

Last Updated: July 9, 2026
Introduction to Isovaleric Acidemia, IVA
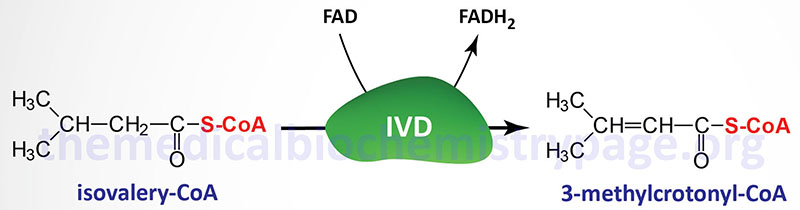
Isovaleric acidemia (IVA) is an autosomal recessive disorder that is a member of the family of disorders referred to as the organic acidemias. Indeed, IVA was the first inborn error of metabolism to be characterized as an organic acidemia. Isovaleric acid is an intermediate in the catabolism of the branch-chain amino acid, leucine that accumulates as a result of mutations in the gene encoding isovaleryl-CoA dehydrogenase (IVD).
Infants that are suffering from the most severe form of isovaleric acidemia, that are diagnosed by an acute episodic encephalopathy, exhibit a characteristic odor of “sweaty feet”. This characteristic odor results from the accumulation of unconjugated isovaleric acid. Screening infants for IVA occurs as a part of a typical new born screening panel for metabolic disorders.
Molecular Biology of Isovaleric Acidemia
Isovaleric acidemia results from mutations in the gene (IVD) encoding isovaleryl-CoA dehydrogenase. Isovaleryl-CoA dehydrogenase is a member of the family of acyl-CoA dehydrogenases (ACAD), several of which are involved in mitochondrial β-oxidation of fatty acids, and as such is also known as acyl-CoA dehydrogenase 2 (ACAD2).
The IVD gene is located on chromosome 15q15.1 and is composed of 15 exons that generate seven alternatively spliced mRNAs, each of which encodes a unique protein isoform. One of the IVD encoded proteins (isoform 3) initiates translation from an alternative in-frame AUG codon.
Analysis of the IVD gene in numerous IVA patients has shown that mutations include missense and nonsense mutations. These mutations result in five distinct classes of enzyme variants that differ in subunit size. These mutant IVD enzymes exhibit residual enzyme activity that is on the order of 0% to 3% of wild-type enzyme activity.
Clinical Features of Isovaleric Acidemia
Isovaleric acidemia is divided into two distinct symptomatic forms, a severe lethal form and a chronic intermittent form. The severe lethal form of IVA is the most common. In the severe neonatal lethal form infants suffer from massive metabolic acidosis within a few days of birth accompanied by hyperammonemia, encephalopathy, lethargy, poor feeding, vomiting, and the characteristic “sweaty foot” odor. Metabolic ketoacidosis with an unexplained anion gap is typical in infants suffering from the severe lethal form of IVA. Early neonatal presentation is highly associated with mortality. If properly diagnosed, infants have far better outcomes with as many as 65% of infants attaining near normal cognitive development.
In the chronic intermittent form afflicted individuals suffer from periodic attacks of severe ketoacidosis with the intervening periods being symptom free. The chronic intermittent form presents later in childhood with developmental delay in addition to the recurrent episodes of ketoacidosis. The frequency of metabolic decompensation is more frequent in the neonatal period, decreasing with age.
Metabolic Analysis in Isovaleric Acidemia
Analysis of organic acids in IVA patients typically finds metabolic byproducts of isovaleryl-CoA such as isovalerylglycine and 3-hydroxyisovaleric acid. Additional abnormal levels of metabolites can include lactic acid, 3-hydroxybutyric acid, acetoacetic acid, 3-hydroxyisovaleric acid, and isovalerylglutamic acid. Isovaleryl-CoA is generally conjugated and excreted as nontoxic isovalerylglycine.
During events which trigger episodes of metabolic decompensation, that includes infections, pregnancy, or increased protein consumption or breakdown, the concentration of isovaleryl-CoA exceeds the capacity of the liver to esterify metabolites resulting in accumulation of isovaleryl-CoA, isovaleric acid, and 3-hydroxyisovaleric acid. Isovaleryl-CoA inhibits the activity of N-acetylglutamate synthase (NAGS), resulting in a form of secondary NAGS deficiency. NAGS is required to generate the critical activator (N-acetylglutamate) of the urea cycle enzyme, carbamoyl phosphate synthetase 1 (CPS1). Lack of active CPS1 results in impairment of the urea cycle resulting in the hyperammonemia typical in IVA infants. Accumulation of isovaleryl-carnitine is also found in IVA infants.
Treatment of Isovaleric Acidemia
Treatment of IVA includes protein restriction to reduce the load of the toxic metabolites derived from the catabolism of leucine. In addition, supplementation with glycine and/or carnitine promotes the synthesis and excretion of the less toxic isovaleryl conjugates. Infants should also be fed a leucine restricted metabolic formula. Common childhood illnesses in IVA patients should be attended to with prompt preventive measures to prevent poor outcomes and to reduce morbidity and mortality.