
Last Updated July 13, 2026
Gangliosides
Gangliosides are members of the glycosphingolipid family of glycolipids. Gangliosides are glycosphingolipids that are defined by the presence of N-acetylneuraminic acid (NANA; also known as sialic acid) in varying amounts. The specific names for gangliosides are a key to their structure. The first letter in the nomenclature, G, refers to ganglioside. The following letters, A, M, D, T, Q, and P, indicate that the molecule contains none (absent or asialo), mono-, di-, tri, quatra(tetra), or poly(more than 4)-sialic acid residues. The numerical designations, 1, 2, and 3, refer to the carbohydrate sequence that is attached to ceramide. See the Sphingolipid Metabolism and the Ceramides page for details on the synthesis and structures of the gangliosides.
Introduction to Tay-Sachs Disease
Tay-Sachs disease is an autosomal recessive disease that is a member of a family of disorders identified as the GM2 gangliosidoses which themselves belong to the large family of lysosomal storage disorders (lysosomal storage diseases). The GM2 gangliosidotic diseases are severe psycho-motor developmental disorders caused by the inability to properly degrade membrane associated gangliosides of the GM2 family (see Figure below) as a result of pathogenic variants in the gene (HEXA) encoding the α-subunit of the lysosomal enzyme, β-hexosaminidase (commonly called hexosaminidase A, HexA). Because neural cell membranes are enriched in GM2 gangliosides, the inability to degrade this class of sphingolipid results in neural cell death. In addition to Tay-Sachs disease the family includes Sandhoff disease and GM2 activator deficiency.
The eponymic term, Tay-Sachs disease, is reserved for the infantile forms of GM2 gangliosidotic diseases. The infantile forms are very severe and infants will not normally survive beyond 2 years of age. There are numerous pathogenic variants of the HEXA encoded α-subunit that encompass a clinical spectrum from subacute to chronic forms of disease in which symptoms may be delayed for many years.
The frequency of Tay-Sachs disease in certain populations is quite high. In Ashkenazi Jews, who represents the largest population of ethnic Jews, the occurrence is as high as 1 in 4000 live births. For this reason it is often recommended that all potential parents of this ethnicity be diagnosed to determine whether or not they are carriers of Tay-Sachs alleles since this frequency is 1 in 30 within this population.
Hexosaminidases
GM2 ganglioside degradation requires the enzyme β-hexosaminidase and the GM2 activator protein (encoded by the GM2A gene). β-Hexosaminidase is a dimer composed of 2 subunits, either the α subunit (encoded by the HEXA gene) and/or the β subunit (encoded by the HEXB gene). The various isoforms of β-hexosaminidase result from the combination of α and β subunits. The β-hexosaminidase S (HexS) enzyme is a homodimer of αα, the β-hexosaminidase A (HexA) enzyme is a heterodimer of αβ, and the β-hexosaminidase B (HexB) enzyme is a homodimer of ββ. It is the α-subunit that carries out the catalysis and only the HexA form of β-hexosaminidase can catalyze the cleavage of GM2 gangliosides (the bond cleaved is shown by the arrow in the Figure below). The activator first binds to GM2 gangliosides followed by hexosaminidase and then digestion occurs. The degradation of GM2 gangliosides takes place in the lysosomes. Failure to degrade these sphingolipids results in the lysosomes becoming engorged filling the cell, eventually choking off normal cellular functions. Because of the deposition of abnormal sphingolipids in the lysosomes the GM2 gangliosidotic diseases are also referred to as lysosomal storage disorders.
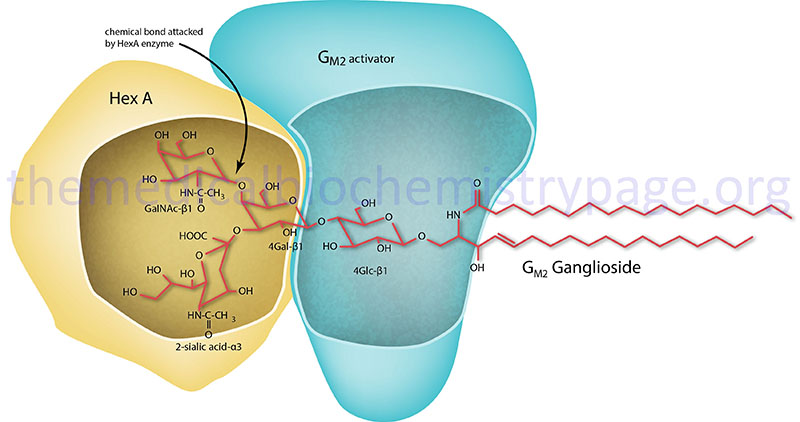
Molecular Biology of Tay-Sachs Disease
The HEXA gene resides on chromosome 15q23 spanning 35 kb and composed of 14 exons that generate two alternatively spliced mRNAs encoding precursor proteins of 540 amino acid (isoform 1) and 529 amino acids (isoform 2). The HEXA isoform 1 protein is likely processed to the same functional size as the isoform 2 protein.
As of 2025 there have been 180 pathogenic variants identified in the HEXA gene resulting in Tay-Sachs disease. Of these pathogenic variants, 65 have been shown to result in single nucleotide changes, the rest are deletions or insertions of varying size. Pathogenic variants in the HEXA gene will result in defective synthesis of the α-subunit of β-hexosaminidases and thus, defective formation of the HexA and HexS forms of the enzyme. The non-mutant β-subunit, encoded by the HEXB gene, can still form the functional HexB isoform of the enzyme.
The most common pathogenic variant in the HEXA gene in individuals of Ashkenazi Jewish ethnicity is a four nucleotide (TATC) insertion in exon 11 resulting in a frame-shift variant. This frame-shift variant results in the recognition of a translation stop codon downstream of the insertion resulting in a non-functional α-subunit of both the HexA and HexS isoforms of β-hexosaminidase. In addition, the insertion and recognition of the aberrant stop codon causes the mRNA to be degraded by the nonsense mediate mRNA decay (NMD) process.
Clinical Features of Classical Tay-Sachs Disease
Tay-Sachs disease results from defects in the HEXA gene encoding the α-subunit of β-hexosaminidase. As such, Tay-Sachs disease and variants are defective in the HexA form of the enzyme but not the HexB form. Deficiencies in the HEXB gene result in Sandhoff disease and deficiencies in the GM2A gene result in GM2 activator deficiency diseases (also referred to as the AB variant of Tay-Sachs disease.
The clinical phenotypes associated with the infantile forms of Tay-Sachs disease, Sandhoff disease and GM2 activator deficiency are for the most part indistinguishable. In fact, the initial discrimination of Sandhoff disease from Tay-Sachs disease was accomplished by enzyme assay not by clinical differential.
Infants with Tay-Sachs disease appear normal at birth. Symptoms usually begin with mild motor weakness by 3 to 5 months of age. Parents will begin to notice that their afflicted child has a dull response to outside stimuli. Another early symptom is an exaggerated startle response (sudden extension of arms and legs) to sharp sounds. By 6 to 10 months of age infants will begin to show regression of prior acquired motor and mental skills. It is the loss of these activities that will normally prompt parents to seek a medical opinion. A progressive loss in visual attentiveness may lead to an ophthalmological consultation which will reveal macular pallor and the presence of the characteristic “cherry-red spot” in area of the fovea centralis of the eye (see Figure below).
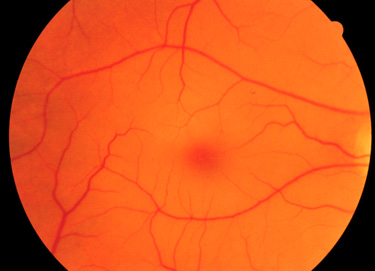
At around 8 to 10 months of age the symptoms of Tay-Sachs disease begin to progress rapidly. Infants are progressively non-responsive to parental stimulation. The exaggerated startle response becomes quite pronounced. Most frightening to parents is the onset of seizures which initially can be controlled by anti-seizure medication. However, the seizures become progressively more severe and are very frequent by the end of the first year. Psycho-motor deterioration increases by the second year and invariably leads to decerebrate posturing (typical of patients in persistent vegetative states), difficulty in swallowing and increased seizure activity. Ultimately, the patient will progress to an unresponsive vegetative state with death resulting from bronchopneumonia resulting from aspiration in conjunction with a depressed cough.
Late-Onset Tay-Sachs Disease
Late-onset Tay-Sachs disease (LOTS), is a clinical subtype of the GM2 gangliosidoses. LOTS is characterized by dysarthria (motor speech dysfunction), dysmetria (inability to judge distance), and ataxia (loss of muscle coordination) resulting from involvement of the cerebellum and proximal muscle weakness and atrophy resulting from involvement of the anterior horn cells. LOTS is an infrequent disorder and the diagnosis is often missed or delayed. The majority of LOTS patients develop signs of either cerebellar or anterior motor neuron involvement. LOTS patients may also display psychotic episodes.
Most patients with LOTS are compound heterozygotes that harbor the Gly 269 to Ser (G269S) pathogenic variant in the HEXA gene along with another deleterious allele. The G269S pathogenic variant is associated with residual β-hexosaminidase activity. Deleterious alleles most often found in individuals with LOTS, who have the G269S allele, are the R504C allele, the TATC1278 null allele, the IVS2 null allele, and individuals who are homozygous for the G269S allele.