

Last Updated: July 13, 2026
Essential Fructosuria
Essential fructosuria is a benign metabolic disorder caused by the lack of fructokinase (formally termed ketohexokinase, KHK) which is normally present in the liver, pancreatic islets and kidney cortex. The fructosuria of this disease depends on the time and amount of fructose and sucrose intake. Since the disorder is asymptomatic and harmless it may go undiagnosed.
Molecular Biology of Essential Fructosuria
There are two forms of ketohexokinase (KHK) in mammals that result from alternative splicing of the KHK gene. These two isoforms are called KHK-A (fructokinase A) and KHK-C (fructokinase C). The KHK gene is located on chromosome 2p23.3 and is composed of 9 exons that encode the two alternatively spliced mRNAs encoding KHK-A and KHK-C.
The majority of pathogenic variants in the KHK resulting in essential fructosuria are missense variants. Almost all patients harbor one of two common variants which are a substitution of the glycine at amino acid position 40 for an arginine (G40R) or the substitution of the alanine at amino acid position 43 for a threonine (A43T).
Hereditary Fructose Intolerance
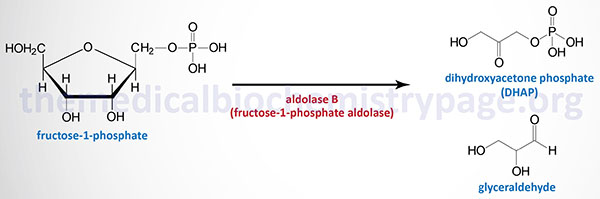
Hereditary fructose intolerance (HFI) is a potentially lethal autosomal recessive disorder resulting from a lack of aldolase B which is normally expressed in the liver, small intestine, and kidney cortex.
The incidence of HFI is on the order of 1 in 20,000 live births. The disorder is characterized by severe hypoglycemia and vomiting following fructose intake. Prolonged intake of fructose by infants with this defect leads to vomiting, poor feeding, jaundice, hepatomegaly, hemorrhage, and eventually hepatic failure and death. Patients will remain symptom free on a diet devoid of fructose and sucrose.
HFI patients will also develop metabolism dysfunction-associated fatty liver disease (MAFLD). MAFLD is also known as metabolic dysfunction-associated steatotic liver disease, MASLD. MAFLD was formerly referred to as non-alcoholic fatty liver disease, NAFLD. In HFI patients, the MAFLD is not associated with obesity and insulin resistance as is the case in the more commonly occurring forms of MAFLD. In addition, HFI patients present with hypoglycemia, whereas in obesity-associated MAFLD there is an associated hyperglycemia.
The biochemistry of aldolase B deficiency is complex due to the fact that this enzyme can catalyze three distinct reactions. Under normal conditions, the tissues that express aldolase B utilize this enzyme for the cleavage of fructose-1,6-bisphosphate (F1,6BP) within the context of glycolysis. Like aldolase A during gluconeogenesis, aldolase B can also catalyze the condensation of dihydroxyacetone phosphate (DHAP) and glyceraldehyde-3-phosphate to form F1,6BP in the gluconeogenic direction. Since the liver, kidney, and small intestine all contribute to endogenous glucose production (gluconeogenesis), this action of aldolase B is physiologically significant. With consumption of sucrose or fructose, the ability of aldolase B to cleave hepatic fructose-1-phosphate, generated via the fructokinase reaction, to glyceraldehyde and DHAP becomes physiologically relevant.
The primary cause of the manifesting symptoms in HFI is the trapping of inorganic phosphate (Pi) in fructose-1-phosphate and the consequent reduction in the pool of ATP via the fructokinase reaction. The trapping of the inorganic phosphate pool and ATP depletion leads to global reduction in all cellular processes that rely on phosphorylation or ATP. The loss of the inorganic phosphate pool impairs glycogen breakdown due to the role of Pi as a substrate for the phosphorolytic action of hepatic glycogen phosphorylase. This, therefore, contributes to the severe hypoglycemia upon ingestion of fructose or sucrose.
Also contributing to the severe hypoglycemia in HFI patients is the increased levels of fructose-1-phosphate (F1P). Hepatic glucose is phosphorylated by glucokinase, a member of the hexokinase family of enzymes, which is specific for glucose as its substrate. The activity of glucokinase is regulated by the protein identified as glucokinase regulatory protein (GKRP) encoded by the GCKR gene. During the fasting state, glucokinase is “held” in the nucleus by interaction with GKRP. This localization prevents glucokinase access to cytosolic glucose until it is released from GKRP.
Fructose-1-phosphate, derived from the action of hepatic fructokinase phosphorylating fructose, stimulates the release of glucokinase from GKRP. Indeed, the ability of F1P to stimulate release of glucokinase from GKRP ultimately contributes to the potentially lethal hypoglycemia associated with HFI. This latter effect results from inappropriate release of glucokinase to the cytosol leading to the phosphorylation of glucose, thereby, trapping the glucose within hepatocytes. The release of glucokinase from GKRP is also regulated by phosphorylation of GKRP. The phosphorylation of GKRP occurs through the action of AMPK whose activity rises as the energy charge falls (increasing AMP levels). Therefore, since the energy charge falls rapidly upon hepatic fructose metabolism there is a rapid release of glucokinase from GKRP and increased trapping of glucose within hepatocytes.
The depletion of the inorganic phosphate pool in HFI also activates AMP deaminase resulting in increased nucleotide catabolism. This latter effect is the cause of the hyperuricemia associated with HFI. As the level of fructose-1-phosphate increases, it inhibits the fructokinase reaction in a feedback mechanism. Inhibition of fructokinase leads to reduced hepatic fructose uptake contributing to the fructosemia of HFI.
Molecular Biology of Hereditary Fructose Intolerance
Aldolase B is encoded by the ALDOB gene. The ALDOB gene is located on chromosome 9q31.1 and is composed of 9 exons that encode a 364 amino acid protein. More than 50 different pathogenic variants have been identified in the ALDOB gene in patients with hereditary fructose intolerance. Most pathogenic variants in the ALDOB gene are missense variants.
Very nearly half of all HFI patients harbor a specific pathogenic variant that results in the change of alanine at position 149 with proline (A149P). Two additional common missense variants found in HFI patients are the A150P and A174D variants.
Fructose-1,6-Bisphosphatase Deficiency
Hereditary fructose-1,6-bisphosphatase (F1,6BPase) deficiency is a very rare autosomal recessive disease. The disease results from inherited defects in the gene (FBP1) encoding the hepatic form of F1,6BPase.
Loss of fully functional FBP1 results in severely impaired hepatic gluconeogenesis and leads to episodes of hypoglycemia, apnea, hyperventilation, ketosis and lactic acidosis, seizures, hepatomegaly, hyperlipidemia, hepatosteatosis, and liver damage. The severity of these symptoms increases when carbohydrate is insufficient in the diet. These symptoms can take on a lethal course in neonates particularly in the absence of carbohydrate intake. Later in life episodes are triggered by fasting and febrile infections. Because of the existence of two FBP genes (FBP1 and FBP2) it is not possible to utilize assays for F1,6BPase deficiency in the white blood cells of suspected patients as a sole means of diagnosis, patients must undergo molecular analysis of the FBP1 gene.
Molecular Biology of Fructose-1,6-Bisphosphatase Deficiency
Humans express two F1,6BPase genes with one expressing the liver version of the enzyme (the FBP1 gene) and the other expressing a muscle version of the enzyme (the FBP2 gene). The FBP1 gene is located on chromosome 9q22.32 and is composed of 8 exons that generate two alternatively spliced mRNAs, both of which encode the same 338 amino acid protein. Exon 1 of the FBP1 gene is non-coding. In patients with F1,6BPase deficiency deletions of exons 2, 3, and 8, as well as deletion of exons 3 through 7 have been identified. In addition, deletion of the entire FBP1 gene has been found in some patients. Common missense, nonsense, and frameshift variants have also been identified in the FBP1 gene in F1,6BPase deficient patients.
The FBP2 gene is located at the same chromosomal location as the FBP1 gene but is composed of 7 exons that encode a protein of 339 amino acids. The liver and muscle F1,6BPase enzymes share 77% amino acid sequence identity.