
Last Updated: July 13, 2026
Introduction of Homocyst(e)inemia/Homocyst(e)inuria
Homocysteinemias (homocystinemias) represent a family of autosomal recessive disorders resulting from defects in several of the genes involved in the conversion of methionine to cysteine. As the name implies, these disorders result in elevated levels of homocysteine and homocystine in the blood, where the elevated urine output of the metabolite is referred to as homocysteinurina or homocystinuria. The most common nomenclature for these disorders is homocystinuria. In addition to homocysteinuria/homocystinuria, patients excrete elevated levels of methionine and metabolites of homocysteine.
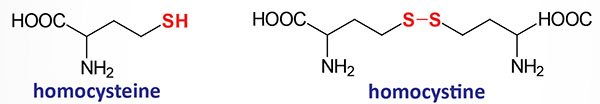
Homocystine is a disulfide-bonded homodimer of two homocysteines. This is similar to the formation of cystine from two cysteines as diagrammed in the Cystinuria page.
Three forms of homocysteinemia/homocystinuria have been classified based on the underlying cause.
Homocystinuria type 1 results from defects in the gene (CBS) encoding cystathionine β-synthase or from deficiency in the required cofactor derived from vitamin B6, pyridoxal phosphate, PLP.
Homocystinuria type 2 results from defects in the synthesis of methylcobalamin in the context of the reaction catalyzed by methionine synthase. Methionine synthase is also called homocysteine methyltransferase and is encoded by the MTR (5-methyltetrahydrofolate-homocysteine methyltransferase) gene. In addition to methylcobalamin, methionine synthase requires 5-methyl-THF as a cofactor.
Homocystinuria type 3 results from pathogenic variants in the gene (MTHFR) encoding methylenetetrahydrofolate reductase. The MTHFR encoded enzyme converts N5,N10-methylenetetrahydrofolate (5,10-methylene-THF) to N5-methyltetrahydrofolate (5-methyl-THF).
Molecular Biology of Classic Homocystinuria
The most common causes of homocystinuria (classic homocystinuria; also identified as homocystinuria type 1) are pathogenic variants in the gene (CBS) encoding cystathionine β-synthase. Because the CBS encoded enzyme requires pyridoxal phosphate (PLP) as a cofactor, deficiency in vitamin B6 is also associated with homocystinuria type 1.
The CBS gene is located on chromosome 21q22.3 and is composed of 23 exons that generate five alternatively spliced mRNAs. These five mRNAs encode proteins of 551 amino acids (isoform 1) and 446 amino acids (isoform 2).
More than 150 pathogenic variants in the CBS gene have been identified as causing homocystinuria. The majority of these pathogenic variants are missense variants. The two most commonly identified pathogenic variants in the CBS gene, in homocystinuria patients, are the I278T (Ile at position 278 changed to Thr) and G307S (Gly at position 307 changed to Ser) variants.
Although much rarer than pathogenic variants in the CBS gene, pathogenic variants in the MTHFR (methylenetetrahydrofolate reductase), MTR (5-methyltetrahydrofolate-homocysteine methyltransferase), MTRR (5-methyltetrahydrofolate-homocysteine methyltransferase reductase), and MMADHC (metabolism of cobalamin associated D) genes are also associated with homocystinuria.
Pathogenic variants in the MTHFR gene were originally associated with a form of homocysteinemia referred to as hyperhomocysteinemia. This form of homocysteinemia/homocystinuria is commonly referred to as homocystinuria type 3. The most common pathogenic variant in the MTHFR gene that results in what was originally identified as hyperhomocysteinemia is a C to T change at nucleotide position 677 (C677T) that results in substitution of Ala for Val at amino acid position 222 (A222V).
The MTR encoded enzyme is most commonly called methionine synthase. Pathogenic variants in the MTR gene are also associated with inherited hyperhomocysteinemia. In the case of the MTR gene the most common pathogenic variant is a change of an A for a G at nucleotide position 2756 (A2756G). The form of homocystinuria caused by pathogenic variants in the MTR gene is often referred to as homocystinuria type 2.
The MTRR encoded enzyme is commonly called methionine synthase reductase.
The MMADHC encoded enzyme is also identified as methylmalonic aciduria and homocystinuria, cblD type. Pathogenic variants in the MMADHC gene are more commonly associated with methylmalonic acidemia. Homocystinuria resulting from defects in methylcobalamin synthesis is commonly referred to as homocystinuria type 2.
Clinical Spectrum of the Homocyst(e)inemias/Homocyst(e)iurias
Classic homocystinuria is often associated with intellectual impairment, although the complete syndrome is multifaceted and many individuals with this disease are mentally normal, while others experience variable levels of developmental delay along with learning problems.
Common symptoms of homocystinuria are dislocated optic lenses (ectopia lentis), osteoporosis, lengthening and thinning of the long bones, and an increased risk of abnormal blood clotting (thromboembolism). The nature of the lens dislocation (subluxation), evident in homocysteinemia patients, can serve as a differential diagnostic tool. Lens dislocation is also found in patients suffering from Marfan syndrome, which is a connective tissue disorder caused by defects in the fibrillin gene (FBN1). In homocystinuria the lens subluxation occurs in a downward and inward direction, whereas in Marfan syndrome the lens subluxation occurs upward and outward. Some instances of genetic homocysteinemia respond favorably to pyridoxine therapy suggesting, that in these cases the defect in CBS is a decreased affinity for the cofactor, pyridoxal phosphate.
Homocysteinemia can also result from vitamin deficiencies due to the role of the co-factor forms of B6, B12, and folate in the overall metabolism of methionine. Vitamin B6 (as pyridoxal phosphate) is required for the activity of CBS and cystathionine γ-lyase and the homocysteinemia that results with B6 deficiency is also associated with elevated methionine levels in the blood. Vitamin B12 and folate (as N5-methyl-THF) are required for the methionine synthase reaction so a deficiency of either vitamin can result in homocysteinemia presenting with reduced levels of plasma methionine. The enzyme methylmalonyl-CoA mutase also requires B12 and so a homocysteinuria resulting from a deficiency in this vitamin is also associated with methylmalonic academia. Indeed, the measurement of serum methionine and methylmalonic acid in cases of homocysteinemia (homocystinuria) allows for a differential diagnosis of the nutritional (non-genetic) cause.
Another related disorder, sometimes referred to as hyperhomocysteinemia, is most often associated with manifestation of symptoms much later in life. The characteristic pathology of hyperhomocysteinemia is coronary artery disease (CAD) and an increased risk for deep vein thromboses, DVT. In addition there is an increased risk for mild cognitive impairment and dementia. Evidence suggests a link between hyperhomocysteinemia and Alzheimer disease. Similar to the dietary causes of homocystinuria, hyperhomocysteinemia can result from deficiencies in vitamin B6, vitamin B12, and folate. An inherited form of hyperhomocysteinemia results from pathogenic variants in the methylene tetrahydrofolate reductase (MTHFR) gene. The most common pathogenic variant in this gene that results in hyperhomocysteinemia is a C to T change at nucleotide position 677 (C677T). Pathogenic variants in the methionine synthase (MTR) gene are also associated with inherited hyperhomocysteinemia. In the case of the MTR gene the most common pathogenic variant is a change of an A for a G at nucleotide position 2756 (A2756G).
Elevated levels of homocysteine in the blood have been shown to correlate with cardiovascular dysfunction. The role of homocysteine in cardiovascular disease is related to its ability to induce a state of inflammation. Homocysteine serves as a negatively charged surface that attracts the contact phase of the intrinsic pathway of blood coagulation. Activation of the intrinsic coagulation cascade leads to inappropriate thrombolytic events as well as resulting in increases in inflammatory cytokine release from leukocytes that are activated as a result of the pro-coagulant state. Therefore, it is important to ensure that proper function of the methionine synthase reaction is maintained. Although it would be assumed that increased intake of vitamin B12 should lead to increased conversion of homocysteine to methionine and thus, reduced levels of circulating homocysteine, controlled studies have shown that this does not occur.