
Last Updated: January 5, 2026
Introduction to the Tyrosinemias
Inherited tyrosinemias, as the name implies, are characterized by the accumulation of tyrosine in the blood and tissues. Three distinct types of tyrosinemia have been characterized, all of which are due to defects in the pathway of tyrosine catabolism.
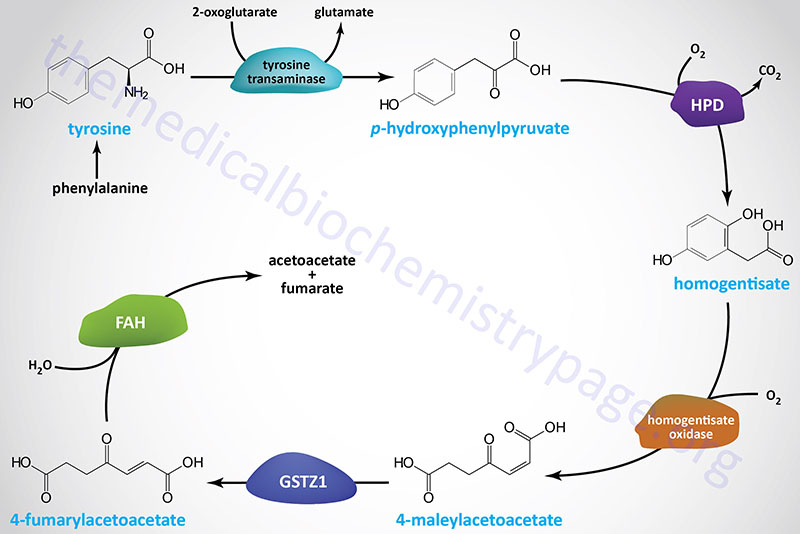
Tyrosinemia type 1 (TYRSN1) is an autosomal recessive disease and represents the most severe form of the tyrosinemias. Tyrosinemia type 1, is a potentially fatal childhood disorder associated with liver failure, painful neurologic crises, rickets, and hepatocarcinoma. This disorder is caused by a deficiency of fumarylacetoacetate hydrolase which catalyzes the final reaction of tyrosine catabolism. If untreated, death typically occurs at less than 2 years of age, with some chronic forms allowing longer survival. It has a prevalence of about 1 in 100,000 live births in the general population.
Tyrosinemia type 2 (TYRSN2) is an autosomal recessive disease characterized as an oculocutaneous syndrome. Tyrosinemia type 2 is also known as Richner-Hanhart syndrome. Tyrosinemia type 2 presents with hyperkeratotic plaques on the hands and soles of the feet and photophobia due to deposition of tyrosine crystals within the cornea. This disorder is caused by a deficiency of tyrosine aminotransferase which catalyzes the first reaction of tyrosine catabolism.
Tyrosinemia type 3 (TYRSN3) is an extremely rare autosomal recessive disorder associated with ataxia and mild intellectual impairment. This disorder is caused by a deficiency of 4-hydroxyphenylpyruvate dioxygenase which catalyzes the second reaction of tyrosine catabolism.
Mutations the HGD gene, that encodes homogentisate oxidase (homogentisate 1,2-dioxygenase), also represents a disorder of phenylalanine and tyrosine catabolism. However, the consequences of HGD mutations are relatively benign and are the cause of the disorder called alkaptonuria.
Diagnosis of patients with tyrosinemia can made by detection of elevated tyrosine in the blood through plasma amino acid chromatography and measurement of tyrosine metabolites in the urine by organic acid analysis. Due to the hepatocellular pathology in tyrosinemia type 1 there will also be an associated elevation in plasma methionine. Urine organic acids show elevated 4-hydroxyphenyl organic acids in each type of tyrosinemia. In the case of tyrosinemia type 1 there will also be elevated levels of succinylacetone, a particularly toxic metabolite.
In addition to measurement of amino acid levels and organic acid levels in tyrosinemia patients, diagnosis can be confirmed by enzyme or molecular studies. The latter is particularly true for testing in the case of tyrosinemia type 1.
Molecular Biology of the Tyrosinemias
Tyrosinemia type 1 (TYRSN1) results from mutations in the gene (FAH) encoding fumarylacetoacetate hydrolase. The FAH gene is located on chromosome 15q25.1 and is composed of 15 exons that generate three alternatively spliced mRNAs, each of which encodes the same 419 amino acid protein. Mutations in the FAH gene, that are associated with tyrosinemia type 1, include missense and nonsense mutations and splice site mutations. Almost all of the mutations in the FAH gene are single nucleotide changes. In the Saguenay-Lac-Saint-Jean region of Quebec Canada 100% of tyrosinemia type 1 patients harbor a mutation in the splice donor site in intron 12. In this region of Canada almost 1 in very 1,800 children are born with tyrosinemia type 1. The carrier frequency of the splice donor mutation in this population is 1 in 25.
Tyrosinemia type 2 (TYRSN2) results from mutations in the gene (TAT) encoding tyrosine transaminase. The TAT gene is located on chromosome 16q22.2 and is composed of 12 exons that generate a protein of 454 amino acids. Expression of the TAT gene is exclusive to the liver. Mutations in the TAT gene that result in tyrosinemia type 2 include large deletions, missense and nonsense mutations, frameshift mutations, and splice variants.
Tyrosinemia type 3 (TYRSN3) results from mutations in the gene (HPD) encoding 4-hydroxyphenylpyruvate dioxygenase. The HPD gene is located on chromosome 12q24.31 and is composed of 17 exons that generate two alternatively spliced mRNAs encoding proteins of 393 amino acids (isoform 1) and 354 amino acids (isoform 2). Mutations in the HPD gene associated with tyrosinemia type 3 include missense and nonsense mutations.
Clinical Features of the Tyrosinemias
Tyrosinemia type 1 (TYRSN1)
Tyrosinemia type 1 is characterized by hypertyrosinemia and progressive liver disease. Clinically this form of tyrosinemia can be divided into two classes, those presenting prior to 6 months of age with severe liver pathology and those presenting after 6 months of age with mild liver and renal pathology. Secondary renal tubular dysfunction in the chronic forms of tyrosinemia type 1 leads to generalized aminoaciduria, loss of phosphate, and, renal tubular acidosis. The loss of phosphate contributes to the development of rickets, referred to as hypophosphatemic rickets.
Infants with tyrosinemia type 1 have often been described as possessing an odor of rotten mushrooms or of boiled cabbage.
In infants under 6 months of age with acute liver dysfunction, markedly prolonged prothrombin and partial thromboplastin times (PT and PTT, respectively) are found. Measurement for clotting factors will detect normal levels of factors V and VIII but decreased levels of prothrombin (factor II) and factors VII, IX, XI, and XII.
Tyrosinemia type 1 patients also present with dramatically elevated levels of the liver produced protein, α-fetoprotein (AFP). Normally the levels of AFP are high at birth but fall shortly after such that by the age of 1 year levels are very low and remain so. The characteristic odor of tyrosinemia type 1 infants taken together with elevated AFP can lead to the clinical recognition of the disorder as tyrosinemia type 1 from other causes of acute liver failure. Measurements for serum methionine and tyrosine as well as urine succinylacetone allow for a definitive diagnosis of tyrosinemia type 1 to be made.
Defects in the FAH gene result in the accumulation of the upstream tyrosine metabolites, maleylacetoacetate and fumarylacetoacetate, as well as their metabolic by-products succinylacetone and succinylacetoacetate. These compounds, especially fumarylacetoacetate, are toxic to cells, particularly liver and kidney, explaining the pathology associated with this disease. Succinylacetone is also a potent inhibitor of the tyrosine catabolic enzyme, 4-hydroxyphenylpyruvate dioxygenase (encoded by the HPD gene) and the heme biosynthetic enzyme, δ-aminolevulinic acid (ALA) dehydratase [also called porphobilinogen (PBG) synthase]. The inhibition of ALA dehydratase leads to accumulation of δ-aminolevulinic acid (ALA). ALA is neurotoxic and thought to be the cause of the acute porphyria-like pathology in tyrosinemia type 1.
Tyrosinemia type 2 (TYRSN2)
Tyrosinemia type 2 is associated with intellectual impairment, painful corneal eruptions, photophobia, keratitis, and painful palmoplantar hyperkeratosis. This constellation of symptoms explains the association of the name oculocutaneous tyrosinemia with tyrosinemia type 2. The ocular pathology of tyrosinemia type 2 usually manifests in the first year of life. The cutaneous pathology is typified by dramatic yellowish thickening associated with the hyperkeratosis. Developmental delay has been ascribed to tyrosinemia type 2, however, not all patients manifest with this phenotype.
Diagnosis of tyrosinemia type 2 includes measurement of plasma amino acids and urine organic acids. The levels of tyrosine in the blood of tyrosinemia type 2 patients is the highest of all three forms of tyrosinemia. Plasma tyrosine level will typically be greater than 500 mM (normal levels are 30-120 μM), whereas and other amino acids, including methionine and phenylalanine, will be normal.
Tyrosinemia type 3 (TYRSN3)
Tyrosinemia type 3 is associated with mild intellectual impairment and/or convulsions but these patients do not display hepatic damage nor ocular pathology as is characteristic of tyrosinemia type 1 and type 2. Tyrosinemia type 3 is associated with lower tyrosine levels (350-650 mM) than either tyrosinemia type 1 or type 2. Tyrosinemia type 3 patients have increased excretion of 4-hydroxyphenylpyruvate, 4-hydroxyphenyllactate, and 4-hydroxyphenylacetate.
An autosomal dominant variant of HPD mutation results in the disorder referred to as hawkinsinuria. The name was coined from the identification of a novel sulfur containing amino acid, 2-cystenyl-1,4-dihydroxycyclohexenylacetate [(2-L-cystein-S-yl-1,4-dihydroxycyclohex-5-en-1-yl)-acetic acid], in the urine of a patient exhibiting metabolic acidosis and tyrosinemia. The novel amino acid was named hawkinsin. Hawkinsin is formed as a result of the action of glutathione on the intermediate, 1,2-epoxyphenyl acetic acid, of the 4-hydroxyphenylpyruvate dioxygenase catalyzed reaction.
Individuals with hawkinsinuria continue to excrete hawkinsin throughout their lives but any pathology resolves upon phenylalanine and tyrosine restriction and after around 18 months the restrictions are no longer required. The mutation in the HPD gene that cause hawkinsinuria is a missense mutation that converts the alanine at position 33 to a threonine, designated A33T
Treatment of Tyrosinemias
Therapy consists of a diet low in phenylalanine and tyrosine for each of the tyrosinemias. However, dietary restrictions alone have not been shown to be effective at managing the acute stage of tyrosinemia type 1. In some cases of tyrosinemia type 1, liver transplantation is required, but this carries its own set of risks. Dietary restriction of phenylalanine and tyrosine has been shown to be very effective in the management of the pathology of tyrosinemia type 2.
In 1992 it was discovered the the compound, 2-(2-nitro-4-trifluoromethylbenzoyl)-1,3-cyclohexanedione (NTBC), was an effective inhibitor of 4-hydroxyphenylpyruvate dioxygenase, the HPD encoded enzyme. Inhibition of 4-hydroxyphenylpyruvate dioxygenase prevents the downstream substrate of the FAH encoded enzyme from being generated which in turn reduces the intracellular accumulation of fumarylacetoacetate. As indicated above, fumarylacetoacetate is particularly toxic to hepatocytes. for tyrosinemia type 1.
NTBC is called nitisinone and is sold under the trade name of Orfadin. Since the institution of the use of nitisinone for the treatment of tyrosinemia type 1 it has been found that over 90% of treated patients have shown dramatic improvement. Even with the use of nitisinone, tyrosinemia type 1 patients require dietary restriction of phenylalanine and tyrosine. Tyrosine has low solubility and by inhibiting the HPD encoded enzyme with nitisinone, without dietary restriction, tyrosine crystals can deposit in tissues, in particular the cornea leading to inflammation in the eye. Although the course of disease in nitisinone treated children is significantly reduced, these patients still require long-term monitoring for the possible development of hepatocarcinoma.