
Last Updated: October 31, 2025
Introduction to Wilson Disease
Wilson disease (WD) is inherited as an autosomal recessive disorder of copper homeostasis. The disease is characterized by excessive copper deposition primarily in the liver and brain. These sites of copper deposition are reflective of the major manifestations of Wilson disease. Wilson disease is named for Dr. Samuel Alexander Kinnier Wilson who first described the disorder in 1912. The frequency of Wilson disease ranges from 1:5,000 to 1:30,000 live births.
Molecular Biology of Wilson Disease
Wilson disease is the result of defects in the P-type ATPase protein that is primarily responsible for copper homeostasis effected by the liver. This protein is encoded by the ATPase, Cu2+-transporting, beta polypeptide (ATP7B) gene. The ATP7B gene is located on chromosome 13q14.3 spanning 80 kbp and composed of 28 exons that generate 41 alternatively spliced mRNAs, that collectively encode 30 distinct protein isoforms.
Numerous mutations have been identified in the ATP7B gene resulting in Wilson disease. The majority of these mutations are clustered in the transmembrane domains of the encoded protein. In Europeans and North Americans two mutations account for 38% of the observed mutations in this disease. These two mutations are a substitution of glutamine for histidine at amino acid 1069 (H1069Q) and arginine for glycine at amino acid 1267 (G1267R).
The molecular basis for the observed phenotypic variations in Wilson disease are complex and involve both environmental influences and the effects of modifier genes such as COMMD1 (copper metabolism MURR1 domain-containing protein 1). The MURR1 domain was first identified in a murine protein and refers to: mouse U2af1-rs1 region. An additional ATP7B modifier protein is the chaperone protein ATOX1 (antioxidant protein 1). An additional locus affecting the phenotype of Wilson disease is apolipoprotein E ε3/3. Individuals harboring this particular apoE allele have a delayed onset in the presentation of Wilson disease symptoms.
The ATP7B protein contains an ATPase domain, a hinge domain, a phosphorylation site, and six copper-binding sites. The ATP7B gene is expressed predominantly in the liver, but also in the kidney and placenta. Lower levels of expression are also detectable in the brain, heart, and lungs. The structure of the ATP7B encoded protein is highly similar to that of the ATP7A encoded protein which is disrupted in another copper-transport defect disease called Menkes disease.
Function of the ATP7B Encoded Transporter
The ATP7B protein is localized to the trans-Golgi network where it transports copper into the lumen of the Golgi. When copper levels rise, ATP7B is translocated to vesicular compartments closely associated with the plasma membrane where it can transport copper into these compartments. The copper in these vesicles can then be released by exocytosis.
When copper is transferred from intestinal enterocytes to the plasma, through the action of the ATP7A protein, it is bound to albumin and delivered to the liver. In the liver the copper is transferred to intracellular storage sites by the chaperone ATOX1. Copper is stored intracellularly bound to the protein metallothionein. Any copper in excess of the binding capacity of metallothionein is excreted into the biliary canaliculi through the transport action of ATP7B.
ATP7B also facilitates the transfer of copper to the copper-dependent ferroxidase, ceruloplasmin. Ceruloplasmin that does not have bound copper is referred to as apoceruloplasmin. Copper-containing ceruloplasmin, produced in the liver, is then released to the blood. In all other tissues the form of ceruloplasmin is a plasma membrane GPI-linked form that is responsible for iron efflux from the cell. Greater than 90% of the copper in the blood is bound to ceruloplasmin. Due to its function in overall iron homeostasis, loss of ceruloplasmin activity in Wilson (and Menkes) disease is associated with defective iron homeostasis in critical tissues such as the brain and kidneys. Deficiencies in ATP7B result in increased levels of apoceruloplasmin and a toxic build-up of copper in hepatocytes due to defective transfer to the biliary canaliculi.
Clinical Features of Wilson Disease
The majority of patients with Wilson disease present with hepatic or neuropsychiatric symptoms. All other patients, representing about 20% of all Wilson disease cases, manifest with symptoms that are attributable to involvement of other organs. Hepatic presentation in Wilson disease occurs in late childhood or adolescence. The symptoms in these patients include hepatic failure, acute hepatitis, or progressive chronic liver disease. Patients that manifest neurological symptoms of Wilson disease present in the second to third decade of life. In these patients there will be extrapyramidal, cerebellar and cerebral-related symptoms such as Parkinsonian tremors, diminished facial expressions and movement, dystonia, and choreoathetosis. About 30% of Wilson disease patients will exhibit psychiatric disturbances that include changes in behavior, personality changes, depression, attention deficit hyperactivity disorder, paranoid psychosis, suicidal tendencies, and impulsivity.
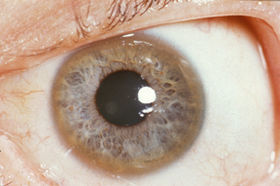
As the disease progresses copper deposition leads to vacuolar degeneration in the proximal tubular cells of the kidneys which causes a renal Fanconi syndrome (substances that are normally absorbed by the kidney are excreted in the urine). Acute release of copper into the circulation results in damage to red blood cells resulting in hemolysis. The most significant sign in the diagnosis of Wilson disease results from the deposition of copper in Descemet’s membrane of the cornea. These golden-brown deposits can be seen with a slit-lamp and are Kayser-Fleischer rings (see image below).
Treatment of Wilson Disease
Treatment of Wilson disease is aimed at reducing the toxic concentration of copper. This can be accomplished with copper chelating agents such as trientine and D-penicillamine. Once the copper levels have been reduced the chelating agents can be reduced or eliminated and zinc salt used to prevent systemic absorption of copper. Patients with progressive liver failure will require liver transplantation. Liver transplantation is also required in patients that do not respond to copper chelation therapy.