
Last Updated: November 9, 2025
Introduction to Maroteaux-Lamy Syndrome
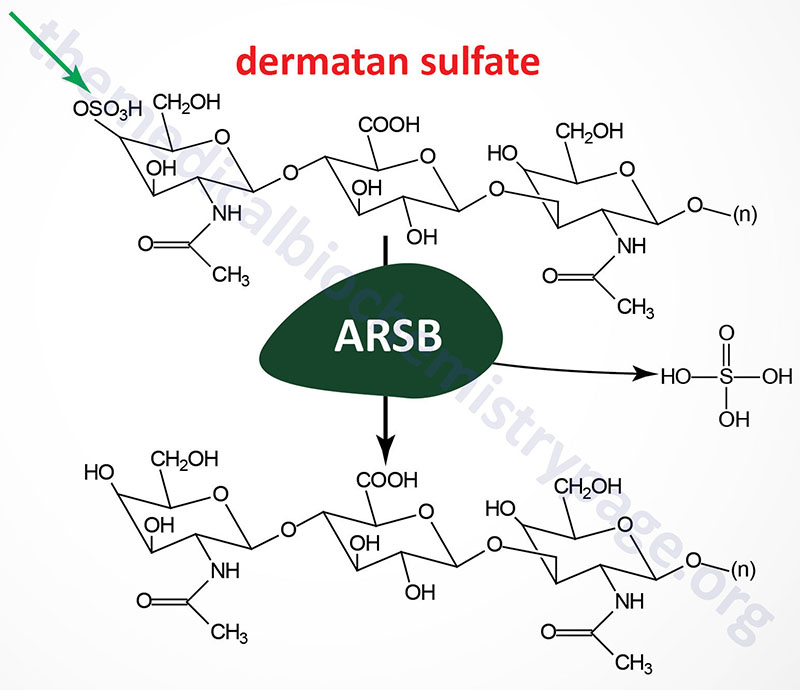
Maroteaux-Lamy syndrome (mucopolysaccharidosis type VI; MPS VI) is an autosomal recessive disorder that belongs to the subfamily of lysosomal storage disorders that were historically identified as the mucopolysaccharidoses. This disorder is characterized by the lysosomal accumulation of dermatan sulfates as a consequence of deficiencies in the lysosomal hydrolase N-acetylgalactosamine 4-sulfatase (arylsulfatase B). Arylsulfatase B is a member of a family of sulfatases, many of which are necessary for the degradation of the glycosaminoglycans.
Molecular Biology of Maroteaux-Lamy Syndrome
Arylsulfatase B is encoded by the ARSB gene. The ARSB gene is located on chromosome 5q14.1 spanning 206 kb and is composed of 17 exons that generate two alternatively spliced mRNAs. The longer precursor protein (isoform a) is a 533 amino acid glycoprotein. The N-terminal region of arylsulfatase B is highly similar to the same domains of arylsulfatase A (cerebroside 3-sulfatase) and arylsulfatase C (steroid sulfatase). Deficiencies in the latter result in ichthyosis.
As of 2025 a total of 137 pathogenic mutations have been identified in the ARSB gene resulting in Maroteaux-Lamy syndrome (MPS VI). The majority of the mutations are frameshift and nonsense mutations leading to loss of expression of functional arylsulfatase B protein.
Clinical Features of Maroteaux-Lamy Syndrome
Maroteaux-Lamy syndrome (MPS VI) was first recognized as a Hurler syndrome-like disorder. The distinction between Hurler and Maroteaux-Lamy syndromes was made on the basis of normal intelligence and lack of heparan sulfate accumulation and excretion in the latter. Although mental development proceeds normally in Maroteaux-Lamy patients, the progression of physical and visual impairments ultimately impedes psychomotor abilities.
The clinical features and severity in Maroteaux-Lamy patients is variable, but usually include short stature, hepatosplenomegaly, stiff joints, corneal clouding, cardiac abnormalities, facial dysmorphism and dystosis multiplex. Dystosis multiplex refers to a constellation of skeletal abnormalities that are characterized by an enlarged skull, thickened calvarium, premature closure of lamboid and sagittal sutures, shallow orbits, enlarged J-shaped sella turcica (a saddle-shaped skull structure into which sits the bottom of the pituitary gland), and abnormal spacing of the teeth with dentigerous cysts. There is anterior hypoplasia of the lumbar vertebrae, the long bone diaphyses are enlarged and an irregular appearance of the metaphyses. The epiphyseal centers not well developed, the pelvis is poorly formed with small femoral heads and coxa valga. The clavicles are short, thick and irregular and the ribs are oar shaped. Phalanges are shortened and trapezoidal in shape.
Hepatosplenomegaly is always present in Maroteaux-Lamy patients after the age of 6. In patients with the most severe forms of the disease there is a shortened trunk, protuberant abdomen and prominent lumbar lordosis (excessive inward curvature of the lower spine). Because of the cardiac involvement, most patients with the severe forms of the disease will die of heart failure by the second or third decade.