
Last Updated: April 1, 2026
Introduction to the Synthesis of Triglycerides
Fatty acids are stored for future use as triglycerides (TG; also termed triacylglycerides, TAG) in all cells, but primarily in adipocytes of adipose tissue. Triglycerides constitute molecules of glycerol to which three fatty acids have been esterified. The fatty acids present in triglycerides are predominantly saturated fatty acids. The fatty acids incorporated into triglycerides are all activated to acyl-CoAs through the action of various acyl-CoA synthetases.
There are three major pathways for the synthesis of triglycerides referred to as the glycerol-3-phosphate (G3P) pathway, the dihydroxyacetone phosphate (DHAP) pathway, and the monoacylglycerol pathway. The glycerol-3-phosphate pathway of triglyceride synthesis is referred to as the Kennedy pathway after Eugene Kennedy who first worked out the pathway of triglyceride synthesis and published his results in 1960.
The glycerol-3-phosphate pathway is the major pathway (90%) utilized for triglyceride synthesis in hepatocytes. However, under conditions of insulin stimulation of the liver, glycolysis is highly active as a means to drive the excess glucose carbons into fatty acid synthesis. Under these conditions the DHAP pathway using the DHAP derived from glycolysis is the major triglyceride synthesis pathway in the liver. Within adipocytes, which lack expression of glycerol kinase, the DHAP derived from glycolysis is absolutely required for triglyceride synthesis thus, the DHAP pathway is the predominant triglyceride synthesis pathway in these cells. The monoacylglycerol pathway is used almost exclusively in enterocytes of the small intestines for the synthesis of triglycerides from dietary fatty acids.
Glyceroneogenesis and Triglyceride Synthesis
Although the DHAP derived from glycolysis is believed to be the primary source of the glycerol-3-phosphate used as the backbone for triglyceride synthesis in adipocytes, evidence clearly demonstrates that pyruvate (and by their relationship to pyruvate, alanine and lactate) can serve as the source of glycerol-3-phosphate via a process referred to as glyceroneogenesis.
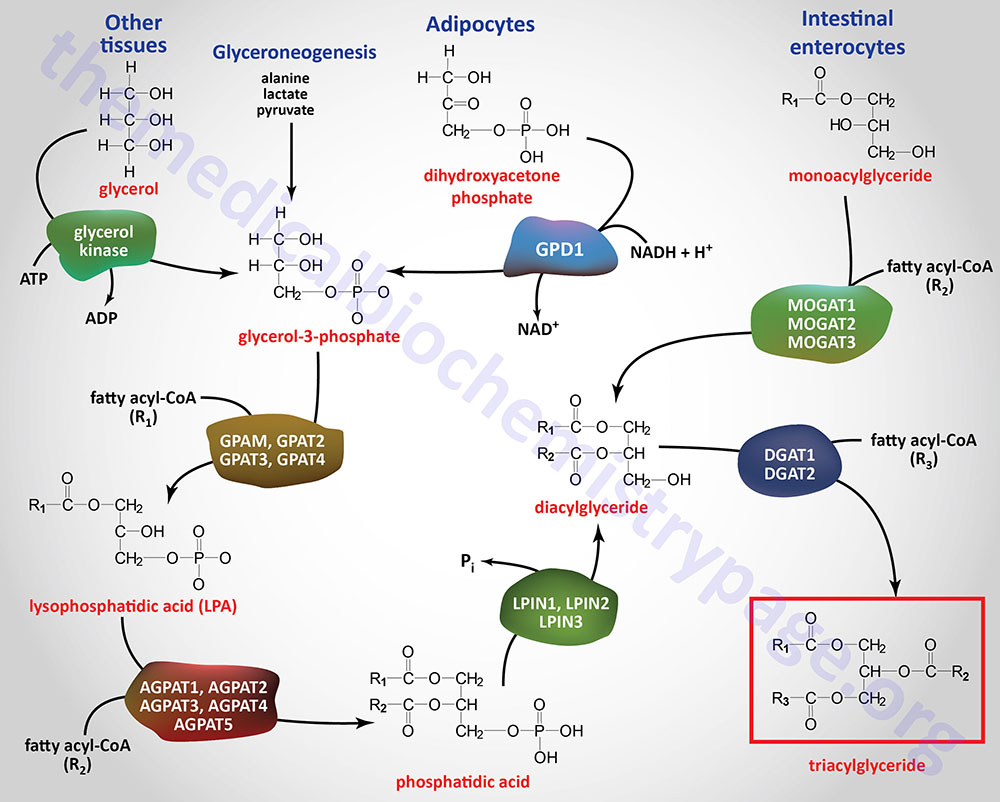
Glyceroneogenesis is essentially a diversion from the pathway of gluconeogenesis. Indeed, the pathways of gluconeogenesis and glyceroneogenesis share a common set of reactions, such that these pathways cannot be functionally separated. The cytosolic form of phosphoenolpyruvate carboxykinase (PEPCK-c: encoded by the PCK1 gene) catalyzes the GTP-dependent decarboxylation of oxaloacetate to form phosphoenolpyruvate (PEP) which is important in the process of gluconeogenesis. The PCK1 encoded enzyme is a key regulatory enzyme in glyceroneogenesis.
In the process of glyceroneogenesis in adipose tissue, the carbons of pyruvate (or lactate or alanine) enter the TCA cycle and contribute to the formation of malate. The malate can be transported to the cytosol where the cytosolic malate dehydrogenase reduces malate to oxaloacetate (OAA). As indicated, OAA is a substrate for PEPCK-c which generates PEP. Since the enzymes of glycolysis from DHAP to PEP are reversible, the PEP generated from pyruvate can be diverted to DHAP synthesis. The DHAP is then reduced to glycerol-3-phosphate via the action of the cytoplasmic form of glycerol-3-phosphate dehydrogenase (encoded by the GPD1 gene). The resulting glycerol-3-phosphate can then be used as the backbone for triglyceride synthesis.
The processes of glyceroneogenesis allow adipose tissue to generate triglycerides during periods periods of starvation where the delivery of glucose to adipose tissue would be severely limiting. Glyceroneogenesis is not only important for the synthesis of triglycerides in adipose tissue, but is functional in the liver as well.
Glycerol-3-Phosphate as Triglyceride Precursor
The glycerol backbone of triglycerides is activated by phosphorylation at the C-3 (sn-3) position by glycerol kinase generating glycerol-3-phosphate. Glycerol kinase is encoded by the GK gene. The GK gene is located on the X chromosome (Xp21.2) and is composed of 24 exons that generate four alternatively spliced mRNAs, each of which encode a distinct protein isoform.
Since expression of the GK gene is absent in adipocytes, these cells require the reduction of glycolytic DHAP to glycerol-3-phosphate via the action of cytosolic glycerol-3-phosphate dehydrogenase
The acylation of glycerol-3-phosphate is the major pathway for triglyceride synthesis in most human cells. Once formed, glycerol-3-phosphate is acylated at the C-1 (sn-1) position by one of a family of glycerol-3-phosphate acyltransferase (GPAT) enzymes that are expressed in either the mitochondria or in the endoplasmic reticulum, ER. The products of the acylation reactions are lysophosphatidic acids (LPA or lysoPA). Lysophosphatidic acids are themselves potent bioactive lipids. The LPA generated through the actions of the various GPAT enzymes serve, not only as substrates for triglyceride synthesis, but also for the synthesis of phospholipids.
The GPAT family includes the two mitochondrial enzymes (encoded by the GPAM and GPAT2 genes) and the two ER enzymes (encoded by the GPAT3 and GPAT4 genes). The GPAT3 and GPAT4 genes belong to the larger 1-acylglycerol-3-phosphate O-acyltransferase (AGPAT) family.
The GPAM gene is located on chromosome 10q25.2 and is composed of 26 exons that generate two alternatively spliced mRNAs encoding the same 828 amino acid precursor protein. GPAM is also known as GPAT1. The highest levels of expression of the GPAM gene are seen in liver and adipose tissues. Expression of the GPAM gene decreases during fasting and increases following food intake, the latter the result of effects of insulin. GPAM exhibits a preference for palmitic acid (C16:0).
The GPAT2 gene is located on chromosome 2q11.1 and is composed of 23 exons that generate nine alternatively spliced mRNAs, that collectively encode seven distinct protein isoforms. Expression of the GPAT2 gene is nearly exclusive to the germ cells of the testes. Substrate preference for GPAT2 appears to be for arachidonic acid (C20:4).
The GPAT3 gene is located on chromosome 4q21.23 and is composed of 17 exons that generate three alternatively spliced mRNAs all of which encode the same 434 amino acid protein. Confusion results from the fact that GPAT3 has also been identified as AGPAT8, AGPAT9, and AGPAT10 where the latter two designations have also been associated with LPCAT1. Expression of GPAT3 increases during adipogenesis indicating that the encoded enzyme plays a central role in triglyceride storage in adipose tissue.
The GPAT4 gene is located on chromosome 8p11.21 and is composed of 17 exons that generate three alternatively spliced mRNAs that collectively encode two distinct protein isoforms. Isoform 1 is 456 amino acids and isoform 2 is 248 amino acids. GPAT4 is also known as AGPAT6 and lysophosphatidic acid acyltransferase-zeta (LPAATζ). Expression of the GPAT4 gene is highest in the liver and in white and brown adipose tissue, WAT and BAT, respectively. GPAT4 exhibits a preference of monounsaturated fatty acids (MUFA).
All of the GPAT enzymes are members of the large family of 1-acylglycerol-3-phosphate O-acyltransferases (AGPAT) as outlined in the following section.
Acylglycerophosphate acyltransferases: AGPAT
Following formation of a lysophosphatidic acid, various 1-acylglycerol-3-phosphate O-acyltransferases (AGPAT) add another CoA-activated fatty acid to the sn-2 position generating 1,2-diacylglycerol phosphates which are commonly identified as phosphatidic acids (PA). Several enzymes of the AGPAT family are also referred to as lysophospholipid acyltransferases (LPLAT) as well as lysophosphatidic acid acyltransferases (LPAAT). Several enzymes in the LPLAT family are involved in the synthesis of the glycerophopholipids from lysophosphatidic acids and from lysophospholipids as discussed in the Synthesis of Phospholipids page.
There are five major 1-acylglycerol-3-phosphate O-acyltransferase encoding genes identified as AGPAT1–AGPAT5 that function in the synthesis of triglycerides. All of these AGPAT enzymes are integral membrane proteins with the AGPAT1 and AGPAT2 enzymes specifically localized to the ER.
The AGPAT1 gene is located on chromosome 6p21.32 and is composed of 11 exons that generate five alternatively spliced mRNAs, four of which encode the same 283 amino acid protein. The AGPAT1 encoded enzyme is also known as LPLAT1 and as LPAATα (alpha). The AGPAT1 gene is ubiquitously expressed. The AGPAT1 enzyme is predominantly localized to the endoplasmic reticulum, ER.
The AGPAT2 gene is located on chromosome 9q34.3 and is composed of 7 exons that generate two alternatively spliced mRNAs encoding two different isoforms of the enzyme. The AGPAT2 encoded enzyme is also known as LPLAT2 as well as LPAATβ (beta). The AGPAT2 enzyme is predominantly localized to the endoplasmic reticulum, ER. Mutations in the AGPAT2 gene result in congenital generalized lipodystrophy 1 (CGL1), also known as Berardinelli-Seip syndrome. This disorder is an autosomal recessive disorder characterized by marked lack of adipose tissue at birth, severe insulin resistance, hypertriglyceridemia, hepatic steatosis, and early onset of diabetes.
The AGPAT3 gene is located on chromosome 21q22.3 and is composed of 22 exons that generate five alternatively spliced mRNAs, four of which encode the same 376 amino acid protein. The AGPAT3 encoded enzyme is also known LPLAT3 as well as LPAATγ (gamma). The AGPAT3 gene is ubiquitously expressed with highest levels in heart, adipose tissue, and liver. The AGPAT3 enzyme is predominantly localized to the endoplasmic reticulum (ER) where it is involved in retrograde transport processes from the Golgi back to the ER.
The AGPAT4 gene is located on chromosome 6q26 and is composed of 9 exons that encode a 378 amino acid protein. The AGPAT4 encoded enzyme is also known as LPLAT4 as well as LPAATδ (delta). The AGPAT4 gene is predominantly expressed in the brain and muscle. The encoded protein localizes to the outer membrane of the mitochondria and to the trans-Golgi network.
The AGPAT5 gene is located on chromosome 8p23.1 and is composed of 9 exons that encode a 364 amino acid protein. The AGPAT5 encoded enzyme is also known as LPLAT5 as well as LPAATε (epsilon).
Humans express six additional genes in the large AGPAT family, two of which are the GPAT3 (also known as AGPAT9) and GPAT4 (also known as AGPAT6) genes indicated above. Four of the AGPAT family member genes encode enzymes involved in the synthesis of glycerophospholipids (phospholipids).
The 11 genes that constitute the mammalian AGPAT family are categorized into three groups identified as group I, II, and II. Group III is further divided into three subgroups, IIIa, IIIb, and IIIc. Group I genes include AGPAT1 and AGPAT2. Group II genes include GPAT3 (AGPAT9), GPAT4 (AGPAT6), LPCAT1 (AGPAT10), LPCAT2 (AGPAT11), and LPCAT4 (AGPAT7). Group IIIa genes include AGPAT3 and AGPAT4. Group IIIb comprises the LCLAT1 (AGPAT8) gene. Group IIIc comprises the AGPAT5 gene.
Monoacylglycerides as Triglyceride Precursors
Intestinal monoacylglycerides (MAG; also termed monoglycerides), derived from the hydrolysis of dietary fats, can also serve as substrates for the synthesis of triglycerides which can then be incorporated into chylomicrons. The intestinal synthesis of a triglyceride from a MAG first involves MAG acylation carried out by one of a family of monoacylglycerol O-acyltransferases (MOGAT). Humans express three MOGAT genes identified as MOGAT1, MOGAT2, and MOGAT3.
The MOGAT1 gene is located on chromosome 2q36.1 and is composed of 6 exons that encode a 335 amino acid protein.
The MOGAT2 gene is located on chromosome 11q13.5 and is composed of 8 exons that encode a 334 amino acid protein.
The MOGAT3 gene is located on chromosome 7q22.1 and is composed of 8 exons that generate two alternatively spliced mRNAs encoding two distinct isoforms of the enzyme.
The resultant diacylglycerides (DAG; also termed diglycerides), in intestinal enterocytes, are then converted to triglycerides via the action of the DGAT enzymes outlined in the next section.
Diacylglycerides as Triglyceride Precursors
The phosphate of a phosphatidic acid is removed, by phosphatidic acid phosphatase (PAP1), to yield 1,2-diacylglycerols (DAG; also termed diglycerides). The PAP1 enzyme is encoded by a gene that is a member of the lipin family (see Lipin Genes: Triglyceride Synthesis and Transcriptional Regulation section below) identified as the LPIN1 gene. The DAG, along with a fatty acyl-CoA, serve as substrates for a diacylglycerol O-acyltransferase (DGAT) enzyme forming a triglyceride. Humans express two DGAT encoding genes identified as DGAT1 and DGAT2.
The DGAT1 gene is located on chromosome 8q24.3 and is composed of 21 exons that encode a 488 amino acid protein. The DGAT1 encoded enzyme primarily utilizes exogenous (dietary) fatty acids for the synthesis of triglycerides.
The DGAT2 gene is located on chromosome 11q13.5 and is composed of 10 exons that generate two alternatively spliced mRNAs encoding two isoforms of the enzyme. The DGAT2 gene is expressed at highest levels in the liver and utilizes newly synthesized fatty acids as its substrates for the synthesis of triglycerides.
The DGAT2 gene belongs to a family of related genes identified as the diacylglycerol O-acyltransferase 2 family. The DGAT2 family is composed of eight genes that includes the three monoacylglycerol O-acyltransferase genes described in the previous section. One of the eight DGAT2 family genes is a non-functional pseudogene. The three MOGAT genes were previously identified as diacylglycerol O-acyltransferase 2-like 1 (DGAT2L1), DGAT2L2, and DGAT2L3.
Role of DGAT2 in Hepatic Steatosis
Evidence in mice has demonstrated that inhibition, or deletion, of DGAT2 results in a reduction in the level of triglycerides in the liver. This work has led to the development, and testing, of orally administered DGAT2 inhibitors for the treatment of non-alcoholic fatty liver disease (NAFLD) and non-alcoholic steatohepatitis (NASH). NAFLD and NASH are now referred to as metabolic dysfunction-associated fatty (or steatotic) liver disease (MAFLD or MASLD) and metabolic dysfunction-associated steatohepatitis (MASH).
One such DGAT2 inhibitor, ervogastat, when co-administered with an oral inhibitor of acetyl-CoA carboxylase (ACC), clesacostat, has been granted fast-track status by the US FDA for approval for use in humans with MASH (NASH).
When DGAT2 was inhibited or deleted in mice it was found that in addition to reduced levels of the DGAT2 encoded enzyme, there was reduction in the expression of SREBP-1c-regulated genes. This result suggested a link between DGAT2 and the activity of the SREBP transcription factor. Expression of several SREBP-1c target genes, including those encoding acyl-CoA synthetase short-chain family member 2 (ACSS2), ATP-citrate lyase (ACLY), acetyl-CoA carboxylase 1 (ACACA), acetyl-CoA carboxylase 2 (ACACB), fatty acid synthase (FASN), and patatin-like phospholipase domain containing 3 (PNPLA3), are significantly reduced when DGAT2 is inhibited. The PNPLA3 encoded protein belongs to the inducible PLA2 (iPLA2) family of PLA2 enzymes and it is commonly called adiponutrin. Expression of the PNPLA3 gene localized to the liver and adipose tissue. Unlike the effects of DGAT2 inhibition of SREPB-1c activity, there is no significant effect on the activity of the related transcription factor, SREBP-2.
The role of DGAT2 in the activity of SREBP-1c involves the cleavage of SREBP-1c and thus, activation of its transcription factor activity. However, inhibition of DGAT2 is not associated with a reduction in the content of cholesterol in the membranes of the endoplasmic reticulum (ER) indicating that DGAT2 does not contribute to SREBP-1c activity through modulation of the role of cholesterol in the activity of SCAP, the protein that retains SREBP proteins in ER preventing migration to the Golgi where cleavage normally takes place. Inhibition of DGAT2 is associated with increased phosphatidylethanolamine (PE) content in the ER membrane. The alteration in PE content contributes to the suppression of SREBP-1c cleavage thereby resulting in reduced expression of SREBP-1c target genes.
Glycerolipid-Fatty Acid Cycle
The glycerolipid-fatty acid cycle represents a central pathway of overall energy homeostasis. Th cycle integrates glucose and lipid metabolism and also generates bioactive lipid molecules, the lysophosphatidic acids (LPA or LysoPA). When fuels are abundant, excess glucose and free fatty acids are diverted into triglyceride and glycerolipid synthesis. The glucose is metabolized to glycerol-3-phosphate and the fatty acids are esterified to this molecule to generate triglycerides. However, in the context of the glycerolipid-fatty acid cycle the synthesis of lysoPA, phosphatidic acid (PA), and phospholipids also takes place.
The glycerolipid-fatty acid cycle consists of both lipid synthesis (lipogenesis) and lipid metabolism (lipolysis) segments and as such serves to generate complex bioactive lipids that control many biological processes. These biological processes include, but are not limited to, the synthesis, secretion, and function of insulin. Indeed, the glycerolipid-fatty acid cycle is disturbed in obesity and type 2 diabetes.
During lipogenesis reactions the glycerol-3-phopshate derived from glycolysis is esterified, as described above, to generate triglycerides. As indicate, during triglyceride synthesis the intermediates, lysoPA, PA, and diacylglycerides all constitute bioactive lipids. When triglycerides are broke down, in the context of lipolysis reactions, diacylglycerides, monoacylglycerides, free fatty acids, and glycerol are generated.
The function of the glycerolipid-fatty acid cycle can be regulated through the dephosphorylation of glycerol-3-phosphate to glycerol. This dephosphorylation is catalyzed by an enzyme identified as phosphoglycolate phopshatase. Phosphoglycolate phosphatase is encoded by the PGP gene. The PGP gene is located on chromosome 16p13.3 and is composed of 2 exons that encode a 321 amino acid protein. Expression of the PGP gene is ubiquitous with highest level seen in testes, heart, skeletal muscle, and pancreas. The high level of expression of PGP in heart and skeletal muscle is likely to ensure that toxic levels of lipids do not accumulate in these two tissues that prefer to oxidize fatty acids for energy production.
The level of phosphoglycolate phosphatase activity is directly correlated to the control of glycolysis, lipogenesis, lipolysis, fatty acid oxidation, mitochondrial energy metabolism, and the cellular redox state, particularly in pancreatic β-cells and hepatocytes. In addition, the activity of phosphoglycolate phosphatase is involved in the regulation of glucose-stimulated insulin secretion (GSIS), the response of pancreatic β-cells to stress, and in control of gluconeogenesis in hepatocytes.
Lipin Genes: Triglyceride Synthesis and Transcriptional Regulation
In addition to its role in overall TAG and phospholipid homeostasis, phosphatidic acid phosphatase (PAP1: encoded by the LPIN1 gene) has been shown to possess additional important activities. The lipin-1 encoding gene (LPIN1) was originally identified as being mutated in a line of mice possessing a fatty liver dystrophy syndrome. This mouse line is identified as the fld mouse.
Subsequent to the identification of the LPIN1 gene in the fld mouse, two additional related genes were identified. These three lipin genes are identified as LPIN1, LPIN2, and LPIN3. All characterized lipin proteins possess phosphatidic acid phosphatase activity that is dependent upon Mg2+ or Mn2+ and phosphatidic acid as the substrate.
The LPIN1 gene is located on chromosome 2p25.1 and is composed of 29 exons that generate 13 alternatively spliced mRNAs that collectively encode nine distinct protein isoforms.
The LPIN2 gene is located on chromosome 18p11.31 and is composed of 25 exons that generate three alternatively spliced mRNAs, each of which encode the same 896 amino acid protein. Mutations in the LPIN2 gene are associated with Majeed syndrome which is characterized by chronic recurrent osteomyelitis, cutaneous inflammation, recurrent fever, and congenital dyserythropoietic anemia.
The LPIN3 gene is located on chromosome 20q12 and is composed of 24 exons that generate two alternatively spliced mRNAs, both of which encode distinct protein isoforms.
In addition to the obvious role of lipin-1 in triglyceride synthesis, evidence indicates that the protein is also required for the development of mature adipocytes, coordination of peripheral tissue glucose and fatty acid storage and utilization, and serves as a transcriptional co-activator. The latter function has significance to diabetes as it has been shown that some of the effects of the thiazolidinedione (TZD) class of drugs used to treat the hyperglycemia associated with type 2 diabetes are exerted via the effects of lipin-1. Although the lipin proteins do not contain DNA-binding motifs they have protein-interaction domains that allow them to form complexes with nuclear receptors and function as transcriptional regulators.
Lipin-1 has been shown to interact with peroxisome proliferator-activated receptor-γ [PPARγ] co-activator 1α (PGC-1α) and PPARα leading to enhanced gene expression. Lipin-1 also is known to interact with additional members of the nuclear receptor family including the glucocorticoid receptor (GR) and hepatocyte nuclear factor-4α (HNF-4α).
Lipin-1 also induces the expression of the adipogenic transcription factors PPARγ and CCAAT-enhancer-binding protein α (C/EBPα). The functions of lipin-1α and lipin-1β appear to be complimentary with respect to adipocyte differentiation. Lipin-1α induces genes that promote adipocyte differentiation while lipin-1β induces the expression of lipid synthesizing genes such as fatty acid synthase (FAS) and diacylglycerol acyltransferase (DGAT). The interactions of lipin-1 with PPARα and PGC-1α leads to increased expression of fatty acid oxidizing genes such as carnitine palmitoyltransferase 1 (CPT1: encoded by the CPT1A gene), acyl-CoA oxidases (peroxisomal fatty acid oxidation pathway), and medium-chain acyl-CoA dehydrogenase (MCAD: encoded by the ACADM gene).