
Last Updated: December 16, 2025
The Pyruvate Dehydrogenase Complex (PDHc)
As the name of the disorder implies, the pyruvate dehydrogenase complex deficiencies represent a family of disorders caused by mutations in genes encoding subunits of the pyruvate dehydrogenase complex, PDHc.
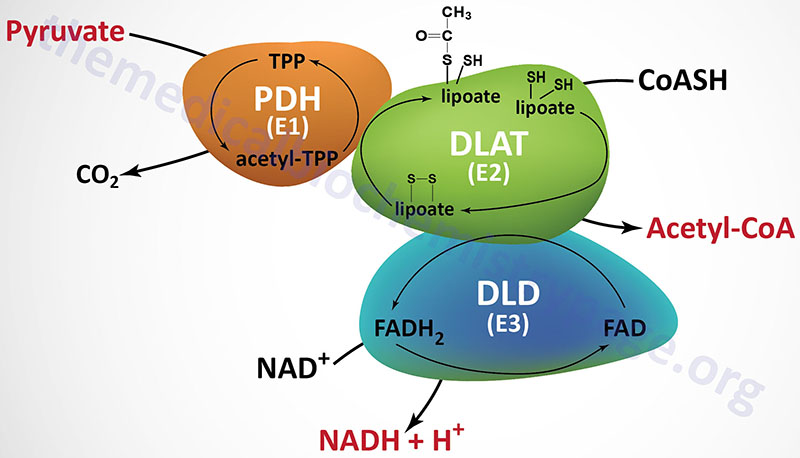
PDHc is a mitochondrial enzyme complex responsible for the oxidation of pyruvate to acetyl-CoA. The acetyl-CoA can then be fully oxidized to CO2 and H2O in the TCA cycle. The PDHc is composed of multiple copies of three distinct enzyme activities: pyruvate dehydrogenase (PDH, identified as the E1 component), dihydrolipoamide S-acetyltransferase, (DLAT, identified as the E2 component), and dihydrolipoamide dehydrogenase, (DLD, identified as the E3 component).
The PDH activity (E1) is a heterotetrameric complex composed of two α (alpha) and two β (beta) subunits.
The α subunits are primarily encoded by the PDHA1 gene. The PDHA1 gene is located on chromosome Xp22.12 and is composed of 12 exons that generate four alternatively spliced mRNAs. The predominant PDHA1 derived mRNA (isoform 1) encodes a precursor protein of 390 amino acids. Another PDH α-subunit gene (PDHA2) encodes a testis-specific isoform.
The β subunits are encoded by the PDHB gene. The PDHB gene is located on chromosome 3p14.3 and is composed of 10 exons that generate three alternatively spliced mRNAs, each of which encode a distinct protein isoform.
The DLAT gene is located on chromosome 11q23.1 and it is composed of 14 exons that generate 13 alternatively spliced mRNAs. Each of these 13 mRNAs encode distinct protein isoforms of the DLAT enzyme.
The DLD gene is located on chromosome 7q31.1 and it is composed of 14 exons that generate four alternatively spliced mRNAs with the largest mRNA encoding a protein of 509 amino acids. The DLD encoded proteins function within the PDHc as homodimeric subunits. The DLD encoded proteins are also functional components of the 2-oxoglutarate dehydrogenase complex (more commonly known as α-ketoglutarate dehydrogenase) of the TCA cycle, and of the branched-chain keto-acid dehydrogenase complex (BCKD, an amino acid metabolism enzyme complex).
In addition to the catalytic components of the PDHc there is a non-catalytic component termed component X (PDHX) of the PDHc which is also called E3 binding protein. The PDHc component X protein tethers the E3 subunits of the PDHc to the E2 subunits. The PDHX gene is located on chromosome 11p13 and is composed of 12 exons that generate three alternatively spliced mRNAs, each of which encode distinct protein isoforms. The protein encoded by the isoform 1 mRNA is the largest of the three proteins (501 amino acids). The protein encoded by the isoform 2 mRNA is translated from an upstream in-frame AUG codon and the protein encoded by the isoform 3 mRNA lacks 227 amino acids that are present in the central region of the protein encoded by the isoform 1 mRNA.
Genetics of Pyruvate Dehydrogenase Complex Deficiencies
Given that the PDHc is localized to the mitochondria and that all of the protein components are encoded by nuclear genes, deficiencies of PDHc activity are referred to as nuclear-encoded mitochondrial disorders. PDHc deficiencies represent a major cause of neonatal encephalopathies associated with primary lactic acidosis. Elevated lactate accompanied by elevated pyruvate with a normal lactate-to-pyruvate ratio is a hallmark of PDHc deficiency.
PDHc deficiency can be caused by mutations in any of the genes encoding the principal subunits of the complex: the E1α encoding PDHA1 gene, the E1β encoding PDHB gene, the E2 encoding DLAT gene, or the E3 encoding DLD gene. In addition, PDHc deficiency is associated with mutations in the PDHX gene, the pyruvate dehydrogenase phosphatase catalytic subunit 1 (PPM2C) encoding gene PDP1, and the lipoic acid synthetase encoding gene LIAS.
The PDP1 gene is located on chromosome 8q22.1 and is composed of 6 exons that generate four alternatively spliced mRNAs that collectively encode two distinct protein isoforms.
The LIAS gene is located on chromosome 4p14 and is composed of 11 exons that generate six alternatively spliced mRNAs, each of which encode a distinct protein isoform.
The most common (84%) causes of PDHc deficiency are mutations in the PDHA1 gene that encodes the E1α subunit. As indicated above, the PDHA1 gene is localized to the X chromosome. Although genetically defined as an X-linked recessive disorder, PDHA1 deficiency is considered an X-linked dominant condition given that both males and females manifest the symptoms of PDHc deficiency. Normally, females are very rarely affected by X-linked recessive disorders but this is not the case with PDHA1 mutations. The reason for this is that there is skewing of X chromosome inactivation such that females with one mutant PDHA1 gene appear to have the X chromosome harboring the wild-type PDHA1 gene inactivated at a higher rate than predicted based upon the normal random X-inactivation process.
At least 37 different mutations, including deletions, insertions, and point mutations have been identified in the PDHA1 gene. Most missense mutations are found in exons 1-9 and most frameshift mutations are found in exons 10 and 11. Most of the reported mutations in the PDHA1 gene were unique. Males and females with PDHA1 mutations have been found to be equally represented. However, almost all of the missense mutations have been identified in males and almost all of the deletions or insertions have been identified in females. It is speculated that insertions and deletions in hemizygous males may cause intrauterine death.
With respect to the PDHA1 gene the most frequently reported mutations were found at amino acid positions 263, 302, and 378 with most of the mutations being missense mutations. Again, males account for the majority (83%) of patients harboring missense mutations in PDHA1. One of the most significant amino acid positions in PDHA1, with respect to disease, is amino acid position 378. Mutations in this amino acid may be particularly lethal resulting in death within the first two years of life. In contrast, patients harboring missense mutations at either position 263 or 302 appeared to have the lowest rate of lethality.
Mutations in the PDHB gene, that encodes the E1β subunit of the PDHc, account for only a small percentage of cases of PDHc deficiency. Though the severity of enzyme deficiency is similar to PDHA1, PDHB mutations also present with considerable phenotypic variability and severity.
When cells from PDHc deficient patients were analyzed in culture a rather striking finding was observed. In PDHc deficient fibroblasts it was found that there is overexpression of hypoxia inducible factor 1α (HIF1α). HIF1α is a transcription factor that regulates the expression of numerous genes involved in critical pathways of growth control and cell metabolism such as those encoding glucose transporters and most enzymes in the pathway of glycolysis. Although the precise mechanism for the overexpression of HIF1α in PDHc deficiency is not fully understood, it may be due to increased levels of pyruvate which leads to stabilization of HIF1α by inhibiting its degradation by cytoplasmic prolylhydroxylases. The increased levels of HIF1α create a positive feedback loop in PDHc deficiency due to the HIF1α-mediated increase in glycolytic flux.
Clinical Features of Pyruvate Dehydrogenase Complex Disorders
In patients with PDH deficiency the major presentations encompass metabolic and neurologic pathologies with both occurring with equal frequency. The metabolic form presents as severe lactic acidosis in the newborn period which very often leads to lethality. PDHc deficiency accounts for about 15% of known causes of congenital lactic acidosis. The common neurologic pathologies in PDH deficient patients are hypotonia, lethargy, spasticity, intellectual impairment, and the development of seizures. Between these two extremes of metabolic lethality and severe neurological deficit there is a continuous spectrum of intermediate forms characterized by intermittent episodes of lactic acidosis associated with cerebellar ataxia. The symptoms of many patients with PDH deficiency fit into the category of Leigh syndrome. Leigh syndrome represents a large family of related disorders characterized by subacute necrotizing encephalopathy. Leigh syndrome patients present in the first year of life with and characteristically develop rapidly progressing psychomotor impairment and death within the first 2-3 years of life.
With respect to PDH deficiency, the more severe the PDHc defect, the more severe is the accompanying clinical illness. In addition, the earlier the onset of symptoms of PDH deficiency, the more rapid will be the progression of the disease resulting in more widespread damage in the central nervous system. Patients with less than 15–20% of normal PDHc activity generally present with lactic acidosis of infancy and severe neurological deficits. Patients with 40–50% residual PDHc activity have a milder illness in which intermittent ataxia is the most prominent neurological symptom.
Therapeutic Intervention for PDHc Disorders
There is currently no effective treatment for patients with PDHc deficiency. One strategy that has had variable success is the use of a ketogenic diet, supplementation with thiamine, and administration of dichloroacetate. In typical ketogenic diets the majority of fat is in the form of long-chain saturated fatty acids in triglycerides. The reduced glucose intake in the ketogenic diet result in a shift away from glycolysis, particularly in the brain, and toward the utilization of the ketones. The high fat content of the ketogenic diet drives ketone synthesis in the liver. The benefit of thiamine supplementation is in maximizing any residual PDHc activity, a complex that requires thiamine pyrophosphate in addition to several other vitamin-derived co-factors. Dichloroacetate inhibits the pyruvate dehydrogenase kinases (PDK) which normally phosphorylate and inhibit the PDHc. Thus, inhibition of the PDKs will prevent any inhibition of residual PDH activity.