
Last Updated: July 13, 2026
Introduction to VLCAD Deficiency
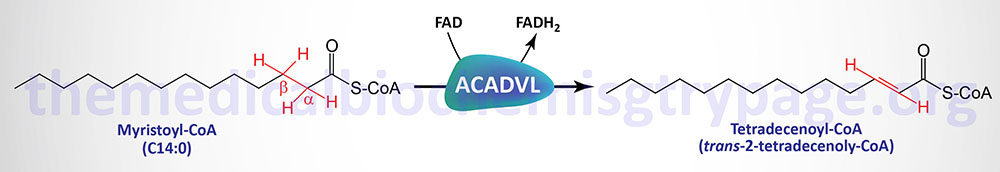
The very long-chain acyl-CoA dehydrogenase (VLCAD) enzyme is a member of the fatty acyl-CoA dehydrogenase family. The fatty acyl-CoA dehydrogenases are FAD-dependent enzymes involved in the first step of the mitochondrial fatty acid β-oxidation pathway. Each of these dehydrogenases has a range of substrate specificities determined by the length of the fatty acid.
Very long-chain acyl-CoA dehydrogenase (also known as ACAD6) prefers fats of 16–24 carbons and is inactive on any fatty acid less than 12 carbons. VLCAD deficiency is inherited as an autosomal recessive disorder resulting from pathogenic variants in the ACADVL gene.
Short-chain acyl-CoA dehydrogenase (SCAD, also called butyryl-CoA dehydrogenase) prefers fats of 4–6 carbons in length. Medium-chain acyl-CoA dehydrogenase (MCAD; also known as ACAD1) prefers fats of 6–16 carbons in length with maximal activity for C10 saturated fatty acyl-CoAs.
Long-chain acyl-CoA dehydrogenase (LCAD; also known as ACAD4) prefers fats of 10–18 carbons in length with maximal activity for C12 saturated fatty acyl-CoAs. The LCAD enzyme is encoded by the ACADL gene but due to very low level of expression in humans, the encoded enzyme does not appreciably, if at all, contribute to mitochondrial fatty acid β-oxidation. Indeed, mitochondrial oxidation of long-chain fatty acids begins through the actions of VLCAD.
Molecular Biology of VLCAD Deficiency
The ACADVL gene is located on chromosome 17p13.1 and is composed of 22 exons that generate four alternatively spliced mRNAs each of which encode distinct protein isoforms. The isoform 4 encoding ACADVL mRNA does not initiate translation efficiently and is, therefore, a likely mRNA candidate for nonsense mediated decay, NMD. Unlike the localization of the SCAD and MCAD enzymes to the mitochondrial matrix, VLCAD is an inner mitochondrial membrane-localized enzyme
Pathogenic variants in the ACADVL gene resulting in VLCAD deficiency include nonsense variants, missense variants, small intragenic deletions/insertions, and splice site variants. The most common pathogenic variant in the ACADVL gene results in the substitution of the Val at amino acid position 283 for an Ala (V283A). In Arab populations a nonsense variant at nucleotide 65, that introduces a stop codon at amino acid position 22, is highly common.
The worldwide incidence of VLCAD deficiency occurs with a frequency of between 1:30,000 to 1:100,000 live births. The original identification of VLCAD deficiency was described in 1985 and was ascribed to pathogenic variants in the ACADL gene which encodes long-chain acyl-CoA dehydrogenase (LCAD). However, subsequent analyses demonstrated that the disorder was in fact due to pathogenic variants in the ACADVL gene. All patients who had previously been diagnosed as suffering from LCAD deficiency were reclassified as being VLCAD deficient.
Clinical Features of VLCAD Deficiency
VLCAD deficiency manifests with three heterogeneous clinical phenotypes. The infantile form, which is often lethal, is associated with severe early onset cardiac and multi-organ failure presenting in the first months of life. Additional symptoms of this form of VLCAD deficiency include hypoglycemia, hepatomegaly, hypotonia, dilated or hypertrophic cardiomyopathy, and arrhythmia. The cardiomyopathy and arrhythmias associated with the infantile form of VLCAD deficiency are often lethal. Approximately 80% of patients with the severe early onset form of VLCAD deficiency harbor null variants in the ACADVL gene resulting in no enzyme activity.
The childhood onset form of VLCAD deficiency presents with hypoketotic hypoglycemia, hepatic dysfunction, and rhabdomyolysis with little likelihood of cardiac pathology. This form of VLCAD deficiency is often referred to as the hepatic form. The hepatic for of VLCAD deficiency usually presents in early childhood and the associated hypoketotic hypoglycemia and hepatomegaly look similar to the infantile presentation of MCAD deficiency.
The late onset form of VLCAD deficiency presents with episodic myopathy and intermittent rhabdomyolysis that is provoked by exercise. The late onset form of VLCAD deficiency is the most common. The common V283A variant in the ACADVL gene is often found in patients with non-cardiac phenotypes such as the milder childhood forms and adult forms of VLCSD deficiency.
Diagnosis of VLCAD deficiency during newborn screening, or upon clinical presentation of symptoms, is accomplished by measuring plasma for elevated acylcarnitines. The key metabolites that are most often abnormal in VLCAD deficiency are C14:2, C14:1, C14:0, and C12:1 acylcarnitines. Determination of VLCAD deficiency through measurement of plasma acylcarnitine levels is most sensitive when the blood is collected from a patient during a period of metabolic stress, such as fasting. In addition to metabolic testing VLCAD deficiency can be diagnosed by molecular analysis for pathogenic variants in the ACADVL gene.
Treatment of VLCAD Deficiency
Treatment of VLCAD deficiency includes the use of intravenous glucose as an energy source, intervention for cardiac rhythm disturbances, and the monitoring of rhabdomyolysis. Acute episodes of rhabdomyolysis are treated with ample hydration and the use of drugs, such as acetazolamide, to alkalize the urine to protect renal function and to prevent acute renal failure secondary to myoglobinuria. Diet modification and intensive supportive care can often result in the reversal of cardiac dysfunction in VLCAD deficiency patients.
Patients afflicted with the more severe forms of VLCAD deficiency are typically placed on a low-fat formula, with supplemental calories (15%-18% of total calories) provided through medium-chain triglycerides (MCT). Inclusion of MCT in the diet benefits older VLCAD deficiency patients who have exercise intolerance.
Another avenue of therapy that shows some benefit for exercise tolerance in VLCAD deficiency is the inclusion of triglycerides with odd-chain fatty acids in their diets. The artificially produced triglyceride is called triheptanoin and it contains three seven-carbon saturated fatty acids. The released odd chain fatty acids provide anaplerotic substrates for the TCA cycle so this treatment is referred to as the anaplerotic diet.
In addition to being used in the treatment of VLCAD deficiency, triheptanoin has been used in patients with pyruvate carboxylase deficiency and with CPT-2 deficiency. Metabolism of the seven-carbon fatty acids in triheptanoin also results in increased production of ketones (β-ketopentanoate and β-hydroxypentanoate) which easily cross the blood-brain barrier (BBB) and are utilized by neural tissues. The use of supplemental dietary carnitine in the treatment of VLCAD deficiency is not recommended due to results obtained from studies in a mouse model of VLCAD deficiency. In these studies the accumulation of long-chain acylcarnitines was associated with myocardial toxicity.