

Last Updated: June 3, 2026
Introduction to Carnitine Deficiency, Systemic Primary (CDSP)
Carnitine deficiencies lead to an inability to transport long-chain fatty acids (as fatty acylcarnitines) into the mitochondria for oxidation. Carnitine deficiencies can occur in newborns and particularly in pre-term infants. Carnitine deficiencies are also found in patients undergoing hemodialysis or those affected by any one of various organic acidemias/acidurias (e.g. propionic acidemia or isovaleric acidemia).
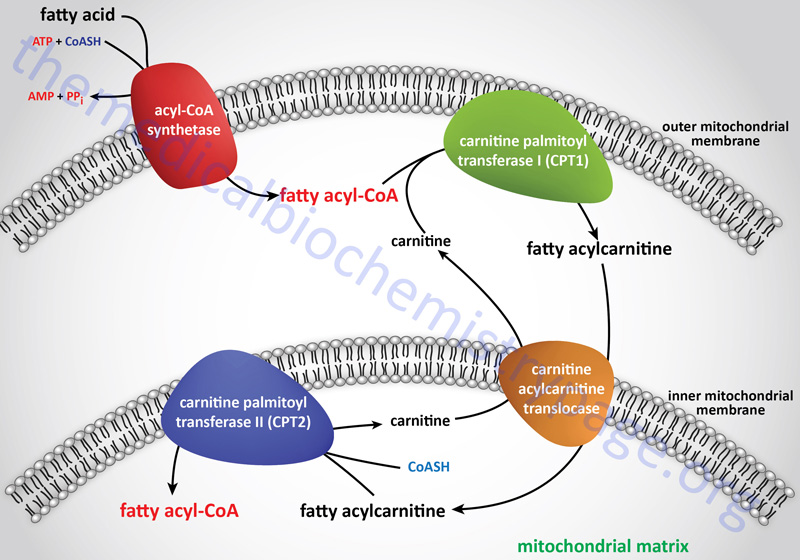
Deficiencies in carnitine are defined as either primary or secondary. Primary carnitine deficiencies are due to defects in carnitine transport into cells or due to defects in synthesis from lysine, although the latter are very rare with only a few cases identified worldwide. Mutations in the major carnitine transporter, encoded by the SLC22A5 gene [also known as zwitterion/cation transporter 2 (OCTN2)], are the predominant causes of primary carnitine deficiency. This disorder is most correctly referred to as carnitine deficiency, systemic primary (CDSP).
Secondary carnitine deficiencies are most commonly the result of trapping of carnitine in fatty acylcarnitine due to defects in mitochondrial fatty acid β-oxidation enzymes such as is the case in medium-chain acyl-CoA dehydrogenase deficiency (MCADD).
Primary carnitine deficiency (carnitine deficiency, systemic primary: CDSP) is an autosomal recessive disorder that results from the lack of function of the carnitine transporter, OCTN2. Within the US, Europe, and Japan the estimated frequency of carnitine deficiency, primary systemic is 1:40,000 live births. The frequency in Australia has been estimated to be between 1:37,000 and 1:100,000.
Due to loss of functional OCTN2, primary carnitine deficiency patients exhibit urinary carnitine wasting. The loss of carnitine in the urine leads to very low serum carnitine levels ranging from 0 mM to 5 mM where the normal value should be 25 mM–50 mM. In addition to low serum levels of carnitine these patients have decreased intracellular carnitine accumulation.
As indicated, the major function of carnitine is to facilitate the transport of long-chain fatty acids (as fatty acylcarnitines) into the matrix of the mitochondria where the carnitine is subsequently exchanged for coenzyme A. Once inside the mitochondria the fatty acyl-CoAs can be oxidized for energy production. Therefore, primary carnitine deficiency results in defective fatty acid β-oxidation. The lack of adequate energy production, particularly in the liver, leads to deficient glucose synthesis via gluconeogenesis and a consequent increase in glucose oxidation. Due to the increase in glucose oxidation for energy production there is the potential for severe hypoglycemia. The hypoglycemia stimulates glucagon release from the pancreas and epinephrine release from the adrenal glands leading to increased fatty acid release from adipose tissue. Due to the lack of oxidation, the fatty acids accumulate in the liver, skeletal muscle, and heart resulting in hepatic steatosis, myopathy, and cardiomyopathy, respectively.
Molecular Biology of Primary Carnitine Deficiency
The SLC22A5 (OCTN2) gene is located on chromosome 5q31.1 and is composed of 11 exons that generate two alternatively spliced mRNAs encoding proteins of 581 amino acids (isoform a) and 557 amino acids (isoform b).
Over 100 mutations have been identified in the SLC22A5 gene resulting in carnitine deficiency, systemic primary. Most of the mutations in SLC22A5 are missenses mutations but nonsense, splice site, and deletion/insertion mutations have also been identified.
The most commonly identified mutation is the change of the C residue at position 136 to a T (c.136C>T). This mutation results in the change of proline at amino acid position 46 to a serine and this mutation is designated P46S (p.P46S). Most families with members suffering from carnitine deficiency, systemic primary have been identified to harbor private mutations. In a few cases the mutations causing the disorder have been found to occur at mutation-prone DNA sequences in the SLC22A5 gene.
Clinical Features of Primary Carnitine Deficiency
The typical metabolic presentation in patients with primary carnitine deficiency occurs most frequently before 2 years of age. Infants with primary carnitine deficiency exhibit poor feeding and can become lethargic and minimally responsive. The presentation of primary carnitine deficiency is quite variable with respect to the age of onset, the severity of the symptoms, and the involved organs.
The inability to utilize fat for energy during stress or fasting can trigger acute metabolic decompensations early in life. In most cases, due to the fatty infiltration of the liver, these infants will present with hepatomegaly. Blood laboratory studies will find hypoglycemia and hyperammonemia while urinalysis will show hypoketonuria. Liver function enzymes (AST and ALT) will be variably elevated and creatine kinase (CK) may also be found to be mildly elevated.
Failure to treat primary carnitine deficiency infants promptly with intravenous glucose during periods of metabolic decompensation will result in progression to coma and death. In older patients with primary carnitine deficiency there is a strong likelihood for skeletal myopathy, cardiomyopathy sometimes associated with hypotonia, and arrhythmias leading to sudden death.
The key diagnostic for primary carnitine deficiency, systemic primary is the measurement of plasma carnitine levels. Free and acylated carnitine are extremely reduced where free carnitine is usually <5 mM (normal 25 mM–50 mM). Urine organic acids do not show any consistent anomaly, although in some patients a non-specific dicarboxylic aciduria has been reported. Following assessment of serum carnitine levels a diagnosis of carnitine deficiency, systemic primary is confirmed by demonstrating reduced carnitine transport in skin fibroblasts that have been isolated from the patient. The level of carnitine transport in cultured patient fibroblasts is correlated to the severity of the mutation in the SLC22A5 gene. Nonsense mutations have been found to be associated with the absence of any carnitine transport activity.
Treatment of Primary Carnitine Deficiency
Treatment of primary carnitine deficiency patients with dietary carnitine supplementation (100–400 mg/kg/day) has been shown to be highly beneficial, particularly if started before irreversible organ damage occurs. The precise dose of carnitine to be utilized should be adapted to each individual patient and determine by serial measurements of plasma carnitine levels. The long term prognosis for carnitine deficiency, systemic primary patients is favorable provided the patients remain on carnitine supplements.