thyroid stimulating hormone (TSH). The actions of the thyroid hormones maintain optimal lipid and carbohydrate metabolic homeostasis. Although the thyroid gland is not essential for life, hypothyroidism in fetal life and in early childhood results in reduced growth (dwarfism) as well as severe intellectual impairment. Hypothyroidism in adulthood is associated with reduced resistance to cold as well as intellectual and physical impairment. At the other end of the spectrum, hyperthyroidism in adults is associated with excessive heat generation, metabolic wasting, cardiac dysfunction (tachycardia), tremors and anxiety.
Steroid Hormone Biosynthesis Reactions
The particular steroid hormone class synthesized by a given cell type depends upon its complement of peptide hormone receptors, its response to peptide hormone stimulation and its genetically expressed complement of enzymes. The following indicates which peptide hormone is responsible for stimulating the synthesis of which steroid hormone:
- Luteinizing Hormone (LH): progesterone and testosterone
- Adrenocorticotropic hormone (ACTH): cortisol
- Follicle Stimulating Hormone (FSH): estradiol
- Angiotensin II/III: aldosterone
Role of ACTH in Adrenal Steroid Hormone Synthesis
ACTH is a 39 amino acid peptide that is derived by post-translational modification from the 241 amino acid proprotein, proopiomelanocortin, POMC. ACTH is the main physiologically active product of the actions of the hypothalamic releasing hormone, corticotropin releasing hormone, (CRH), on the anterior pituitary. Negative feedback on ACTH secretion is exerted by cortisol at both the hypothalamic and anterior pituitary levels. Thus, the primary product of the systemic actions of ACTH, cortisol, regulates the further actions of this corticotropic hormone.
The biological role of ACTH is to stimulate the production of adrenal cortex steroids, principally the glucocorticoids cortisol and corticosterone. ACTH also stimulates the adrenal cortex to produce the mineralocorticoid, aldosterone as well as the androgen, androstenedione.
ACTH exerts its effects on the adrenal cortex by binding to a specific receptor that is a member of the melanocortin receptor family. The ACTH receptor is identified as MC2R for melanocortin-2 receptor. The ACTH receptor is a Gs-type G-protein coupled receptor (GPCR). The activity of the MC2R is dependent upon a a small accessory protein, called melanocortin receptor accessory protein (MRAP). The function of MRAP is to regulate trafficking of MC2R to the plasma membrane and to enhance ACTH binding and activation of MC2R. The binding of ACTH to MC2R binding triggers activation of adenylate cyclase, elevation of cAMP, and increased PKA activity of adrenal cortex tissue.
Several targets of the ACTH activated PKA are hormone sensitive lipase (HSL), steroidogenic acute regulatory protein (StAR), and CYP11A1 [also called P450-linked side chain-cleaving enzyme (P450ssc), 20,22-desmolase, or cholesterol desmolase]. Activation of HSL increases the de-esterification of cholesterol esters generating free cholesterol. When originally characterized, the adrenal cortical cholesterol de-esterifying enzyme was called cholesterol esterase, and later demonstrated to be HSL. The activation of StAR results in increased transport of free cholesterol into the mitochondria where steroid hormone synthesis is initiated. The activation of CYP11A1 results in increased conversion of cholesterol to pregnenolone.
StAR and Mitochondrial Cholesterol Transport
In order for cholesterol to be used for steroid hormone biosynthesis it must be in the free state. This initial step is catalyzed by hormone sensitive lipase (HSL) which removes the fatty acid esterification from cholesterol esters to generate free cholesterol. This is the same hormone sensitive lipase that is expressed in adipocytes and is encoded by the LIPE gene. The generation of free cholesterol from cholesterol esters within adrenal cortical cells, and other steroidogenic tissues, was originally thought to be the function of an enzyme activity termed cholesterol esterase.
The free cholesterol must then be transported from the outer mitochondrial membrane to the inner membrane. The transport process is mediated by steroidogenic acute regulatory protein (StAR) and this transport process represents the rate-limiting step in steroidogenesis. Expression of the STAR gene is exclusive to the adult adrenal glands, ovary, and testis with highest levels of expression within adrenal cortical cells. The StAR protein is the founding member of a large family of proteins that all possess a STeroidogenic Acute Regulatory protein-related lipid Transfer (START) domain.
Transcriptional Regulation of STAR Gene
A major regulator of the transcription of the gene (STAR) encoding StAR is ACTH. When ACTH binds to its receptor (MC2R), a Gs-type GPCR, there is activation of cAMP production. Although the STAR gene lacks a consensus binding site for canonical cAMP-activated transcription mediated by cAMP response element-binding protein (CREB) family member transcription factors, cAMP does indeed result in increased STAR transcription. The cAMP-responsive region within the promoter region of the STAR gene contains a highly conserved overlapping motif (5′-TGACTGATGA-3′) that has been shown to interact with CREM (CREB/CRE modulator), ATF1 (activating transcription factor 1), activator protein 1 (AP-1), and the CCAAT/enhancer-binding protein β (C/EBPβ) families of proteins. CREM, ATF1, and CREB constitute a subfamily of the basic leucine zipper (bZIP) transcription factors. These transcription factors exert their transcriptional regulation by binding as homo- or heterodimers to the cAMP response element in regulatory regions of target genes.
START Domain Protein Family
The STaR protein is a member of a large family of proteins that contain an α/β helix-grip structure forming a hydrophobic pocket for lipid binding. This structural domain is referred to as the STeroidogenic Acute Regulatory protein-related lipid Transfer (START) domain. The START domain is composed of ~210 amino acids and is found in proteins that bind sterols, phospholipids, bile acids, and ceramides. The human START domain-containing protein family consists of 15 members with most being encoded by genes with the STARD nomenclature. Two members of the START domain protein family are acyl-CoA thioesterases (ACOT11, and ACOT12). Details of the START domain protein family and their functions are discussed in the Cholesterol: Synthesis, Metabolism, and Regulation page.
Post-Translational Modification of STaR Activity
The activity of StAR is regulated by post-translational modifications including phosphorylation. The ACTH-mediated activation of PKA plays a central role in the regulation of StAR activity in the adrenal cortex. There are at least two consensus PKA phosphorylation sites in the StAR protein, Ser57 and Ser195. Of these two sites, the phosphorylation of StAR on Ser195 by PKA is essential to render the protein fully active in its capacity to support cholesterol transfer into the mitochondria. The significance of Ser195 phosphorylation to StAR activity is demonstrated by the fact that mutation of Ser195 is one of many point mutations in the human STAR gene that are associated with congenital lipoid adrenal hyperplasia.
Steroid Hormone Enzyme Nomenclature
The various hydroxylases involved in the synthesis of the steroid hormones have a nomenclature that indicates the site of hydroxylation (e.g. 17α-hydroxylase introduces a hydroxyl group to carbon 17). These hydroxylase enzymes are members of the cytochrome P450 class of enzymes and as such also have a nomenclature indicative of the site of hydroxylation in addition to being identified as P450 class enzymes (e.g. the 17α-hydroxylase is also identified as P450c17).
The officially preferred nomenclature for the cytochrome P450 class of enzymes is to use the prefix CYP. Thus, 17α-hydroxylase is identified as CYP17A1. Another example is the gene encoding the P450ssc enzyme (also called 20,22-desmolase or cholesterol desmolase) which is identified as CYP11A1. There are currently 57 identified CYP genes in the human genome.
Table of the Primary Enzyme Activities of Steroid Hormone Biosynthesis
| Common Name(s) | Gene ID | Activities | Primary Site of Expression/Comments |
| steroidogenic acute regulatory protein | STAR | mediates transport of cholesterol from outer mitochondrial membrane to the inner membrane | all steroidogenic tissues except placenta and brain; located on chromosome 8p11.23 and is composed of 7 exons that encode a 285 amino acid protein |
| desmolase, P450ssc | CYP11A1 | cholesterol-20,22-desmolase | steroidogenic tissues; located on chromosome 15q24.1 and is composed of 10 exons that generate two alternatively spliced mRNAs; isoform a protein is 521 amino acids and represents the functional enzyme; the isoform b protein lacks a mitochondrial targeting sequences and thus, likely plays no role in the P450ssc complex. |
| 3β-hydroxysteroid dehydrogenase type 1 | HSD3B1 | 3β-hydroxysteroid dehydrogenase | placental for progesterone synthesis; dual function enzyme; approved name is hydroxy-delta-5-steroid dehydrogenase, 3 beta- and steroid delta-isomerase 1; located on chromosome 1p12 and is composed of 4 exons that generate two alternatively spliced mRNAs, both of which encoded the same 373 amino acid protein; expression predominates in the placenta with very low levels in adrenal glands and small intestine |
| 3β-hydroxysteroid dehydrogenase type 2 | HSD3B2 | 3β-hydroxysteroid dehydrogenase | steroidogenic tissues; dual function enzyme; approved name is hydroxy-delta-5-steroid dehydrogenase, 3 beta- and steroid delta-isomerase 2; the two activities are 3β-hydroxysteroid dehydrogenase and Δ4,5-isomerase; located on chromosome 1p12 and is composed of 5 exons that generate two alternatively spliced mRNAs, both of which encoded the same 372 amino acid protein; expression of the HSD3B2 gene is exclusive to the adrenal glands |
| P450c11 | CYP11B1 | 11β-hydroxylase | only in zona fasciculata and zona reticularis of adrenal cortex; located on chromosome 8q24.3 and is composed of 9 exons that generate two alternatively spliced mRNAs encoding precursor proteins of 503 amino acids (isoform 1) and 437 amino acids (isoform 2) |
| P450c17 | CYP17A1 | two activities: 17α-hydroxylase and 17,20-lyase | steroidogenic tissues; located on chromosome 10q24.32 and is composed of 8 exons that encode a 508 amino acid precursor protein |
| P450c21: 21-hydroxylase | CYP21A2 | two substrate binding sites; one for 17α-hydroxyprogesterone and one for progesterone | not expressed in the zona reticularis; located on chromosome 6p21.33 and is composed of 10 exons that generate four alternatively spliced mRNAs that collectively encode three distinct protein isoforms |
| aldosterone synthase | CYP11B2 | 18α-hydroxylase | exclusive to zona glomerulosa of adrenal cortex; located on chromosome 8q24.3 and is composed of 9 exons that encode a 503 amino acid precursor protein |
| estrogen synthetase | CYP19A1 | aromatase | gonads, brain, adrenals, adipose tissue, bone, with highest levels seen in placenta; located on chromosome 15q21.2 and is composed of 18 exons that generate eleven different mRNAs through the use of alternative promoters and alternative mRNA splicing all of which encode the same 503 amino acid protein |
| 17β-hydroxysteroid dehydrogenase type 3 | HSD17B3 | 17-ketoreductase | steroidogenic tissues; humans express 18 enzymes of the 17β-hydroxysteroid dehydrogenase type, all of which are members of the short chain dehydrogenase/reductase (SDR) superfamily; this family of enzymes is involved in several reactions of steroid hormone synthesis including adrenal steroids, and male and female sex hormones; located on chromosome 9q22.32 and is composed of 15 exons that encode a 310 amino acid protein |
| sulfotransferase | SULT2A1 | sulfotransferase | liver, adrenals; located on chromosome 19q13.33 and is composed of 6 exons that encode a 285 amino acid protein |
| 5α-reductase type 3 | SRD5A3 | 5α-reductase | steroidogenic tissues; primary enzyme carrying out the testosterone to DHT conversion; located on chromosome 4q12 and is composed of 6 exons that encode a 318 amino acid protein |
Cholesterol Desmolase: CYP11A1 (P450 Side Chain Cleavage Enzyme)
The first reaction in converting cholesterol to C18, C19 and C21 steroids involves the cleavage of a 6-carbon group from cholesterol and is the principal committing, regulated, and rate-limiting step in steroid biosynthesis. The enzyme system that catalyzes the cleavage reaction is known as P450-linked side chain cleaving enzyme (P450ssc) or 20,22-desmolase, or cholesterol desmolase, The P540ssc enzyme is localized to the inner mitochondrial membrane of steroid-producing cells, but not in significant quantities in other cells.
Mitochondrial 20,22-desmolase (P450ssc) is a complex enzyme system consisting of a cytochrome P450 family member enzyme and the proteins, ferredoxin reductase (also known as adrenodoxin reductase) and ferredoxin 1 (also known as adrenodoxin). Ferredoxin reductase and adrenodoxin reductase are components of numerous cytochrome P450 enzyme complexes in addition to P450ssc. The cytochrome P450 enzyme of P450ssc is encoded by the CYP11A1 gene (see the Cytochrome P450 (CYP) Enzymes page for description of CYP nomenclature).
The ferredoxin reductase gene (FDXR) is located on chromosome 17q25.1 and is composed of 14 exons that generate seven alternatively spliced mRNAs, each of which encodes a distinct protein isoform.
Humans express two genes encoding ferredoxin, FDX1 and FDX2.
The ferredoxin 1 gene (FDX1) is located on chromosome 11q22.3 and is composed of 5 exons that encode a 184 amino acid precursor protein that is targeted to the mitochondria. The FDX1 encoded protein is the one involved in steroid biosynthesis.
The FDX2 gene is located on chromosome 19p13.2 and is composed of 5 exons that encode a 186 amino acid precursor protein that is also targeted to the mitochondria. The FDX2 encoded protein is involved in the synthesis of heme A (heme a) as well as several Fe-S domain containing proteins.
The overall cholesterol side-chain cleavage occurs through a series of three reactions all catalyzed by the desmolase complex. The activity of each of these components is increased by two principal cAMP- and PKA-dependent processes. First, principally in response to the actions of ACTH in adrenal cortical cells, the level of cAMP rises. Secondly the increased cAMP results in the stimulated activity of PKA. Activated PKA phosphorylates a number of proteins involved in steroid hormone biosynthesis such as the steroidogenic acute regulatory protein (StAR) and hormone sensitive lipase (encoded by the LIPE gene).
Hormone-sensitive lipase (HSL) is the neutral cholesteryl ester hydrolase (NCEH) activity in steroidogenic cells. The activation of hormone sensitive lipase results in the removal of the fatty acid esterification at the C-3 position of cholesterol leading to increased concentrations of free cholesterol. It is free cholesterol that is the substrate for mitochondrial 20,22-desmolase (P450ssc).
Regulation of CYP11A1 Expression
Long-term regulation of steroid synthesis is also effected at the level of the gene (CYP11A1) encoding the CYP component of the 20,22-desmolase complex. This gene contains a cAMP response element (CRE) that binds the transcription factor identified as cAMP-response element-binding protein, CREB (encoded by the CREB1 gene).
Humans express seven genes in the CREB subfamily of the basic leucine zipper (bZIP) family of transcription factors. The seven CREB gene are identified as CREB1, CREB3, CREB5, and CREB3-like 1, 2, 3, and 4 (CREB3L1, CREB3L2, CREB3L3, and CREB3L4). The CREB3L3 encoded protein is commonly identified as CREBH. The CREB1 encoded proteins are most closely related in structure and function to two additional transcription factors called cAMP response element modulator (CREM) and activating transcription factor 1 (ATF-1). Another member of the activating transcription factor (ATF) family, ATF-4, was originally identified as CREB2.
CREB is phosphorylated on Ser133 by PKA as well as several other kinases such as calmodulin-dependent protein kinase (CaMK) and members of the mitogen-activated protein kinase (MAPK) family. Phosphorylation of CREB occurs in the nucleus which allows interaction with the co-activator, CREB-binding protein (CBP), and binding to CREs in target genes such as CYP11A1. The consequences of CREB binding to CYP11A1 are increased rates of 20,22-desmolase RNA transcription, thereby leading to increased levels of the enzyme.
Steroid Hormones of the Adrenal Cortex
The adrenal cortex is responsible for production of three major classes of steroid hormones: glucocorticoids, which regulate carbohydrate metabolism; mineralocorticoids, which regulate the body levels of sodium and potassium; and androgens, whose actions are similar to that of steroids produced by the male gonads. Cholesterol, acquired from the diet or from LDL, or produced de novo in adrenal cortical cells, serves as the precursor for all of the adrenal steroid hormones. Cholesterol uptake from the blood occurs through the binding of LDL to the LDL receptor. Chronic stimulation of the adrenal cortex by ACTH leads to increased LDL receptor gene expression resulting in increased receptor density.
Adrenal Cortical Domains in Steroid Hormone Synthesis
The adrenal cortex is composed of three main tissue regions and in order of their depth from the surface of the cortex these regions are identified as the zona glomerulosa, the zona fasciculata, and the zona reticularis. Although the pathway to pregnenolone synthesis is the same in all zones of the cortex, the zones are histologically and enzymatically distinct, with the exact steroid hormone product dependent on the enzymes present in the cells of each zone.
Mnemonics for remembering the zones of the adrenal cortex or which adrenal hormones are synthesized are “Go Find Rex” and “Make Good Sex”, respectively. In the first mnemonic the first letter of each word is the guide where G is glomerulosa, F is fasciculata, and R is reticularis. The first letters of the words in the second mnemonic are also the guide where M is mineralocorticoids, G is glucocorticoids, and S is sex hormones (androgens). Another useful mnemonic is “sweet, salt, sex” where sweet refers to glucocorticoids and their role in glucose metabolism, salt refers to aldosterone and its role in sodium homeostasis, and sex refers to the androgenic hormones.
Figure Showing Reactions of Adrenal Cortical Steroid Hormone Synthesis
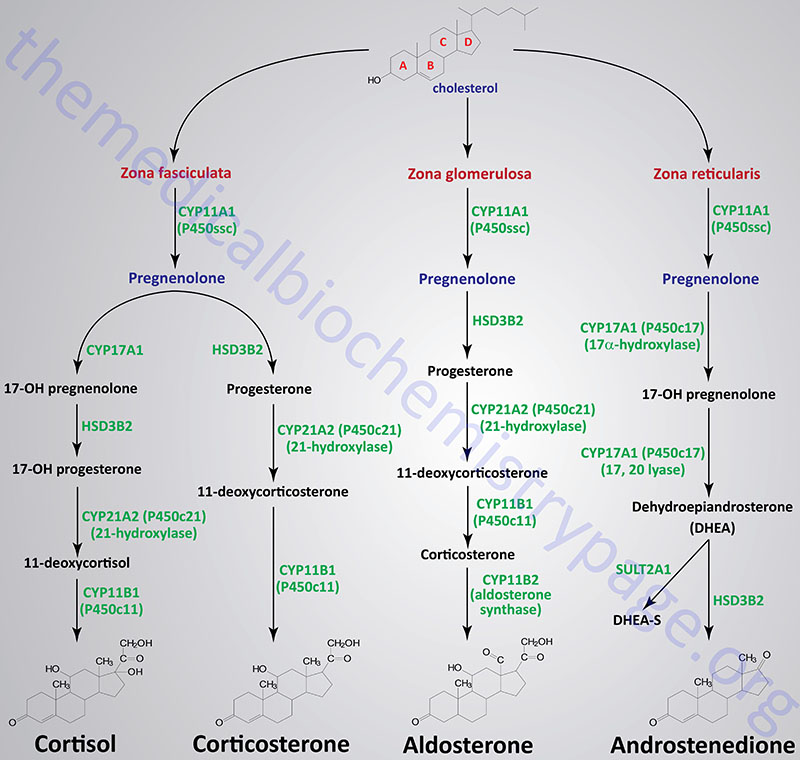
Reactions of the Zona Fasciculata
The primary steroid hormones produced by adrenal cortical zona fasciculata cells are the glucocorticoids, cortisol and corticosterone. Pregnenolone is converted to these two glucocorticoids by two distinct pathways of enzymes. As noted earlier, P450ssc (CYP11A1) is a mitochondrial activity, whose product, pregnenolone, moves to the cytosol, where it is converted either to 11-deoxycortisol or 11-deoxycorticosterone. The latter compounds then re-enter the mitochondria for conversion to cortisol and corticosterone, respectively.
In the pathway for the synthesis of cortisol, pregnenolone is converted to 17α-hydroxypregnenolone (17α-OH pregnenolone, or simply 17-OH pregnenolone) via the bi-functional enzyme encoded by the CYP17A1 gene. This enzyme possesses both 17α-hydroxylase and 17,20-lyase activities.
In the next reaction of this pathway 17-hydroxypregnenolone is converted to 17α-hydroxyprogesterone (17α-OH progesterone or often written simply as 17-hydroxyprogesterone) via the bi-functional enzyme encoded by the HSD3B2 gene. The two activities of this enzyme are 3β-hydroxysteroid dehydrogenase and Δ4,5-isomerase.
The next step in cortisol synthesis is catalyzed by the enzyme encoded by the CYP21A2 gene. The CYP21A2 encoded enzyme is more commonly called 21-hydroxylase. The 21-hydroxylase enzyme possesses two different substrate binding sites, one for 17α-hydroxyprogesterone and one for progesterone. In the pathway of cortisol synthesis 21-hydroxylase converts 17α-hydroxyprogesterone to 11-deoxycortisol. Mutations in the CYP21A2 gene are the most common causes of congenital adrenal hyperplasia.
The synthesis of cortisol from 11-deoxycortisol is catalyzed by the enzyme encoded by the CYP11B1 gene, steroid 11β-hydroxylase.
In the pathway for the synthesis of corticosterone, progesterone is converted to 11-deoxycorticosterone (simply referred to as deoxycorticosterone; DOC) by 21-hydroxylase.
The conversion of deoxycorticosterone to corticosterone is catalyzed by steroid 11β-hydroxylase. Cells of the zona fasciculata lack aldosterone synthase (CYP11B2) that converts corticosterone to aldosterone.
Reactions of the Zona Glomerulosa
The primary steroid hormone produced by adrenal cortical zona glomerulosa cells is the mineralocorticoid, aldosterone. As in the synthesis of cortisol, conversion of pregnenolone to progesterone requires the bi-functional enzyme encoded by the HSD3B2 gene (3β-hydroxysteroid dehydrogenase and Δ4,5-isomerase activities). Progesterone is converted to 11-deoxycorticosterone (commonly called deoxycorticosterone: DOC) by the CYP21A2 encoded enzyme, 21-hydroxylase. This is the same reaction occurring in zona fasciculata cells in the synthesis of corticosterone. Deoxycorticosterone is converted to corticosterone by the CYP11B1 encoded enzyme.
Corticosterone then enters the mitochondria where it is converted to aldosterone in a two-step reaction catalyzed by aldosterone synthase. Aldosterone synthase is encoded by the CYP11B2 gene. The CYP11B2 encoded enzyme is bi-functional possessing steroid 11β-hydroxylase and steroid 18-hydroxylase (aldosterone synthase) activities. The 18-hydroxylase activity converts corticosterone to 18-hydroxycorticosterone and then to aldosterone.
Zona glomerulosa cells do not express the CYP17A1 gene that encodes the enzyme that converts pregnenolone and progesterone to their C17 hydroxylated analogs. Thus, the pathways to the glucocorticoids and the androgens are blocked in these cells. Zona glomerulosa cells are unique in the adrenal cortex in that they are the only cells expressing the enzyme responsible for converting corticosterone to aldosterone, the principal and most potent mineralocorticoid. This enzyme is P450c18 (or 18α-hydroxylase, CYP11B2), also more commonly called aldosterone synthase. The result is that the zona glomerulosa is mainly responsible for the conversion of cholesterol to the weak mineralocorticoid, corticosterone and the principal mineralocorticoid, aldosterone.
Reactions of the Zona Reticularis
The primary steroid hormones produced by adrenal cortical zona reticularis cells are the androgens, androstenedione and dehydroepiandrosterone. As noted earlier, P450ssc (CYP11A1) is a mitochondrial activity. Its product, pregnenolone, moves to the cytosol, where it can be converted into androgens by enzymes of the endoplasmic reticulum.
Pregnenolone is first converted to 17-OH pregnenolone by the bi-functional enzyme encoded by the CYP17A1 gene. This enzyme possesses 17α-hydroxylase and 17,20-lyase activities. The conversion of 17-OH pregnenolone to dehydroepiandrosterone (DHEA) is carried out by the 17,20-lyase activity of the CYP17A1 encoded enzyme. DHEA is then converted to androstenedione by the HSD3B2 encoded bi-functional enzyme. A portion of DHEA is sulfated to DHEA-S via the sulfotransferase encoded by the SULT2A1 gene
Overview of Adrenal Steroid Hormone Functions
The predominant adrenal steroid hormones are the glucocorticoids and the mineralocorticoids. The androgenic hormones produced by the adrenal cortex, although exerting important functions, are most significant in the context of adrenal dysfunction.
The glucocorticoids (discussed in greater detail below) are a class of hormone so called because they are primarily responsible for modulating the metabolism of carbohydrates. In a broad sense the glucocorticoids regulate energy homeostasis, embryonic development and postnatal life, and the responses to stress, thereby directly affecting survival and reproduction.
The mineralocorticoids (discussed in greater detail below) control the excretion of electrolytes (minerals), hence the derivation of the name of this class of hormone. As discussed in detail in the Glucocorticoid Functions section below, the glucocorticoids can exert mineralocorticoid effects through their ability to bind and activate the mineralocorticoid receptor.
The adrenal androgenic hormones, androstenedione and DHEA, circulate bound primarily to sex hormone-binding globulin (SHBG). Although some of the circulating androgen is metabolized in the liver, the majority of interconversion occurs in the gonads (as described below), skin, and adipose tissue. DHEA is rapidly converted to the sulfated form, DHEA-S, in the liver and adrenal cortex. The primary biologically active metabolites of the androgens are testosterone and dihydrotestosterone which function by binding receptors of the nuclear receptor family, thereby effecting changes in gene expression leading to the manifestation of the secondary male sex characteristics.
Glucocorticoid Functions
The glucocorticoids represent a group of steroid hormones whose major function is the modulation of carbohydrate metabolism, hence the derivation of the term, glucocorticoid. The primary glucocorticoid in humans is cortisol. Although modulation of glucose homeostasis is a major glucocorticoid-induced effect, while simultaneously inhibiting all other metabolic pathways not directly involved in glucose production, the glucocorticoids do have other physiologically and biochemically significant functions. Glucocorticoids also regulate overall energy homeostasis, they modulate embryonic and post-natal development, and they modulate stress responses that affect survival and reproduction. Indeed, physiological increases in the production of glucocorticoids is an anticipatory response designed to meet the increased energy demands associated with stress as well as those of major developmental changes occurring during the normal life cycle.
Cortisol inhibits uptake and utilization of glucose resulting in elevations in blood glucose levels. Cortisol acts as an insulin antagonist and also suppresses the release of insulin, both effects leading to reduced glucose uptake and enhanced hepatic gluconeogenesis. The effect of cortisol on blood glucose levels is further enhanced through the increased breakdown of skeletal muscle protein and adipose tissue triglycerides which provides energy and substrates for gluconeogenesis. Cortisol also increases the synthesis of gluconeogenic enzymes, in particular phosphoenolpyruvate carboxykinase (PEPCK, encoded by the PCK1 gene) and glucose-6-phosphatase (G6P, encoded by the G6PC gene). The increased rate of protein metabolism leads to increased urinary nitrogen excretion and the induction of urea cycle enzymes. In order to effectively accomplish these metabolic changes glucocorticoids inhibit several energy-consuming processes such as digestion, and reproduction, as well as inflammatory and other immune responses.
Cortisol is the most important naturally occurring glucocorticoid in humans. As indicated in the Figure above, cortisol is synthesized in the zona fasciculata of the adrenal cortex. When released to the circulation, cortisol is almost entirely bound to protein. A small portion of circulating cortisol is bound to albumin with more than 80% being bound by a specific glycosylated α-globulin called transcortin or corticosteroid-binding globulin (CBG). Transcortin is encoded by the serpin family A member 6 (SERPINA6) gene. Between 5% and 10% of circulating cortisol is free and biologically active. The function of CBG is not only transport of cortisol in the blood but regulated distribution of the hormone into tissues.
Following uptake into tissues, cortisol bioavailability is regulated by two enzymes that function in opposition to one another. One enzyme, 11β-hydroxysteroid dehydrogenase 2 (encoded by the HSD11B2 gene) oxidizes cortisol to its inactive metabolite, cortisone. The other enzyme, 11β-hydroxysteroid dehydrogenase 1 (encoded by the HSD11B1 gene) reduces the 11-oxo group (11-oxoreductase activity) in cortisone and 11-dehydrocorticosterone generating the active glucocorticoids, cortisol and corticosterone, respectively.
Once inside the cell, glucocorticoids exert their effects by direct binding to an intracellular receptor, the glucocorticoid receptor (GR) that is a zinc-finger transcription factor belonging to the nuclear hormone receptor superfamily. Until ligand binds the GR is associated with a chaperone complex that regulates availability to bind ligand. The chaperone complex consists of at least two heat-shock protein (HSP) family proteins, HSP70 and HSP90, and another protein that is a co-chaperone called HOP for HSP70-HSP90 Organizing Protein.
Typical of the structure of nuclear hormone receptors, the GR is composed of three functional domains. The N-terminal domain (NTD) harbors the potent transcriptional regulatory function, most often referred to as the activation function 1 (AF1) domain. The NTD comprises amino acid residues 1–420. The AF1 domain interacts with numerous transcriptional coregulators and the basal transcription factors that are involved in control of expression of glucocorticoid-responsive genes. In addition to harboring the AF1 domain, the NTD is the site of the major post-translational regulation of GR function. There are at least seven sites of Ser phosphorylation in the NTD of the GR.
The DNA-binding domain (DBD), which encompasses the amino acids that form the two zinc-finger domains, is located from amino acids 421–486.
The ligand-binding domain (LBD) resides in the C-terminus of the protein and encompasses amino acids 528–777.
There is a domain between the DBD and the LBD termed the hinge region that harbors two nuclear localization signals identified as NL1 and NL2.
The second activation function domain (AF2) is found in the extreme C-terminal region of the GR. The AF2 domain, like the AF1 domain, also binds to transcriptional coregulators in a ligand-dependent manner.
The GR is encoded by the NR3C1 (nuclear receptor subfamily 3 group C member 1) gene. The NR3C1 gene is located on chromosome 5q31.3 and is composed of 8 protein coding exons (exons 2-9) and 13 variants of exon 1 that differ as a result of upstream promoter elements. The NR3C1 gene also generates at least 15 alternatively spliced mRNAs. Several of these alternative mRNAs are translated from alternative in-frame translation initiation codons. The predominant GR species are identified as GRα (777 amino acids) and GRβ (742 amino acids). Additional GR isoforms resulting from the alternative splicing include GRγ, GR-P, and GR-A. Details are discussed in the Complexity in GR Gene Structure and Expression below.
The NR3C1 gene is constitutively expressed in virtually every cell type, but tissue-specific expression patterns of the alternative GR isoforms result in tissue-specific transcriptional outcomes. It is also important to note that most glucocorticoids bind to the mineralocorticoid receptor (MR encoded by the NR3C2 gene) as well, and as such, can exhibit mineralocorticoid-like activities.
Complexity in GR Gene Structure and Expression
The NR3C1 gene encodes mRNAs that are composed of nine exons, of which exons 2 through 9 encode the protein. As discussed in detail below, exon 2 contains eight conserved AUG codons that result in the ability to generate eight translation initiation variants of the GRα isoform.
So far, 13 variants of human GR exon 1 differing in the upstream promoter regions have been characterized. The NR3C1 promoter region is large and complex with some elements as far as 35 kbp upstream (5′) of the transcriptional start site. The promoters of the NR3C1 gene are identified as A–F and H–J. The NR3C1 promoters posses binding sites for several known transcription factors such as the cAMP response element binding protein (CREB), the estrogen receptor (ER), AP-1, AP-2, NFκB, Sp1, PU.1, and MYB as well as several others.
In addition to the ability for alternative promoter utilization, each promoter displays a distinct level of expression and tissue specificity. The NR3C1 promoter region is highly enriched in GC content (72%) and does not contain a canonical TATA-box nor a CAAT-box. Promoters A and C each direct the transcription of three distinct untranslated exons identified as 1A1–1A3 and 1C1–1C3.
The major GRα and GRβ isoforms are generated as a result of alternative splicing in exon 9. When exon 8 is spliced to the 5′-end of exon 9 the resultant protein is the GRα isoform which represents the classic GR protein mediating the actions of glucocorticoids. When exon 8 is spliced to an internal downstream splice acceptor site in exon 9 the resultant protein is the GRβ isoform. The GRβ isoform contains a shortened LBD and as such is incapable of binding glucocorticoids. The GRβ isoform resides within the nucleus constitutively (i.e. does not require ligand binding for transport to the nucleus as is the case for the GRα isoform). By itself the GRβ isoform is inactive on glucocorticoid-responsive genes. When expressed along with GRα the GRβ isoform serves as a dominant negative inhibitor, antagonizing the actions of GRα on glucocorticoid-responsive genes.
Additional GR isoforms result from other alternative splicing events. The GRγ isoform, which is widely expressed, results from the insertion of a single arginine residue (R452) between the two zinc fingers in the DNA-binding. domain. The GR-A isoform results from the splicing out of exons 5 through 7 such that amino acids 490–674 are deleted. The GR-P isoform results from splicing out of exons 8 and 9. The GRδ isoform results from the splicing out of exon 2 such that amino acids 313–338 are absent (this isoform is also referred to as GRΔ313-338). The GRδ isoform is expressed in several tissues. The significance of the deleted amino acid sequences relates to the fact that there are several sites for phosphorylation of the receptor in that deleted region and these sites are known to be important for the transactivation potential of the receptor. The GR-S1 isoform results from the retention of intron H, that resides between exons 8 and 9, and the splicing out of exons 8 and 9. The GR-NS1 isoform is a variant that contains three nonsynonymous single-nucleotide polymorphisms, two in exon 2 and one in exon 9. The GR-DL1 isoform is a truncated form of the receptor protein that results from a single nucleotide deletion in exon 2.
In addition to the complexity of alternative promoter usage and alternative mRNA splicing, the biological activities of GR isoforms is made even more complex through the utilization of alternative translation initiation sites. There are eight alternative translation initiation sites (all AUG start codons) in exon 2. The alternative translational initiation sites yield eight GRα isoforms harboring various lengths of the NTD. These various GRα isoforms are identified as GRα-A, -B, -C1, -C2, -C3, -D1, -D2, and -D3. The various GRα isoforms result from phenomena referred to as ribosome shunting and/or leaky ribosome scanning. When translation begins from the first initiator methionine codon the classic full-length 777 amino acid GRα isoform results. The significance of these alternative translational initiation sites is that they are tissue specific and the phenomenon is observed is many species. In addition to tissue-specific differences in distribution, the various GRα isoforms also exhibit different transcriptional activities in response to glucocorticoid binding.
The GRα-C isoforms are expressed at highest levels in the pancreas and colon. The GRα-C isoforms exhibit enhanced transcriptional activity in the induction of pro-apoptotic genes and they are more efficient in the recruitment of transcriptional coactivators.
Expression of the GRα-D isoforms are highest in spleen and lungs.
Functions of the GR at Target Genes
In the absence of ligand the glucocorticoid receptor (as well as the mineralocorticoid receptor) remains in the cytosol contained within multiprotein chaperone complexes. The cytosolic GR-containing complex includes heat-shock protein 90 (hsp90), hsp70, the co-chaperone identified as p23 (encoded by the prostaglandin E synthase 3, PTGES3 gene), and various immunophilins (e.g., FKBP51 and FKBP52), all of which prevent its degradation and assist in its maturation. The FKBP51 protein is encoded by the FKBP5 gene while the FKBP52 protein is encoded by the FKBP4 gene. In addition to maintaining the GR in the cytosol, the multiprotein complex facilitates high affinity ligand binding. Upon ligand binding, the GR complex changes its conformation leading to release of the ligand-bound receptor and exposure of the two nuclear localization signals in the GR allowing for rapid transport into the nucleus.
Within the nucleus the ligand-bound GR interacts with hormone response elements (HRE) in target genes. This HRE has a general sequence identified as AGAACAnnnTGTTCT (n represents any nucleotide. Other nuclear receptors, including the mineralocorticoid receptor, the progesterone receptor, and the androgen receptor are known to bind to variants of this same HRE. Examination of numerous glucocorticoid target genes has identified a glucocorticoid response element (GRE) with the sequences GGAACAnnnTGTTCT. The GR binds to the GRE as a homodimer with each of the half sites being bound by one receptor subunit in the homodimer. In addition to GR-mediated activation of gene expression by binding to the GRE, there are target genes whose transcription is repressed by GR binding to target sequences. In these cases the target sequence is referred to as a negative glucocorticoid response element, nGRE. The sequence of the nGRE is CTCC(n)0-2GGAGA and in these elements each half site binds a monomeric GR.
The interaction of GR with GRE causes additional conformational changes to take place in the receptor which facilitates the recruitment of, and interaction with, transcriptional coregulators that assist the transcriptional regulatory actions of the GR. One important family of GR coactivators are the SRC (steroid receptor coactivator) proteins. On the other hand, GR binding the nGRE elements results in the recruitment of, and interaction with, transcriptional corepressors such as nuclear receptor corepressor 1 (NCoR1: encoded by the NCOR1 gene) and silencing mediator of retinoic and thyroid receptors (SMRT: encoded by the NCOR2 gene).
In addition to the direct binding of GR to a GRE, leading to enhanced gene expression, the activated GR has been shown to bind at composite response elements. These composite response elements contain both a GRE half-site and the binding site for another transcription factor (e.g. AP-1, STAT5, ETS, and TEAD1). In these composite transcription factor binding sites GR binds to its GRE and the other transcription factor binds to its appropriate sequence element and the combined interactions result in regulation of gene expression. The GR can also affect transcriptional activity via a mechanism referred to as tethering. In this method the GR is tethered to chromatin by its interaction with other transcription factors that are bound to their cognate recognition sequences in the DNA even though there is no GRE present in the target gene. Tethering of GR has been shown to occur with many transcription factors including CREB (cAMP response element-binding protein), AP-1, NFκB (nuclear factor kappa B), GATA1 [member of family of transcription factors defined by the presence of the consensus sequence WGATAR (W = T or A; R = G or A)], STAT1 (signal transducer and activator of transcription 1), and STAT3.
Anti-Inflammatory Actions of Glucocorticoids
The anti-inflammatory activity of the glucocorticoids is exerted, in part, through inhibition of phospholipase A2 (PLA2) activity with a consequent reduction in the release of arachidonic acid from membrane phospholipids. Arachidonic acid serves as the precursor for the synthesis of various eicosanoids, several of which enhance inflammatory processes. Thus, by inhibiting the release of arachidonic acid, glucocorticoids contribute to a reduction in inflammatory processes.
The immune modulating effects of glucocorticoids are exploited pharmacologically and is the basis for the anti-inflammatory effects of drugs such as prednisone (an intermediate-acting steroid) and dexamethasone (a long-acting steroid). Synthetic glucocorticoids exhibit high affinity for the glucocorticoid receptor and mimic the effects of long-term exposure to high levels of natural glucocorticoids such as cortisol. Thus, the use of synthetic glucocorticoids, such as dexamethasone and prednisone, can lead to severe side effects including hypertension, diabetes, osteoporosis, and glaucoma.
Glucocorticoids inhibit inflammatory processes at the level of transcription and via the enhancement of protein modifications that are anti-inflammatory. Although the anti-inflammatory effects of glucocorticoids is well established, the precise mechanism by which the glucocorticoid receptor regulates the expression of immune system modulating genes is still poorly understood. A generalized model is that glucocorticoids enhance the expression of genes encoding anti-inflammatory proteins while repressing the expression of genes encoding proinflammatory proteins.
Glucocorticoid actions have been well established with respect to transactivation of the expression of the anti-inflammatory genes, ANXA1, DUSP1, and SPHK1. The ANXA1 gene encodes annexin A1 which is a membrane-localized phospholipid-binding proteins. The phospholipid-binding action of annexin A1 inhibits the activity of PLA2. The DUSP1 gene encodes dual specificity phosphatase 1 which, as the name implies, removes phosphate from tyrosine as well as serine and threonine residues. The DUSP1 encoded protein is involved in the regulation of signal transducing kinases of the MAPK family. Since phosphorylation of many proteins in the MAPK signal transduction pathways lead to activation, the removal of phosphate by the DUSP1 encoded enzyme will lead to reduced signaling and in the inflammation process this will lead to decreased inflammation. The SPHK1 gene encodes a sphingosine kinase which is responsible for the synthesis of the bioactive lipid, sphingosine-1-phosphate, S1P. S1P exerts numerous effects, many of which are associate with decreased immune responses.
One of the major effects of glucocorticoids, that leads to reduction in inflammation, is a metabolic reprogramming, specifically within the mitochondria. Glucocorticoids have been shown to increase the activity of both the pyruvate dehydrogenase complex (PDHc) and pyruvate carboxylase, PC. The increase in PC activity is, in part, one of the mechanisms by which glucocorticoids contribute to increased hepatic gluconeogenesis and glucose output by the liver.
Studies have shown that the glucocorticoid receptor (GR) interacts with a regulatory subunit of the PDHc identified as component X (PDHX). The PDHX protein tethers the E3 subunits of the PDHc to the E2 subunits. The GR interacts with the PDHX protein in the cytosol and when glucocorticoids bind the GR the PDHX is released and migrates into the mitochondria resulting in increased levels of PDHc activity. The increased levels of PDHc activity consequently results in increased TCA cycle activity. Several TCA cycle intermediates, including succinate, fumarate, and 2-oxoglutarate (α-ketoglutarate) can function as potent regulators of gene expression. In addition, the increased levels of TCA cycle activity, in response to glucocorticoid actions, leads to increased production itaconate, a metabolite of cis-aconitate. As described in the Pyruvate Dehydrogenase Complex and TCA Cycle page, itaconate is a critical TCA cycle-derived metabolite involved in the modulation of inflammatory processes.
Major Target Tissues for Glucocorticoid Regulation of Metabolism
The major organs that are targets for the metabolic regulatory actions of the glucocorticoids are the liver, adipose tissue, and skeletal muscle. Since the liver is the major organ tasked with the global regulation of glucose homeostasis it is not surprising that this organ is a major cortisol target. Similar to the role of glucagon in the liver, glucocorticoids are essential for the hepatic role of maintaining blood glucose levels during fasted states.
In addition, glucocorticoids exert effects on the liver during periods of stress to ensure adequate glucose is released. Within the liver, two primary target genes for cortisol are the gluconeogenic genes, PCK1 (cytoplasmic phosphoenolpyruvate carboxykinase) and G6PC (glucose-6-phosphatase). Maximal expression of both the PCK1 and G6PC genes requires additional factors including the nuclear receptor, PPARα. Within the liver the PPARA gene, which encodes PPARα, is also a target for cortisol activation.
The energy required for the liver to carry out gluconeogenesis during periods of fasting, or stress, is derived by the oxidation of fatty acids. The principal source of the fatty acids used by the liver is adipose tissue. Glucocorticoid effects on lipid homeostasis in adipose tissue are complex encompassing both increased lipogenesis through adipocyte differentiation and increased lipolysis. Glucocorticoids increase adipose tissue fatty acid release by increasing the expression of the hormone-sensitive lipase gene (symbol: LIPE) and the gene encoding monoglyceride lipase (symbol: MGLL). Under normal physiological conditions glucocorticoids promote preadipocyte differentiation into mature adipocytes and increase dietary fat intake. During glucocorticoid-induced adipocyte differentiation a lipolytic transcriptional program is activated and includes numerous genes involved in triglyceride synthesis, lipid transport, and lipid storage. Triglyceride synthesis genes turned on by cortisol include several AGPAT genes (acylglycerol-3-phosphate acyltransferases) that encode enzymes that incorporate a fatty acid into lysophosphatidic acid and the LPIN1 gene that encodes phosphatidic acid phosphatase that removes the phosphate from phosphatidic acid generating a diglyceride.
In skeletal muscle, glucocorticoids regulate protein and glucose metabolism. Following the consumption of food, insulin release promotes glucose uptake by skeletal muscle where it is stored as glycogen. During periods of fasting, stress, or during exercise, catecholamines (epinephrine) and glucocorticoids stimulate glycogen breakdown. When glucocorticoid levels are elevated, such as during pharmacological treatment with corticosteroids, or under conditions of hypercortisolemia (e.g. Cushing syndrome) there is a resultant insulin resistance, inhibited protein synthesis, and enhanced proteolysis within skeletal muscle. The enhanced proteolysis occurs as a means to provide amino acid carbon skeletons to the liver for gluconeogenesis, a pathway that is highly activated in response to excess glucocorticoid levels. These pathophysiological consequences within skeletal muscle result in muscle weakness and atrophy. Many of the genes that are activated in skeletal muscle by glucocorticoids encode proteins that interfere with insulin signaling in skeletal muscle resulting in the observed decreases in protein synthesis and increases in protein degradation.
Mineralocorticoid Functions
As the name of this class of hormone implies, the mineralocorticoids control the excretion of electrolytes (minerals). The major circulating mineralocorticoid is aldosterone. Aldosterone exerts its primary effects through actions on the kidneys but also functions in the colon and sweat glands. The principal effect of aldosterone is to enhance sodium (Na+) reabsorption in the connecting tubule (CNT) and cortical collecting duct of the nephrons in the kidneys. Within these regions of the nephron aldosterone induces the expression of the Na+,K+-ATPase subunit genes (ATP1A1 and ATP1B1), the genes encoding the subunits (SCNN1A, SCNN1B, and SCNN1C) of the epithelial sodium channel (ENaC), and the SLC12A3 gene (encoding the Na+-Cl– cotransporter, NCC). The net effect of the induction of these transporter genes, by aldosterone, is enhanced Na+ reabsorption as a function of the apical membrane localized ENaC and NCC transporters and delivery to the blood via the action of the basolateral membrane localized Na+,K+-ATPase.
Secondary to the Na+ uptake is efflux of potassium (K+) to the tubular lumen. In addition to K+ excretion, aldosterone enhances the excretion of hydrogen (H+) ions from the collecting duct which is a compensating action to counter the accumulation of the positive charge imparted by increased Na+ reabsorption.
However, the action of aldosterone is also exerted on sweat glands, stomach, and salivary glands to the same effect, i.e. sodium reabsorption. This action is accompanied by the retention of chloride (Cl–) and water resulting in the expansion of extracellular volume leading to increased vascular pressure.
Aldosterone, like all steroid hormones, functions as a ligand activating the transcriptional activity of the mineralocorticoid receptor, MR. The MR, being a member of the nuclear receptor superfamily, is encoded by the NR3C2 (nuclear receptor subfamily 3 group C member 2) gene. The NR3C2 gene is located on chromosome 4q31.23 and is composed of two alternative non-coding exons and eight coding exons (exons 2–9). Alternative splicing yields three mRNAs that encode two distinct protein isoforms. The primary MR protein is composed of 984 amino acids. An additional MR protein, encoded by two of the alternatively spliced mRNAs, is composed of 867 amino acids.
Like all nuclear receptors the MR is composed of an N-terminal domain (NTD), a central DNA-binding domain (DBD), and a C-terminal ligand-binding domain (LBD).
The activation function domain 1 (AF-1) is located in the NTD and a second activation function domain (AF-2) is located at the C-terminal end of the LBD. The AF-1 domain in the MR is actually comprised of two distinct domains identified as AF-1a and AF-1b. The AF-1a domain is located in amino acids 1–167 and AF-1b is located in amino acids 445–602. The AF-1a and AF-1b domains are responsible for the recruitment of various transcriptional coactivators to the MR. The NTD also contains an inhibitory domain that resides between the two AF-1 domains.
The DBD of the MR is encoded by exons 3 and 4 and contains two zinc-finger domains that facilitate receptor dimerization and DNA binding.
The LBD of the MR is highly homologous to the LBD of the GR which explains the ability of glucocorticoids to bind to and activate the MR. In addition, the close homology between the AF-2 domain of the MR, present in the LBD, and that of the GR AF2 domain explains why these two receptors recruit a nearly identical set of transcriptional coactivators. The interaction of transcriptional coactivators with the AF-2 domain occurs in a ligand-binding dependent manner.
In addition to aldosterone, the glucocorticoids, cortisol and corticosterone, bind to and activate the MR. In aldosterone target tissues the expression of the 11β-hydroxysteroid dehydrogenase 2 (HSD11B2) gene ensures that cortisol is deactivated by conversion to cortisone and corticosterone is deactivated by conversion to 11-dehydrocorticosterone. Deoxycorticosterone (DOC; 11-deoxycorticosterone) exhibits some mineralocorticoid action but only about 3% of that of aldosterone.
In the absence of ligand the mineralocorticoid receptor (similarly to the glucocorticoid receptor) remains in the cytosol contained within multiprotein chaperone complexes. The cytosolic MR-containing complex is highly similar to that of the cytosolic GR complexes in that it includes heat-shock protein 90 (hsp90), hsp70, the co-chaperone identified as p23 (encoded by the prostaglandin E synthase 3, PTGES3 gene), and various immunophilins (e.g., FKBP51 and FKBP52), all of which prevent its degradation and assist in its maturation. Upon ligand binding, the MR complex changes its conformation leading to release of the ligand-bound receptor and exposure of the two nuclear localization signals in the MR allowing for rapid transport into the nucleus.
Within the nucleus the ligand bound MR binds to the same type of hormone response element (HRE) to which the GR, progesterone receptor, and androgen receptor binds. As indicated above, this HRE is an imperfect palindrome composed of two 6 bp half sites separated by a 3 bp spacer with the sequences AGAACAnnnTGTTCT. Within the nucleus the MR recruits and interacts with numerous transcriptional coregulators that assist the transcriptional regulatory actions of the MR. The major transcriptional coactivators that interact with the MR include p300/CBP, SRC-1 (steroid receptor coactivator 1), PGC-1α, and ELL.
ELL (Eleven-nineteen Lysine-rich in Leukemia) is so-called as it was a gene on chromosome 19 identified as frequently translocated with the mixed lineage leukemia (MLL) gene on chromosome 11 in acute lymphocytic leukemias, ALL. The ELL gene encodes RNA polymerase II elongation factor that functions as an elongation component of the super elongation complex (SEC), a complex required to regulate small nuclear RNA (snRNA) gene transcription by RNA pol II. The interaction of ELL with the MR represents a selective transcriptional coactivator.
Transcriptional corepressors also interact with the MR including nuclear receptor corepressor 1 (NCoR1: encoded by the NCOR1 gene) and silencing mediator of retinoic and thyroid receptors (SMRT: encoded by the NCOR2 gene).
Initially expression of the aldosterone receptor gene was thought to be restricted to polarized tight epithelial cells such as those present in the nephron of the kidney. However, more recent data demonstrates that the NR3C2 gene is quite ubiquitously expressed and is also expressed in numerous non-epithelial cell types. In additional to the kidneys, the NR3C2 gene is expressed at high levels in the gastrointestinal system, and at moderate levels in the endocrine, reproductive, skeletal, and cardiovascular systems, as well as being expressed at moderate levels in numerous metabolic tissues. Within the central nervous system both the NR3C1 (GR) and NR3C2 (MR) genes are expressed with the NR3C2 expression level being higher than that of NR3C1.
Regulation of Adrenal Steroid Synthesis
Adrenocorticotropic hormone (ACTH), synthesized by corticotropic cells of the anterior pituitary, regulates steroid hormone production in the adrenal cortex, primarily within cells of the zona fasciculata and zona reticularis. ACTH binds to the ACTH receptor present in the plasma membrane of adrenal cortical cells. The ACTH receptor is identified as MC2R for melanocortin-2 receptor. The ACTH receptor is a Gs-type G-protein coupled receptor (GPCR) and ACTH binding triggers activation of adenylate cyclase, elevation of cAMP, and increased PKA. Activation of PKA leads to phosphorylation and activation of hormone sensitive lipase (HSL) leading to increased concentrations of free cholesterol, the substrate for steroid hormone synthesis. The effect of ACTH on the production of cortisol is particularly important, with the result that a classic feedback loop results in cortisol inhibiting ACTH release from the pituitary. The ACTH-cortisol regulatory loop is required to the regulation of the circulating levels of corticotropin releasing hormone (CRH), ACTH, and cortisol.
Aldosterone secretion from the zona glomerulosa is stimulated by an entirely different mechanism. Angiotensin II, and to a lesser extent angiotensin III, stimulate zona glomerulosa cells by binding a plasma membrane G-protein coupled receptor (angiotensin receptor 1; AT1) that is coupled to a Gq-type G-protein that activates phospholipase Cβ (PLCβ).
Angiotensin II is derived from the liver synthesized precursor protein, angiotensinogen, via the concerted actions of kidney-derived renin and the membrane-bound Zn2+-dependent protease, angiotensin-converting enzyme (ACE). Angiotensin III is derived from angiotensin II via the action of another membrane-bound Zn2+-dependent protease, glutamyl aminopeptidase.
Upon angiotensin II binding to AT1 there is a resultant increase in PKC activity and an elevation in intracellular Ca2+ levels. These events lead to increased CYP11A1 (P450ssc) activity and increased production of aldosterone. In the kidney, aldosterone regulates sodium (Na+) retention by stimulating the expression of the mRNA for the Na+/K+–ATPase responsible for the re-accumulation of sodium from the urine.
Potassium is a potent stimulator of aldosterone secretion. Changes in plasma potassium of as little as 0.1mM can cause wide fluctuations (±50%) in plasma levels of aldosterone. Potassium increases aldosterone secretion by depolarizing the plasma membrane of zona glomerulosa cells and opening a voltage-gated calcium channel, with a resultant increase in cytoplasmic calcium and the stimulation of calcium-dependent secretory processes.
Although fasciculata and reticularis cells each have the capability of synthesizing androgens and glucocorticoids, the main pathway normally followed is that leading to glucocorticoid production. However, when genetic defects occur in the three enzyme complexes leading to glucocorticoid production, large amounts of the most important androgen, dehydroepiandrosterone (DHEA), are produced. These mutations lead to hirsutism and other masculinizing changes in secondary sex characteristics in females as is seen in several of the congenital adrenal hyperplasias, CAH.
Pharmacology Affecting Aldosterone
The interplay between renin from the kidney and plasma angiotensinogen is important in regulating plasma aldosterone levels, sodium and potassium levels, and ultimately blood pressure. This hormonal regulatory process is referred to as the renin-angiotensin-aldosterone system, RAAS.
Among the drugs most widely employed to lower blood pressure are the angiotensin converting enzyme (ACE) inhibitors and the angiotensin receptor blockers (ARBs). All drugs that are ACE inhibitors end with the suffix –pril and all drugs that are ARBs end with the suffix –sartan. The ACE inhibitors are potent competitive inhibitors of the enzyme that converts angiotensin I to the physiologically active angiotensin II.
Clinical Significance of Defective Adrenal Steroidogenesis
Defective synthesis of the steroid hormones produced by the adrenal cortex can have profound effects on human development and homeostasis. In 1855 Thomas Addison identified the significance of the “suprarenal capsules” when he reported on the case of a patient who presented with chronic adrenal insufficiency resulting from progressive lesions of the adrenal glands caused by tuberculosis. Adrenal insufficiency is, therefore, referred to as Addison disease. In the absence of steroid hormone replacement therapy, Addison disease can rapidly cause death in a little as 1–2 weeks.
In addition to diseases that result from the total absence of adrenocortical function, there are syndromes that result from hypersecretion of adrenocortical hormones (hypercortisolemia). In 1932 Harvey Cushing reported on several cases of adrenocortical hyperplasia that were the result of basophilic adenomas of the anterior pituitary. Hypercortisolemias that manifest due to adrenocortical hyperplasia are referred to as Cushing syndrome, whereas, hypercortisolemias due to excessive anterior pituitary secretion of ACTH are referred to as Cushing disease.
Despite the characterizations of adrenal insufficiency and adrenal hyperplasia, there remained uncertainty about the relationship between adrenocortical hyperfunction and virilism (premature development of male secondary sex characteristics). In 1942 this confusion was resolved by Fuller Albright when he delineated the differences between children with Cushing syndrome and those with adrenogenital syndromes which are more commonly referred to as congenital adrenal hyperplasias (CAH). The CAH are a group of inherited disorders that result from loss-of-function mutations in one of several genes involved in adrenal steroid hormone synthesis. In the virilizing forms of CAH the mutations result in impairment of cortisol production and the consequent accumulation of steroid intermediates proximal to the defective enzyme. All forms of CAH are inherited in an autosomal recessive manner.
There are two common and at least three rare forms of CAH that result in virilization. The common forms are caused by defects in either CYP21A2 (21-hydroxylase, also identified as just CYP21 or CYP21B) or CYP11B1 (11β-hydroxylase). The majority of CAH cases (90–95%) are the result of defects in CYP21A2 with a frequency of between 1 in 5,000 and 1 in 15,000. Three rare forms of virilizing CAH result from either defects in 3β-hydroxysteroid dehydrogenase (HSD3B2), placental aromatase or P450-oxidoreductase (POR). An additional CAH is caused by mutations that affect either the 17α-hydroxylase, 17,20-lyase or both activities encoded in the CYP17A1 gene. In individuals harboring CYP17A1 mutations that result in severe loss of enzyme activity there is absent sex steroid hormone production accompanied by hypertension resulting from mineralocorticoid excess.
Glucocorticoid Synthesis in the Liver, Adipose Tissue, & Skeletal Muscle
Liver, adipose tissue, and skeletal muscle convert the inactive glucocorticoids, cortisone and 11-dehydrocorticosterone, to the active hormones cortisol and corticosterone via a pathway that is directly controlled by metabolic reactions taking place within the endoplasmic reticulum, ER.
The ER has been recognized for many years as a key organelle responding to changes in nutrient levels. Of particular significance to the role of the ER in nutrient responsiveness are cells of liver (hepatocytes), adipose tissue (adipocytes), and the pancreas (β-cells). Extreme metabolic conditions that include both over feeding and prolonged fasting/starvation result in the activation of ER stress response pathways. One major ER stress induced response to aberrant levels of nutrition is the unfolded protein response (UPR). Activation of the UPR can eventually result in insulin resistance, as is typical in type 2 diabetes, apoptosis, and excess inflammatory responses.
The consumption of excess calories is also associated with the stimulation of the ER-localized pathways of intracellular glucocorticoid activation in many different cells, but particularly in the liver and adipose tissue. The over feeding induced increases in intracellular glucocorticoids plays an important role in the pathology of obesity, the metabolic syndrome, and type 2 diabetes.
With respect to the ER and intracellular glucocorticoid activation, the critical components are glucose-6-phosphate and the ER-localized glucose-6-phosphate dehydrogenase activity, which is referred to as the H form of the glucose-6-phosphate dehydrogenase. The G form of glucose-6-phosphate dehydrogenase is the cytoplasmic enzyme that serves a critical function in the oxidative reactions of the pentose phosphate pathway. The H form of glucose-6-phosphate dehydrogenase activity is identified as hexose-6-phosphate dehydrogenase (encoded by the H6PD gene) and also as glucose 1-dehydrogenase. Whereas the G6PD encoded enzyme resides in the cytosol, the H6PD encoded enzyme resides within the ER and the sarcoplasmic reticulum (SR).
The H6PD gene is located on chromosome 1p36.22 and is composed of 7 exons that generate two alternatively spliced mRNAs encoding precursor proteins of 802 amino acids (isoform 1) and 791 amino acids (isoform 2). The H6PD gene is not expressed in erythrocytes.
Within the ER, hexose-6-phosphate dehydrogenase converts glucose-6-phosphate and NADP+ to 6-phosphogluconate and NADPH in a single step, whereas this process in the cytosol requires two separate enzymes. In addition to glucose-6-phosphate, H6PD can metabolize other hexose-6-phosphates, glucose-6-sulfate, and glucose. One of the primary functions of the ER- and SR-localized NADPH is to maintain redox homeostasis within these organelles. Loss of ER redox homeostasis can lead to ER stress and induction of the unfolded protein response (UPR) which, if severe enough will trigger cell death via the apoptotic pathway.
Another principal function of the NADPH produced by ER-localized hexose-6-phosphate dehydrogenase is to provide the reducing energy to ER-localized reductases, specifically those involved in steroid hormone metabolism, with 11β-hydroxysteroid dehydrogenase 1 (11β-HSD1; encoded by the HSD11B1 gene) being particularly important.
The HSD11B1 gene is located on chromosome 1q32.2 and is composed of 7 exons that generate three alternatively spliced mRNAs all of which encode the same 292 amino acid protein.
The primary function of the HSD11B1 encoded enzyme is to reduce the 11-oxo groups (11-oxoreductase activity) in cortisone and 11-dehydrocorticosterone which generates the active glucocorticoids, cortisol and corticosterone, respectively. Although this enzyme can also inactivate (11β-dehydrogenase activity) cortisol and corticosterone by catalyzing the oxidation reactions converting cortisol to cortisone and corticosterone to 11-dehydrocorticosterone, these inactivating reactions are of minimal significance to intracellular glucocorticoid regulation. The primary determinant of the oxo-reductase activity of HSD11B1 is the ratio of NADPH to NADP+ in the ER. Of clinical significance to the role of ER-localized NADPH is that mutations in the H6PD gene are associated with glucocorticoid deficiency.
Humans express a second 11β-hydroxysteroid dehydrogenase encoding gene identified as HSD11B2. Unlike the enzyme encoded by the HSD11B1 gene, the enzyme encoded by the HSD11B2 gene possesses only the 11β-dehydrogenase activity. The HSD11B2 gene is located on chromosome 16q22.1 and is composed of 7 exons that encode a 405 amino acid protein.
The HSD11B2 gene is expressed primarily in aldosterone-responsive tissues, such as those of the distal tubules of the nephrons of the kidneys. In these tissues the HSD11B2 encoded enzyme is responsible for inactivating cortisol in order to prevent inappropriate activation of the mineralocorticoid receptor (MR). Although the normal receptor for cortisol is the glucocorticoid receptor (GR), the mineralocorticoid receptor has nearly identical affinities for aldosterone (the mineralocorticoid) and cortisol. Mutations in the HSD11B2 gene are associated with an apparent mineralocorticoid excess-induced hypertension due to the lack of ability to inactivate cortisol.
As opposed to the use of NADP+ by the HSD11B1 enzyme in the direction of cortisol and corticosterone oxidation, the HSD11B2 enzyme utilizes NAD+ as its cofactor. In addition to the distal tubule, the HSD11B2 gene is also expressed in the placenta and other tissues whose cells do not express the MR gene. In cells that do not express the MR the function of the HSD11B2 enzyme is to protect those cells from the growth-inhibiting effects of cortisone.
Within adipose tissue and skeletal muscle, both major insulin responsive tissues, the intracellular concentration of glucose-6-phosphate is a direct function of the blood levels of both glucose and insulin. In these tissues, insulin action, through its receptor, results in mobilization of GLUT4 transporters to the plasma membrane leading to enhanced glucose uptake from the blood. In the liver, the uptake of glucose, via the GLUT2 transporter, is solely dependent on glucose levels in the blood and given that the Km of GLUT2 for glucose is high (on the order of 15mM), glucose uptake by the liver only occurs, to a significant degree, during the post-feeding period. As the levels of glucose-6-phosphate rise in these tissues there is a concomitant increase in the activities of the ER-localized proteins that are involved in intracellular glucocorticoid activation. These ER-localized proteins include the H6PD and HSD11B1 encoded proteins, as well as the ER membrane-localized glucose-6-phosphate transporter encoded by the G6PT1 gene. The increase in G6PT1, H6PD, and HSD11B1 activities leads to increased conversion of inactive glucocorticoids to their active forms (cortisol and corticosterone).
Although glucose consumption directly results in increased intracellular glucocorticoid activation, the consumption of fatty acids will indirectly activate this pathway. Free fatty acids are known to interfere with glucose oxidation via the mechanism first proposed by Philip Randle and coworkers in 1963 and now referred to as the glucose-fatty acid cycle. Briefly, the oxidation of fatty acids leads to increased mitochondrial NADH levels which impair the movement of carbon through the TCA cycle resulting in citrate transport to the cytosol which in turn leads to inhibition of the 6-phosphofructo-1-kinase (PFK1) activity of glycolysis. The increased mitochondrial NADH level also inhibits the PDHc reaction further impairing the oxidation of glucose. Thus, one of the effects of over eating, either carbohydrate or lipids, particularly within adipose tissue and skeletal muscle, is enhanced glucocorticoid activation.
When the H6PD gene was knocked out in mice the pathology that resulted included fasting hypoglycemia, low hepatic glycogen content, increased sensitivity to insulin, and decreased negative feedback on the hypothalamic-pituitary-adrenal axis. These results strongly implicate an important role for the triad of G6PT1, H6PD, and HSD11B1 in the metabolic modifications that result in response to feeding. Excess nutrient intake, either in the form of carbohydrate or lipid, can result in increased intracellular glucocorticoid activation, especially in adipose tissue and skeletal muscle, both of which are critical insulin-responsive tissues.
Glucocorticoids have been known for quite some time to induce a state of insulin resistance in both adipose tissue and skeletal muscle. Glucocorticoids interfere with insulin signaling in these tissues resulting in impaired GLUT4 mobilization to the plasma membrane, impaired glucose oxidation, and impaired glycogen synthesis. Within visceral adipose tissue, glucocorticoids stimulate preadipocyte differentiation and triglyceride synthesis. In the liver, glucocorticoids stimulate gluconeogenesis which leads to an exacerbation of the hyperglycemia that is the result of insulin resistance in skeletal muscle and adipose tissue. All of these metabolic disturbances contribute to the development of the metabolic syndrome and the onset of type 2 diabetes.
Gonadal Steroid Hormones
Although many steroids are produced by the testes and the ovaries, the two most important are testosterone and estradiol. These compounds are under tight biosynthetic control, with short and long negative feedback loops that regulate the secretion of follicle stimulating hormone (FSH) and luteinizing hormone (LH) by the pituitary and gonadotropin releasing hormone (GnRH) by the hypothalamus. Low levels of circulating sex hormone reduce feedback inhibition on GnRH synthesis (the long loop), leading to elevated FSH and LH. The latter peptide hormones bind to gonadal tissue and stimulate P450ssc activity, resulting in sex hormone production via cAMP and PKA mediated pathways. The roles of cAMP and PKA in gonadal tissue are the same as that described for glucocorticoid production in the adrenals, but in this case adenylate cyclase activation is coupled to the binding of LH to plasma membrane receptors.
The biosynthetic pathway to sex hormones in male and female gonadal tissue includes the production of the androgens, androstenedione and dehydroepiandrosterone. Testes and ovaries contain an additional enzyme, a 17β-hydroxysteroid dehydrogenase, that enables androgens to be converted to testosterone.
Male Sex Steroid Hormones
In males, LH binds to Leydig cells, stimulating production of the principal Leydig cell hormone, testosterone. Testosterone is secreted to the plasma and also carried to Sertoli cells by androgen binding protein (ABP). In Sertoli cells the Δ4 double bond of testosterone is reduced, by the action of steroid 5α-reductase, producing dihydrotestosterone (DHT).
Testosterone and DHT are carried in the plasma, and delivered to target tissues, by a specific gonadal-steroid binding globulin (GBG). In a number of target tissues, testosterone can be converted to DHT. Dihydrotestosterone is the most potent of the male steroid hormones, with an activity that is 10 times that of testosterone. Because of its relatively lower potency, testosterone is sometimes considered to be a prohormone.
Humans express three different steroid 5α-reductase genes, SRD5A1, SRD5A2, and SRD5A3 where SRD5A3 is the primary enzyme carrying out the testosterone to DHT conversion. Mutations in the SRD5A2 gene are the cause of a form of male pseudohermaphroditism.
In addition to DHT formation, the SRD5A3 encoded enzyme (also called polyprenol reductase) is required for the conversion of polyprenol to dolichol which is necessary for the synthesis of N-linked glycoproteins. Mutations in the SRD5A3 gene are associated with the development of a particular form of congenital disorder of glycosylation (CDG) identified as CDG-Iq.
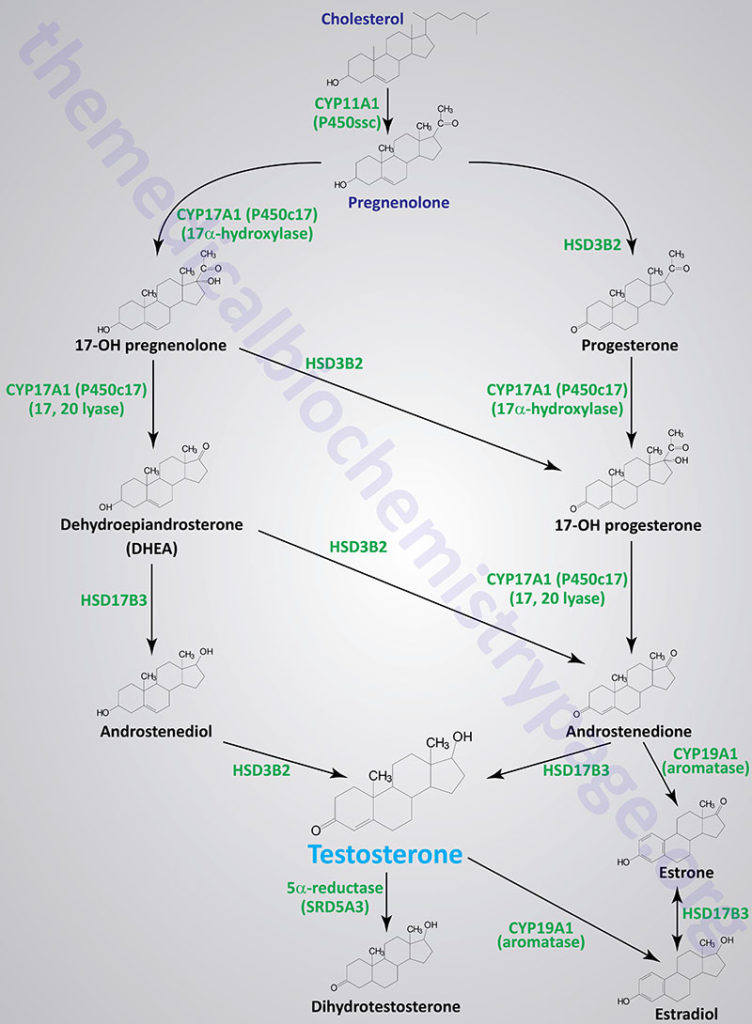
Testosterone is also produced by Sertoli cells but in these cells it is regulated by FSH, again acting through a cAMP- and PKA-regulatory pathway. In addition, FSH stimulates Sertoli cells to secrete androgen-binding protein (ABP), which transports testosterone and DHT from Leydig cells to sites of spermatogenesis. There, testosterone acts to stimulate protein synthesis and sperm development.
The Androgen Receptor
The functions of testosterone and dihydrotestosterone are exerted through their binding to the androgen receptor, AR. The AR is a member of the large family of nuclear receptors that includes the glucocorticoid receptor (GR) and the mineralocorticoid receptor (MR). The AR is encoded by the AR gene that is located on the X chromosome (Xq12) and is composed of 11 exons that generate five alternatively spliced mRNAs.
The AR gene contains two different trinucleotide repeats in the first exon. This exon encodes the N-terminal transcriptional transactivation domain of the AR. These repeats generate polyglutamine (CAG) and polyglycine segments in the AR protein. Expansion of the CAG repeat is pathogenic and the cause of a form of spinal bulbar muscular atrophy (SBMA) commonly called Kennedy disease. The normal size of the CAG repeat is 9-34 repeats and the size found in Kennedy disease patients ranges from 38-62 repeats.
The AR modulates gene expression by interacting with AR response elements (ARE) in target genes. The consensus ARE is a motif (AGAACAnnnTGTTCT) that consists of two hexameric half-sites (5′-AGAACA-3′) often arranged as inverted repeats with three separating nucleotides. Promoter microarray studies have identified more than 1,785 genes that contain the ARE and that are regulated by the actions of AR binding.
In addition to modulation of transcriptional activity through AR binding to target ARE, the AR has been shown to exert non-DNA binding-dependent actions in cells. These effects involve the activation of ERK, AKT, and MAPK signaling pathways. Most of the non-DNA binding effects of AR have been identified via in vitro assays and thus, the physiological relevance of these actions remaining largely unknown.
Functions of Testosterone and Dihydrotestosterone
Dihydrotestosterone exerts numerous effects in the body throughout development, from fetal to adult stages. During embryogenesis the differentiation of male structures is controlled through the concerted actions of DHT, testosterone, and anti-Müllerian hormone (AMH) that inhibit female differentiation.
Peripheral 5α-reductase type 2 (encoded by SRD5A2) converts circulating fetal testosterone to DHT. DHT is essential for the formation of the male external genitalia. DHT promotes proper male differentiation of the urogenital sinus, the genital tubercle, urogenital fold, and labio-scrotal folds. The actions of DHT lead to the formation of the penis, scrotum, and prostate. DHT stimulates gubernacular growth that is required for testicular descent. Indeed, the absence of DHT may lead to ambiguous male external genitalia and undescended testis.
Leydig cells of the testes produce testosterone under the influence of placental human chorionic gonadotropin (hCG). Luteinizing hormone (LH) from the fetal pituitary then takes over testosterone production at around week 16 of gestation. Sex steroids accumulate from testicular production of testosterone in the male fetus and placental production of estrogen in both sexes, causing negative feedback on the fetal pituitary controlling hCG levels in the uterus.
Following birth, the loss of placental estrogen removes negative feedback on the hypothalamic-pituitary-gonadal axis. The loss of the negative feedback results in a transient increase in axis activity in both sexes for the first few months of life. The effect in males leads to a rise in testosterone levels and, as a result, DHT. The negative feedback on the hypothalamic-pituitary-gonadal axis recovers by six months of age keeping the levels of sex hormones low until the onset of the increase in adrenal androgen production referred to as adrenarche.
Adrenarche typically occurs around six years of age in both sexes. At this time there is development of a new layer of adrenal cortical cells, the the zona reticularis, that produces androgens, including testosterone. This increase in testosterone production results in the development of sebaceous and apocrine glands, Testosterone production continues to increase as the zona reticularis continues to mature. Adrenarche results in increased peripheral conversion of testosterone into DHT contributing to pubic hair development.
The onset of puberty is associated with an increase in the activity of the hypothalamic-pituitary-gonadal axis. Hypothalamic secretion of gonadotropin-releasing hormone (GnRH) increases, stimulating pituitary LH secretion, which increases testosterone production from the testes. Increased production of testosterone at puberty is associated with increased conversion to DHT at its target tissues. The actions of this DHT is to promote further growth and maturation of the penis and scrotum. DHT is also the primary androgen responsible for facial hair, body hair, pubic hair, and prostate growth.
Although DHT does not play a significant role in the normal physiology of adult males its effects include prostate enlargement and male pattern hair loss as males age.
Female Sex Steroid Hormones
In females, LH binds to thecal cells of the ovary, where it stimulates the synthesis of androstenedione and testosterone by the usual cAMP- and PKA-regulated pathway. An additional enzyme complex known as aromatase is responsible for the final conversion of the latter two molecules into the estrogens. Aromatase converts androstenedione to estrone and testosterone to estradiol (β-estradiol). Estrone and estradiol are interconvertible via the action of 7-ketoreductase which is also called 17β-hydroxysteroid dehydrogenase type 3 (encoded by the HSD17B3 gene).
Aromatase is encoded by the CYP19A1 gene. Aromatase is a complex endoplasmic reticulum (ER) enzyme found in the ovary and in numerous other tissues in both males and females. Aromatase functions involve hydroxylations and dehydrations that culminate in aromatization of the A ring of the androgens.
The CYP19A1 gene is located on chromosome 15q21.2 and is composed of 18 exons that generate 11 alternatively spliced mRNAs, all of which encode the same 503 amino acid protein.
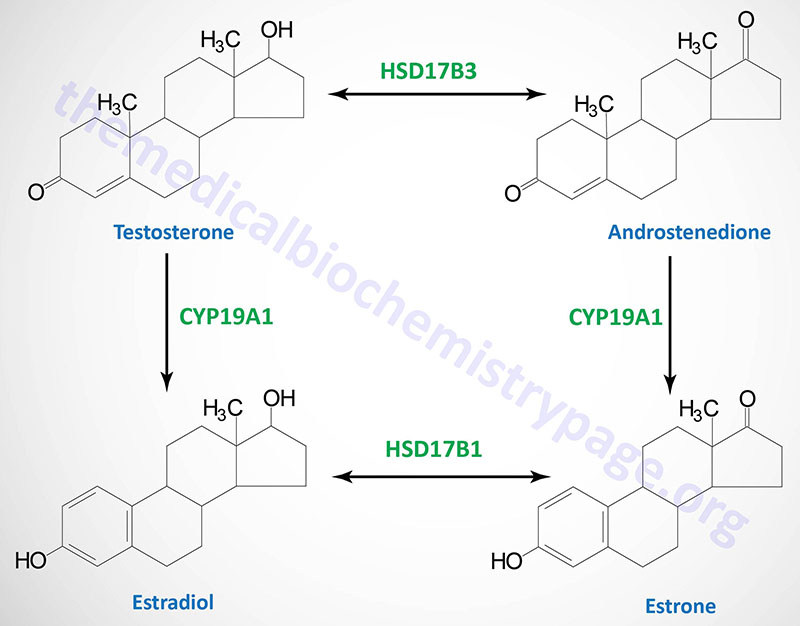
Aromatase activity is also found in granulosa cells, but in these cells the activity is stimulated by FSH. Normally, thecal cell androgens produced in response to LH diffuse to granulosa cells, where granulosa cell aromatase converts these androgens to estrogens. As granulosa cells mature they develop competent large numbers of LH receptors in the plasma membrane and become increasingly responsive to LH, increasing the quantity of estrogen produced from these cells. Granulosa cell estrogens are largely, if not all, secreted into follicular fluid. Thecal cell estrogens are secreted primarily into the circulation, where they are delivered to target tissues by the same globulin (GBG) used to transport testosterone.
The Estrogen Receptor
When discussing the estrogen receptors it is important to understand that both endogenous and exogenous compounds have been identified as being able to bind to and activate these receptors. The primary endogenous estrogenic compounds
are estriol, estrone, and 17β-estradiol, where estriol is the most potent estrogen and 17β-estradiol is the weakest. In addition to these three compounds metabolites of these estrogens have also been shown to regulate the activity of estrogen receptors. Another endogenous compound shown to regulate estrogen receptor activity is 27-hydroxycholesterol. Several exogenous compounds, known as phytoestrogens, such as genistein, daidzein, and equol are known to activate the estrogen receptors.
The major receptors for estrogens are members of the nuclear receptor superfamily. There are two estrogen receptors in this family identified as the estrogen receptor alpha (ERα) and ERβ. The genes encoding these receptors are NR3A1 and NR3A2, respectively. In addition to the nuclear receptors there is an estrogen receptor that is a member of the G-protein coupled receptor (GPCR) family. This receptor was initially identified as GPR30 but is now known as the G-protein coupled estrogen receptor 1 (GPER1).
ERα and ERβ share a significant amino acid sequence homology except in the N-terminal domain which comprises the AF-2 domain. Ligand-bound ERα and ERβ bind to the same DNA sequence termed the estrogen response element (ERE). The ERE is defined by the DNA sequence: GTCAnnnTGACC, where “n” refers to any nucleotide. Even though ERα and ERβ bind to the same ERE and interact with the same coregulators, they exhibit differential tissue distribution and different biological effects.
Several different coactivators (such as NCOA1, NCOA2, NCOA3, CBP, MED1) and corepressors (such as NCOR1, NRIP) can interact with ligand-bound ERα or ERβ and in so doing modify the ability of the receptor to activate or inhibit the expression of target genes.
NCOA1, NCOA2 and NCOA3 are nuclear receptor coactivator 1, 2, and 3, respectively.
CBP is a protein that binds to the cAMP-response element binding protein (CREB) and is closely related to a protein identified as p300. CBP and p300 form one of the histone acetyltransferase families.
MED1 is mediator of RNA polymerase II transcription subunit 1. MED1 is also known as PPARBP.
NCOR1 is nuclear receptor corepressor 1.
NRIP1 is nuclear receptor interacting protein 1.
In addition to their interactions with coactivators and corepressors ERα and ERβ also interact with other transcription factors such as SP1, AP1, and NFκB.
Estrogen-responsive genes include those encoding transcription factors, intracellular signaling molecules, secreted proteins, membrane-associated proteins, enzymes, hormones, and proteases. The large number of estrogen-responsive genes are assumed to directly mediate various estrogen actions in both normal and abnormal (e.g. cancer) tissues.
Functions of Female Sex Hormones
Collectively, the female sex steroid hormones constitute the estrogens and the progestins. The estrogens include estradiol, estrone, and estriol. The progestins include progesterone and 17α-hydroxyprogesterone. Estradiol is the primary estrogen secreted by the ovaries. The majority of estrone in the female body is synthesized from androgens secreted by adrenal cortical cells. Estriol is a very weak estrogen resulting from oxidative reactions on estradiol and estrone. Estradiol is on the order of 12 times more potent as an estrogen than estrone and 80 times more potent than estriol.
Progesterone is the more potent progestin in comparison to 17α-hydroxyprogesterone. In the nonpregnant female secretion of progesterone only occurs at significant levels during the latter half of each ovarian cycle. During pregnancy large amounts of progesterone are produced and secreted by the placenta.
Prior to puberty the synthesis and secretion of the estrogens occurs at minute levels. During puberty the actions of the pituitary gonadotropic hormones stimulate a 20-fold, or higher, increase in estrogen synthesis. The primary functions of the estrogens at puberty are to promote enlargement of the ovaries, the fallopian tubes, the uterus, and the vagina. Within the uterus the estrogens induce changes to the endometrium and increased development of the endometrial glands. Additionally, the estrogen-mediated increases in fat deposition in the external genitalia leads to enlargement of both the labia majora and labia minora. Within the vagina estrogens lead to changes in the epithelial layer making it more resistant to infection and injury.
Estrogens exert several effects related to breast development that relate to the primary function of this tissue being milk production. Estrogens stimulate the development of the stromal tissues of the breast and the development of an extensive ductile system. In addition to the developmental changes, estrogens stimulate fat deposition in the breasts.
Outside the context of the female sex organs and the breasts, the estrogens exert effects on bone development, metabolic processes, and electrolyte balance. In the bone estrogens inhibit osteoclastic activity which promotes bone growth. The effects of estrogens in the bone are, in part, due to activated expression of the gene encoding osteoprotegrin (OPG). Metabolically the estrogens stimulate an increase in whole-body metabolic rate and increase fat deposition in subcutaneous tissues. Protein deposition is also increased in response to the actions of the estrogens. Within the tubules of the kidney the estrogens exert effects similar to those exerted by the adrenal cortical steroid, aldosterone. These effects are sodium and water retention.
Progesterone contributes to the effects of the estrogens in the uterus, fallopian tubes, and the breasts. Progesterone promotes secretory changes to the endometrium of the uterus during the latter half of the ovarian cycle in order to prepare the endometrium for implantation of the fertilized ovum. Within the fallopian tube progesterone promotes increased secretions by the mucosal lining that are necessary for the fertilized ovum as it transit the fallopian tube and prior to implantation. Within the breast progesterone promotes the development of the lobules and alveoli. The effects on the alveoli result in their enlargement and ability to become secretory.
Thyroid Hormones
The thyroid hormones, referred to as the thyronines, are synthesized from the amino acid tyrosine within specialized cells of the thyroid gland. The two major thyroid hormones are triiodothyronine (T3) and thyroxine (T4). Within the periphery the major actions of thyroid hormone are exerted via T3. Synthesis of the thyroid hormones is controlled via the action of the anterior pituitary hormone, thyroid stimulating hormone, TSH (the details of TSH are covered in the Peptide Hormones and Their Receptors page). In addition to pituitary control, synthesis of the thyroid hormones requires iodine uptake into the thyroid gland and incorporation into tyrosine residues in the major thyroid protein, thyroglobulin.
The primary functions for the thyroid hormones are fetal and post-natal development, development of the CNS, modulation of cardiac function through regulation of myocardial contraction and relaxation, renal water clearance, gastrointestinal motility, thermal regulation, energy expenditure, and regulation of lipid metabolism. The thyroid gland also synthesizes the peptide hormone, calcitonin, from parafollicular C cells. However, human calcitonin has no physiological role in humans but the protein is an important marker for thyroid medullary carcinomas.
Table of the Primary Activities of Thyroid Hormone (T3)
| Target Tissue | Primary Effect | Biochemical/Physiological Mechanisms |
| Adipose tissue | catabolic | activation of lipolysis and triglyceride breakdown; increases β-adrenergic receptor density |
| Bone | developmental | promotes bone growth and differentiation |
| Central nervous system | developmental | promotes development of nervous tissue |
| Gastrointestinal system | metabolic | increases carbohydrate absorption |
| Heart | both inotropic and chronotropic | increases density of β1-adrenergic receptors; enhances cardiac responses to catecholamines; enhances ATPase activity of α-myosin heavy chain |
| Liver | metabolic | increases gluconeogenesis and glycogen breakdown; increases cholesterol metabolism; enhances production of LDL receptors |
| Muscle | catabolic | enhances protein breakdown; increases speed of contraction and relaxation; increases β-adrenergic receptor density |
Iodine Homeostasis
Iodine is a critical micronutrient due to its role in the generation of functional thyroid hormones. Dietary intake of iodine is recommended to be 150μg/day for adults and 50-200μg/day for children. In the US, and other developed countries, the use of iodized sodium chloride (salt) ensures an adequate daily intake for most individuals.
The basolateral membrane of thyroid gland cells (thyrocytes) transports iodide into the cell from the circulation. The transporter is called the Na+/I– symporter (NIS) which is encoded by the SLC5A5 gene.
The SLC5A5 gene is located on chromosome 19p13.11 and is composed of 16 exons that encode a 643 amino acid protein.
The NIS transporter move two moles of Na+ and one mole of I– into the thyrocyte. The transporter is able to produce intra-thyrocyte iodide concentrations that are 20-40 fold higher than that in the circulation. In order to continue the uptake of iodide, thyrocytes must transport the Na+ back into the circulation which is catalyzed by a Na+/K+-ATPase.
The expression of the thyrocyte SLC5A5 gene is controlled via the actions of TSH. In addition to regulated expression, TSH controls the migration of NIS into and out of the basolateral membranes of the thyrocyte. Mutations in the SLC5A5 gene result in thyroid dyshormonogenesis type 1 (TDH1). Although the thyroid gland is the primary tissue requiring iodine for its hormonal functions, salivary glands, gastric mucosa, choroid plexus, mammary glands, and the ciliary body of the eye express the SLC5A5 gene.
The incorporation of iodine into tyrosine residues in thyroglobulin occurs in the lumen of thyroid follicles (the colloid) and it is transported across the thyrocyte apical membrane via the action of a Cl–/I–/HCO3– exchanger identified as pendrin (SLC26A4). The SLC26A4 gene is located on chromosome 7q22.3 and is composed of 21 exons that encode a 780 amino acid protein.
The SLC26A4 gene is also expressed in type B intercalated cells of the connecting tubule (CNT) and collecting duct of the renal tubule where it is involved in efflux of HCO3– to the tubular lumen for excretion. Mutations in the SLC26A4 gene are the cause of Pendred syndrome (PDS), also known as thyroid dyshormonogenesis type 2B (TDH2B). PDS is associated with congenital deafness and thyroid dysfunction resulting in goiter.
Thyroid Hormone Synthesis
Chronic stimulation of the thyroid gland, via TSH binding to its receptor on thyrocytes, causes an increase in the synthesis of a major thyroid hormone precursor, thyroglobulin. The thyroglobulin gene (symbol: TG) is located on chromosome 8q24.22 and is composed of 52 exons that encode a 2768 amino acid precursor protein. Mutations in the TG gene are associated with thyroid dyshormonogenesis type 3 (TDH3).
Functional thyroglobulin is a large homodimeric glycoprotein with a molecular weight of 660,000. Although thyroglobulin contains 140 tyrosine residues, only four in each subunit serve as substrates for iodination. Following thyroglobulin synthesis and glycosylation the homodimeric protein is incorporated into exocytotic vesicles. Thyroglobulin is then exocytosed through the apical membrane into the closed lumen of thyroid follicles (the colloid), where it accumulates as the major protein of the thyroid gland and where maturation takes place.
Within the colloid iodide (I–) is oxidized to I+ by thyroid peroxidase (TPO; also called thyroperoxidase) found only in thyroid tissue. The TPO gene is located on chromosome 2p25.3 and is composed of 22 exons that generate six alternatively spliced mRNAs that collectively encode five isoforms of the enzyme. Mutations in the TPO gene are associated with several disorders of thyroid hormone biogenesis which includes congenital hypothyroidism, thyroid hormone organification defect IIA, and congenital goiter.
The oxidation reaction catalyzed by TPO requires hydrogen peroxide (H2O2) which is produced by an NADPH oxidase complex often referred to as thyroid oxidase. TPO and the NADPH oxidase complex are all associated in a large complex at the apical membrane of thyrocytes. The NADPH oxidase is composed multiple subunits encoded by different genes. These genes include dual oxidase 1 (DUOX1) and dual oxidase 2 (DUOX2). Another gene required for the function of the NADPH oxidase complex is DUOXA2 (dual oxidase maturation factor 2) which is involved in the maturation and membrane localization of DUOX2. The activity of the NADPH oxidase is also regulated via the actions of TSH.
The addition of I+ to tyrosine residues of thyroglobulin is catalyzed by TPO at the thyrocyte apical membrane-colloid interface. The products of this reaction are thyroglobulin complexes containing monoiodotyrosyl (MIT) and diiodotyrosyl (DIT) residues. Two molecules of DIT condense to form T4 while a molecule of MIT and one of DIT condense to form T3. Mutations in the TPO gene are associated with thyroid dyshormonogenesis type 2A (THD2A)
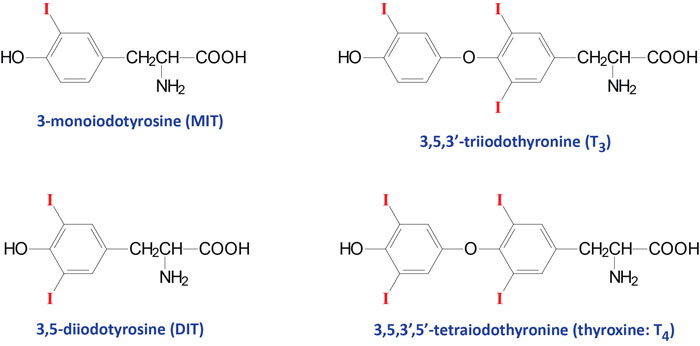
Mature, iodinated thyroglobulin contains approximately three molecules of T4 and one molecule of T3. Following the iodination reactions, thyroglobulin is taken up into vesicles at the colloid-apical membrane interface via a process referred to as pinocytosis. These vesicles then fuse with lysosomes. Lysosomal proteases degrade thyroglobulin releasing T3 and T4, as well as inactive iodotyrosines and amino acids. T3 and T4 are then secreted into the circulation. These compounds are very hydrophobic and require a carrier protein for delivery to target tissues. In the plasma, T3 and T4 are primarily (70%) bound to a carrier glycoprotein known as thyroxin-binding globulin (TBG) and are disseminated throughout the body in this form. In addition to TBG, T3 and T4 can be carried in the blood bound to transthyretin (formerly thyroxine-binding prealbumin) or albumin.
The feedback loop that regulates T3 and T4 production is a single short negative loop, with the T3 and T4 being responsible for down-regulating anterior pituitary TSH secretion. Conversely, continuously secreted hypothalamic thyrotropin-releasing hormone (TRH) is responsible for up-regulating pituitary TSH production. Pituitary thyrotrope secretion of TSH is the net result of the negative effects of T3 and T4 and the positive effect of TRH.
T3 is the more biologically active thyroid hormone and T4 is converted to T3 within peripheral tissues via the actions of a 5′-deiodinase (thyroxine deiodinase type 1; encoded by the DIO1 gene). This deiodinase is also present in the thyroid gland and plays a critical role in overall regulation of iodide homeostasis in this tissue. Deiodination of MIT and DIT also takes place within the thyroid gland. These reactions are catalyzed by an NADPH-dependent flavoprotein (iodotyrosine deiodinase; IYD) which recognizes MIT and DIT but not T3 nor T4. The iodine released from MIT and DIT is reused for hormone biogenesis.
Humans express three different thyroid deiodinase genes identified as DIO1, DIO2, and DIO3. Each of these enzymes contains the modified amino acid, selenocysteine.
The DIO1 gene is located on chromosome 1p32.3 and is composed of 5 exons that generate five alternatively spliced mRNAs. The enzyme encoded by the DIO1 gene, thyroxine deiodinase type I (also called iodothyronine deiodinase type I) is involved in the peripheral tissue conversion of thyroxine (T4) to the more bioactive form, triiodothyronine (T3). In addition to its role in the generation of T3, thyroxine deiodinase I is involved in the catabolism of thyroid hormones.
The DIO2 gene is located on chromosome 14q31.1 and is composed of 7 exons that generate four alternatively spliced mRNAs encoding two distinct protein isoforms. The enzyme encoded by the DIO2 gene, iodothyronine deiodinase type II, is also involved in the conversion of T4 to T3 but does so within the thyroid gland itself. The activity of iodothyronine deiodinase II has been associated with the thyrotoxicosis of Graves disease.
The DIO3 gene is located on chromosome 14q32.31 and is an intronless gene (is a single exon gene) that encodes a protein of 304 amino acids. The enzyme encoded by the DIO3 gene is involved only in the inactivation (catabolism) of T3 and T4. Expression of the DIO3 gene is highest in the female uterus during pregnancy and in fetal and neonatal tissue suggesting a role for this enzyme in the regulation of thyroid hormone levels and functions during early development.
Thyroid Hormone Receptors
Thyroid hormones act by binding to cytosolic receptors of the steroid-thyroid hormone receptor superfamily (nuclear receptors) identified as thyroid hormone receptors (TR). There are two TR receptors designated TRα and TRβ encoded by the THRA and THRB genes, respectively.
The THRA gene is located on chromosome 17q21.1. The THRB is located on chromosome 3p24.2. The mRNAs from both genes are subject to alternative splicing. This results in the TRα1, TRα2, and TRα3 isoforms from the THRA gene and TRβ1 and TRβ2 from the THRB gene. Each of these thyroid hormone receptors possesses the characteristic domains of all members of the nuclear receptor family: ligand-binding domain (LBD), DNA-binding domain (DBD), and activation function domain (AFD).
All of the TR bind to a specific response element in target genes termed the thyroid hormone response element (TRE). The TRE is composed of repeated DNA sequences with different configurations. Evidence indicates that TRs can bind to TREs as monomers or homodimers. However, the major form of the TR bound to a TRE is a heterodimer with retinoid X receptor (RXR). The RXR binding site is upstream of the two directly repeated half-sites of the TRE. The TRE half-sites each contain the sequence T(A/G)AGGTCA as direct repeats separated by a 4 bp spacer. This is referred to as the DR4 element. The RXR response element is GGGGTCA. An important property of TRs is their ability to bind TREs constitutively in the absence of thyroid hormone. In this unliganded state, TR generally represses basal transcription. Binding of thyroid hormone to TR triggers a conformational change in the receptor, resulting in activated transcription of target genes.
Transcriptional activation by TR is mediated not only by ligand binding but by the activity of several coactivator proteins. Steroid receptor coactivator-1 (SRC-1) was the first nuclear receptor coactivator characterized and it has been shown to enhance the activity of ligand-bound TR. The significance of the role of SRC-1 in thyroid hormone function is evident from the fact that loss of this coactivator results in T3 resistance. Several other members of the SRC family of coactivators have been shown to enhance the functions of TR.
Coactivators of the SRC family associate with p300 and CBP. Given that p300 and CBP interact with and mediate the activation of other transcriptional regulation factors it is clear that these proteins are regulators of multiple signal transduction pathways in addition to their roles in steroid/thyroid hormone receptor functions.
Thyroid Hormone Biogenesis Disorders (Congenital Hypothyroidism)
Numerous inherited disorders in the biogenesis of the thyroid hormones have been described. All of these disorders are associated with congenital hypothyroidism. Currently seven distinct gene defects are known that result in this type of disorder. Three of these disorders were indicated in the discussion above, TDH1, TDH2A, and TDH2B.
Table of Thyroid Hormone Biogenesis Disorders
| Dyshormonogenesis Type | Affected Gene | Primary Symptoms / Comments |
| TDH1 | Na+/I– symporter (NIS): SLC5A5 | hypothyroidism appears from birth to early childhood, goiter, most mutations are associated with synthesis of NIS protein harboring residual transporter activity accounting for delay in onset of symptoms |
| TDH2A | thyroid peroxidase, TPO | recurrent goiter, complete iodide release |
| TDH2B | pendrin: SLC26A4 | sensorineural hearing loss, goiter, partial iodide release, enlarged vestibular aqueduct |
| TDH3 | thyroglobulin, TG | large goiters with soft and elastic consistency |
| TDH4 | tyrosine deiodinase, IYD | goiter, continuous iodine and tyrosine loss in the urine, delayed psychomotor development, stunted growth |
| TDH5 | dual oxidase maturation factor 2: DUOXA2 | goiter |
| TDH6 | dual oxidase 2 (DUOX2): thyroid oxidase 2 | partial or defective iodide organification |
Hypo- and Hyperthyroidism
Numerous congenital and acquired forms of hypothyroidism and hyperthyroidism are the result of alterations in the expression, processing, and function of the TSHR. The most common TSHR disorder resulting in hyperthyroidism (thyrotoxicosis) is Graves disease. Graves disease is caused by thyroid-stimulating autoantibodies (TSAb, also called thyroid-stimulating immunoglobulins, TSIs) which bind to and activate the human TSH receptor, leading to the thyrotoxicosis characteristic of this disease. TSAbs bind to the TSH receptor and mimic the TSH stimulation of the thyroid gland by increasing intracellular cAMP. The hyperactivated thyroid then secretes excessive T3 and T4.
Graves disease is classified as a form of thyrotoxicosis, the name for the clinical syndrome resulting from tissues exposed to high levels of thyroid hormones. One theory proposed for the development of the TSAb is that there is a defect in suppressor T cells that allows helper T cells to stimulate B cells to produce thyroid autoantibodies. The clinical features of Graves disease are thyrotoxicosis, goiter (enlarged thyroid gland), an ophthalmopathy in the form of exophthalmos (eyes bulge out), and dermopathy in the form of pretibial myxedema (localized lesions of the skin, primarily in the lower legs, resulting from the deposition of hyaluronic acid).
At the other end of the spectrum are disorders that lead to hypothyroidism. Deficiency in iodine is the most common cause of hypothyroidism worldwide. Indeed the practice of producing iodized table salt was to stem the occurrence of hypothyroidism. When hypothyroidism is evident in conjunction with sufficient iodine intake it is either autoimmune disease (Hashimoto thyroiditis) or the consequences of treatments for hyperthyroidism that are the cause.
In the embryo, thyroid hormone is necessary for normal development and hypothyroidism in the embryo is responsible for cretinism, which is characterized by multiple congenital defects and intellectual impairment. Because the neurological consequences of congenital hypothyroidism are severe, neonatal screening for thyroid hormone levels at birth is routine. Most infants born with congenital hypothyroidism appear normal at birth. However, if left untreated the symptoms will include a thick protruding tongue, poor feeding, prolonged jaundice (which exacerbates the neurological impairment), hypotonia (recognized as “floppy baby syndrome”), episodes of choking, and delayed bone maturation resulting in short stature.
Steroid and Thyroid Hormone Receptors
The receptors, to which steroid and thyroid hormones bind, are ligand-activated proteins that regulate transcription of selected genes. Unlike peptide hormone receptors, that span the plasma membrane and bind ligand outside the cell, steroid/thyroid hormone receptors are found in the cytosol or the nucleus in the absence of ligand. All of these receptors belong to the steroid and thyroid hormone receptor super-family of receptors collectively termed the nuclear receptors.
This large family of receptors includes the androgen receptor (AR, encoded by the AR gene), the progesterone receptor (PR, encoded by the PGR gene), the estrogen receptors (ERα and ERβ, encoded by the ESR1 and ESR2 genes, respectively), the thyroid hormone receptors (TRα and TRβ, encoded by the THRA and THRB genes, respectively), the vitamin D receptor (VDR, encoded by the VDR gene), the retinoic acid receptors (RARα, RARβ, and RARγ, encoded by the RARA, RARB, and RARG genes, respectively), the mineralocorticoid receptor (MR), and the glucocorticoid receptor (GR).
When these receptors bind ligand they undergo a conformational change that renders them activated to recognize and bind to specific nucleotide sequences. These specific nucleotide sequences in the DNA are referred to as hormone-response elements (HREs). When ligand-receptor complexes interact with DNA they alter the transcriptional level (responses can be either activating or repressing) of the associated gene. Thus, the steroid-thyroid family of receptors all have three distinct domains: a ligand-binding domain (LBD), a DNA-binding domain (DBD) and a transcriptional regulatory domain, referred to as the activation function domain (AFD).
Although there is the commonly observed effect of altered transcriptional activity in response to hormone-receptor interaction, there are family member-specific effects with ligand-receptor interaction. Binding of thyroid hormone to its receptor results in release of the receptor from DNA. Several receptors are induced to interact with other transcriptional mediators in response to ligand binding. Binding of glucocorticoid leads to translocation of the ligand-receptor complex from the cytosol to the nucleus.
As indicated above, the receptors for the retinoids (vitamin A and its derivatives) are identified as RAR (for retinoic acid receptors) and exist in at least three subtypes, RARα, RARβ and RARγ. In addition, there is another family of nuclear receptors termed the retinoid X receptors (RXR) that represents a second class of retinoid-responsive transcription factors. The RXR have been shown to enhance the DNA-binding activity of RAR and the thyroid hormone receptors (TRα and TRβ). The RXR represent a class of receptors that bind the retinoid 9-cis-retinoic acid. There are three isotypes of the RXR identified as RXRα, RXRβ, and RXRγ and each of which is encoded by a distinct gene, RXRA, RXRB, and RXRG, respectively. The RXR serve as obligatory heterodimeric partners for numerous members of the nuclear receptor family including PPAR, LXR, and FXR (see below and the Signal Transduction Pathways: Overview page). In the absence of ligand and a heterodimeric binding partner the RXR are bound to hormone response elements (HRE) in DNA and are complexed with corepressor proteins that include a histone deacetylase (HDAC) and silencing mediator of retinoid and thyroid hormone receptor (SMRT) or nuclear receptor corepressor 1 (NCoR).
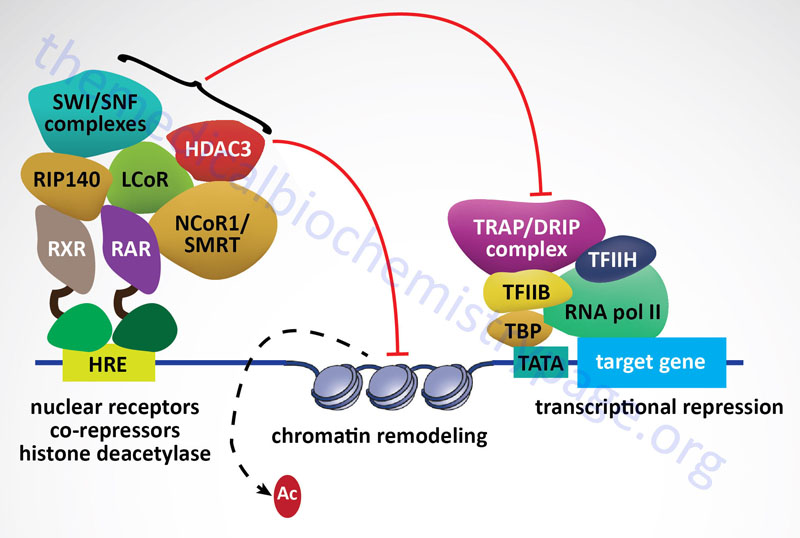
RXRα is widely expressed with highest levels liver, kidney, spleen, placenta, and skin. The critical role for RXRα in development is demonstrated by the fact that null mice are embryonic lethals. RXRβ is important for spermatogenesis and RXRγ has a restricted expression in the brain and muscle. The major difference between the RARs and RXRs is that the former exhibit highest affinity for all-trans-retinoic acid (all-trans-RA) and the latter for 9-cis-RA.
Additional super-family members are the peroxisome proliferator-activated receptors (PPARs). The PPAR family is composed of three family members: PPARα, PPARβ/δ, and PPARγ. Each of these receptors forms a heterodimer with the RXRs. The first family member identified was PPARα and it was found by virtue of it binding to the fibrate class of anti-hyperlipidemic drugs or peroxisome proliferators. Subsequently it was shown that PPARα is the endogenous receptor for polyunsaturated fatty acids. PPARα is highly expressed in the liver, skeletal muscle, heart, and kidney. Its function in the liver is to induce hepatic peroxisomal fatty acid oxidation during periods of fasting.
Expression of PPARα is also seen in macrophage foam cells and vascular endothelium. Its role in these cells is thought to be the activation of anti-inflammatory and anti-atherogenic effects. PPARγ is a master regulator of adipogenesis and is most abundantly expressed in adipose tissue. Low levels of expression are also observed in liver and skeletal muscle.
PPARγ was identified as the target of the thiazolidinedione (TZD) class of insulin-sensitizing drugs. The mechanism of action of the TZDs is a function of the activation of PPARγ activity and the consequent activation of adipocytes leading to increased fat storage and secretion of insulin-sensitizing adipocytokines such as adiponectin.
PPARδ is expressed in most tissues and is involved in the promotion of mitochondrial fatty acid oxidation, energy consumption, and thermogenesis. PPARδ serves as the receptor for polyunsaturated fatty acids and VLDLs. Current pharmacologic targeting of PPARδ is aimed at increasing HDL levels in humans since experiments in animals have shown that increased PPARδ levels result in increased HDL and reduced levels of serum triglycerides.
Genome wide association screening (GWAS) has demonstrated a role for polymorphisms in the PPARγ gene in the etiology of type 2 diabetes. As indicated above, the pharmacologic administration of TZDs is useful in the treatment of the hyperglycemia associated with type 2 diabetes. The TZDs bind to and activate the function of PPARγ ultimately resulting in reductions in circulating triglycerides which secondarily leads to reduced serum glucose levels and subsequently increased insulin sensitivity. It is still not completely clear how impaired PPARγ signaling can affect the sensitivity of the body to insulin or indeed if the observed mutations are a direct or indirect cause of the symptoms of insulin resistance.
In addition to the nuclear receptors discussed here additional family members (discussed in more detail in the Signal Transduction Pathways: Overview page) are the liver X receptors (LXRs, farnesoid X receptors (FXR), the pregnane X receptor (PXR), the estrogen related receptors (ERRα, ERRβ and ERRγ), the retinoid-related orphan receptor (RORα), and the constitutive androstane receptor (CAR).