
Last Updated: March 26, 2026
Introduction to Lipid Synthesis Pathways
All cells possess the capacity to undertake the synthesis of fatty acids and phospholipids as a means to ensure continuous integrity of existing membranes as well as in the synthesis of new membranes. However, the majority of fatty acid biosynthesis occurs in hepatocytes and adipocytes.
Synthesis of triglycerides occurs in most cells but predominantly occurs in intestinal enterocytes for the delivery of dietary fatty acids to the body and hepatocytes of the liver for the delivery of endogenous fatty acids to, primarily, cardiac and skeletal muscle and to adipocytes. Adipocytes can be considered the major cell types tasked with triglyceride synthesis as these are the primary fatty acid storage cells the body.
Fatty acid, triglyceride, and phospholipid synthesis represent reductive biosynthetic processes and as such utilize NADPH as the co-factor for the reductive reactions. The major sources of the NADPH are the dehydrogenases of the Pentose Phosphate Pathway and the the malic enzyme catalyzed oxidation of cytoplasmic malate to pyruvate as outlined in the Figure in the next section and discussed in detail below.
The synthesis of fatty acids takes place in the cytosol. The synthesis of triglycerides takes place within the endoplasmic reticulum, ER. The synthesis of phospholipids occurs on the cytoplasmic face of the membranes of the ER.
Origin of Cytoplasmic Acetyl-CoA
The cytoplasmic acetyl-CoA that is required for fatty acid biosynthesis (and cholesterol biosynthesis) is initially generated in the mitochondria primarily from two sources, the pyruvate dehydrogenase (PDH) reaction and from mitochondrial amino acid catabolism. Indeed, within the liver the carbons of amino acids, via TCA cycle oxidation, significantly contribute to hepatic lipid synthesis and to a much greater extent than do the carbons of glucose.
Mitochondrial acetyl-CoA is also generated from the oxidation of fatty acids, but under conditions of enhanced lipogenesis, fatty acid oxidation is significantly reduced. This latter effect is the result of malonyl-CoA-mediated inhibition of carnitine palmitoyltransferase 1, CPT1. Malonyl-CoA is the product of the rate-limiting enzyme of de novo fatty acid synthesis as discussed in detail below.
In order for these acetyl-CoA units to be utilized for fatty acid synthesis they must be present in the cytoplasm. The shift from glucose oxidation, fatty acid oxidation, and amino acid oxidation, occurs when the need for energy diminishes. This results in reduced oxidation of acetyl-CoA in the TCA cycle and the re-oxidation of reduced NADH and FADH2 via the oxidative phosphorylation pathway. Under these conditions the mitochondrial 2-carbon acetyl units can be stored as fat for future energy demands or diverted into cholesterol biosynthesis.
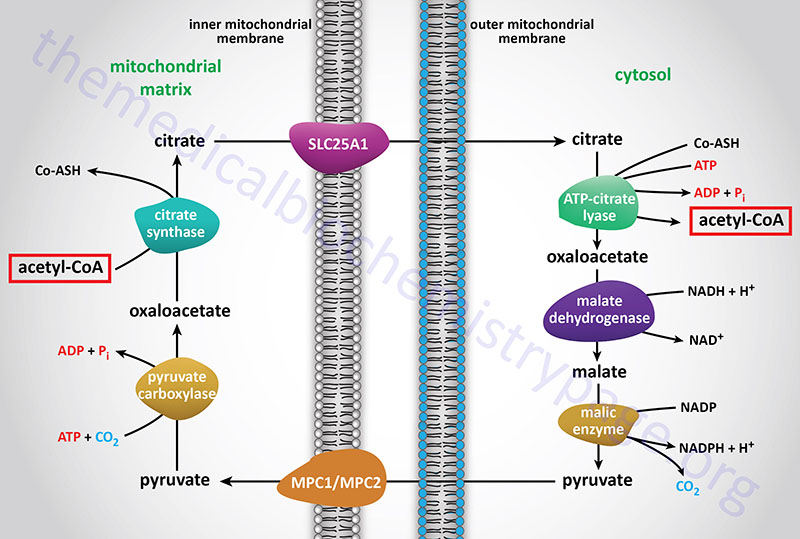
Acetyl-CoA enters the cytoplasm in the form of citrate via the tricarboxylate transport system (encoded by the SLC25A1 gene; see Figure above). In the cytoplasm, citrate and coenzyme A (CoASH) are converted to oxaloacetate and acetyl-CoA by the ATP driven ATP-citrate lyase reaction. This reaction is essentially the reverse of that catalyzed by the TCA cycle enzyme, citrate synthase, except it requires the energy of ATP hydrolysis to drive it forward. The resultant oxaloacetate is converted to malate by cytoplasmic malate dehydrogenase (encoded by the MDH1 gene).
ATP-citrate lyase is encoded by the ACLY gene which is located on chromosome 17q21.2 and is composed of 30 exons that generate four alternatively spliced mRNAs, each of which encodes a unique protein isoform.
Functional ATP-citrate lyase is a homotetrameric complex. ATP-citrate lyase is a critical metabolic enzyme that links glucose metabolism to the processes of both fatty acid synthesis and cholesterol synthesis. In addition to the role of ATP-citrate lyase in cytosolic lipid synthesis, the enzyme is found in the nucleus where it plays a role in epigenetic control of gene expression by providing the acetyl-CoA used in histone acetylation.
The malate produced by this pathway can undergo oxidative decarboxylation by cytoplasmic malic enzyme (encoded by the ME1 gene). The co-enzyme for this reaction is NADP+ generating NADPH. The advantage of this series of reactions for converting mitochondrial acetyl-CoA into cytoplasmic acetyl-CoA is that the NADPH produced by the cytosolic malic enzyme reaction can be a major source of reducing co-factor for the enzymatic activities of the fatty acid synthase (FAS) complex.
Acetate as a Substrate for de novo Fatty Acid Synthesis
Whereas acetyl-CoA, generated within the mitochondria via pyruvate (from glucose oxidation) and amino acid oxidation, represents the primary source of carbon for the pathways of de novo fatty acid synthesis and cholesterol synthesis, both acetate and acetoacetate can contribute to fatty acid synthesis.
Under normal conditions the level of acetate in human serum is < 0.2mM. Normal physiological sources of acetate include bacterial fermentation in the colon which increases significantly when consuming a high fiber diet. This intestinal acetate enters the portal circulation and is taken up by the liver where it is converted to acetyl-CoA. Intracellular generation of acetate is the consequence of the ubiquitously expressed cytosolic enzyme acetyl-CoA hydrolase. The acetate can then be salvaged by reactivation to acetyl-CoA.
In the nervous system, the neurotransmitter acetylcholine (ACh) is degraded to acetate by acetylcholinesterase. In order to replenish the pool of ACh the acetate must be reactivated to acetyl-CoA so that it can participate in the choline acetyltransferase catalyzed reaction.
Acetate is also generated within the nucleus of all cells via the action of histone deacetylases (HDAC). As with the other sources of acetate, this nuclear acetate must be reactivated before it can be oxidized or reused.
Under conditions of prolonged starvation and in type 1 diabetes, the endogenous pathways of acetate production are the main sources for serum acetate. Following the consumption of ethanol, acetate levels can be elevated by as much as 20–fold. Indeed, metabolism of ethanol is a significant contributor the development of hepatic steatosis (fatty liver) which can progress to steatohepatitis and hepatic cancer.
Acetate, from whatever source, is converted to acetyl-CoA by ATP-dependent acetyl-CoA synthetases (AceCS: encoded by acyl-CoA synthetase short-chain family member genes: ACSS). Humans express cytosolic/nuclear and mitochondrial acetyl-CoA synthetases, encoded by the ACSS2 and ACSS1 genes, respectively. Expression of ACSS2 is under the control of SREBP-1c which is itself transcriptionally regulated by PGC-1α. Interestingly, mammalian AceCS are regulated by reversible acetylation catalyzed by the sirtuins, SIRT1 and SIRT3, and the activity of PGC-1α is also regulated by SIRT1 activity. Cytosolic ACSS2 is a target of cytoplasmic SIRT1 whereas, mitochondrial ACSS1 is a target of mitochondrial SIRT3.
N-Acetylaspartate Role in Brain Fatty Acid Synthesis
Neurons in the brain possess unique pathways, relative to other tissues of the body, for the synthesis of lipids such as fatty acids. The acetyl-CoA necessary for cytosolic fatty acid synthesis in cells in the brain, particularly oligodendrocytes, is derived from N-acetylaspartate (NAA), not from citrate as is the case for other tissues. Indeed, NAA is considered the second most abundant metabolite in the brain. NAA is synthesized in neurons from aspartate and acetyl-CoA within the matrix of nerve cell mitochondria via the action of N-acetylaspartate synthetase which is encoded by the NAT8L (N-acetyltransferase 8 like) gene.
The NAT8L gene is located on chromosome 4p16.3 and is composed of 3 exons that encode a 302 amino acid protein. The highest levels of expression of the NAT8L gene are in the brain and adipose tissue.
The NAA produced in neurons is transported to the cytosol of oligodendrocytes via the action of the sodium-dicarboxylate cotransporter encoded by the SLC13A3 gene. Within the cytosol of oligodendrocytes the enzyme, aspartoacylase, hydrolyzes N-acetylaspartate into acetate and aspartate. The acetate can then be converted to acetyl-CoA by ATP-dependent acetyl-CoA synthetases (AceCS: encoded by acyl-CoA synthetase short-chain family member genes: ACSS). Humans express cytosolic/nuclear and mitochondrial acetyl-CoA synthetases, encoded by the ACSS2 and ACSS1 genes, respectively.
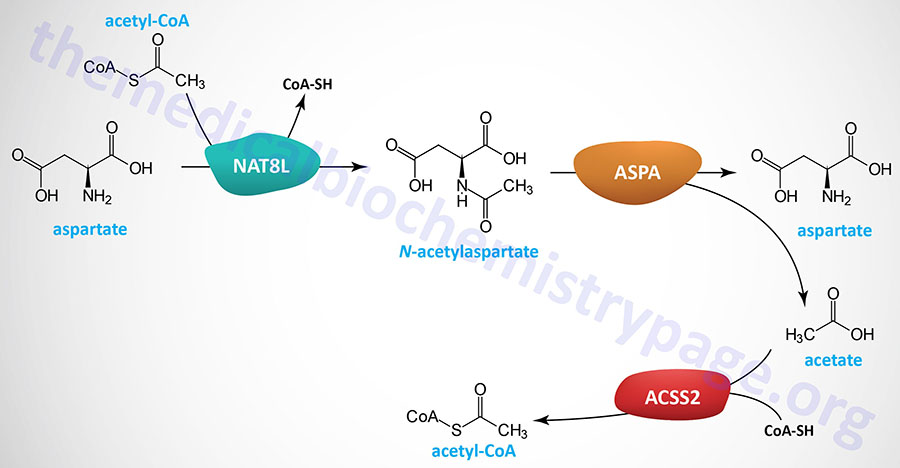
The clinical significance of brain N-acetylaspartate was ascertained from studies of the rare but fatal neonatal leukodystrophy known as Canavan disease. Canavan disease was found to be associated with toxic levels of NAA in the brain due to mutations in the gene (ASPA) encoding aspartoacylase. Aspartoacylase has also been called acylase II and N-acetyl-L-aspartate amidohydrolase.
The ASPA gene is located on chromosome 17p13.2 and is composed of 8 exons that generate two alternatively spliced mRNAs, both of which encoded the same 313 amino acid protein.
Aside from the role of NAA in the mobilization of neuronal acetate from the mitochondria to the cytosol of oligodendrocytes, where it serves in fatty acid biosynthesis, NAA also has osmotic effects in the brain. The role of NAA in osmotic regulation accounts for some of the pathology of Canavan disease which is associated with osmotic dysregulation in the brain which then results in dysmyelination and subcortical vacuolation.
Acetoacetate as a Substrate for de novo Fatty Acid Synthesis
The primary source of acetoacetate is via the hepatic pathway of ketone body synthesis. The oxidation of the branched-chain amino acid leucine also contributes to the production of acetoacetate in the mitochondria. Mitochondrial acetoacetate diffuses across the mitochondrial membrane to the cytosol where the carbons can be utilized in de novo lipogenesis pathways.
Cytosolic acetoacetate is converted to acetoacetyl-CoA via the actions of the cytoplasmic acetoacetyl-CoA synthetase encoded by the AACS gene. The acetoacetyl-CoA is then hydrolyzed into two moles of acetyl-CoA by the cytosolic thiolase encoded by the ACAT2 (acetyl-CoA acetyltransferase 2) gene. These acetyl-CoA molecules can be utilized for fatty acid synthesis as well as for cholesterol synthesis.
Although ketones are not actively utilized by the liver, due to low level expression of the mitochondrially-localized enzyme encoded by the OXCT1 gene, cytosolic acetoacetate can contribute to de novo lipogenesis in the liver through the actions of the AACS and ACAT2 encoded enzymes.
Malic Enzymes
Humans express three malic enzymes, one cytoplasmic that requires NADP+ and two mitochondrial enzymes, one that requires NADP+ and one that requires NAD+.
The NADP+-dependent cytoplasmic enzyme is called malic enzyme 1 and is encoded by the ME1 gene. The ME1 gene is located on chromosome 6q14.2 and is composed of 14 exons that encode a protein of 572 amino acids. The highest level of ME1 expression is in adipose tissue attesting to the major role of the ME1 enzyme in fatty acid synthesis. The NADPH produced by the ME1 catalyzed reaction can be used in biomass producing reductive biosynthetic reactions such as fatty synthesis, cholesterol synthesis, and phospholipid synthesis.
The NAD+-dependent mitochondrial enzyme is called malic enzyme 2 and is encoded by the ME2 gene. The ME2 gene is located on chromosome 18q21.2 and is composed of 16 exons that generate two alternatively spliced mRNAs encoding precursor proteins of 584 amino acids (isoform 1) and 479 amino acids (isoform 2).
The NADP+-dependent mitochondrial enzyme is called malic enzyme 3 and is encoded by the ME3 gene. The ME3 gene is located on chromosome 11q14.2 and is composed of 23 exons that generate five alternatively spliced mRNAs that collectively two protein isoforms, a 604 amino acid protein (isoform 1) and a 559 amino acid protein (isoform 2).
The role of the mitochondrial malic enzymes is principally to provide the cell with an alternate source of pyruvate under conditions where glycolytic flux in reduced. In these circumstances, the pyruvate generated by the actions of ME2 and/or ME3 comes from fumarate precursors such as glutamine.
In neurons, as well as in numerous types of tumor cells, mitochondrial malic enzymes allow for the utilization of the amino acid glutamine as a fuel source. When glutamine is de-aminated by glutaminase the resulting glutamate can also be de-aminated by glutamate dehydrogenase yielding 2-oxoglutarate (α-ketoglutarate) which can then be shunted to malate synthesis in the TCA cycle. The malate can then be decarboxylated to pyruvate via mitochondrial malic enzyme. The pyruvate can then be decarboxylated by the PDHc and the resulting acetyl-CoA can enter the TCA cycle ultimately allowing for glutamine carbons to be oxidized for ATP synthesis.
Within β-cells of the pancreas, this process driven by mitochondrial malic enzyme serves as an important means for the use of amino acid carbon oxidation for the stimulated secretion of insulin. Indeed, this process is energetically equal to glucose-stimulated insulin secretion (GSIS).
The malic enzyme encoded by the ME2 gene also plays a critical role in the progression of certain types of cancers, particularly leukemias. The role of ME2 in these types of cancers relates to its functions in mitochondrial biogenesis.
Reactions of Fatty Acid Synthesis
One might predict that the pathway for the synthesis of fatty acids would be the reversal of the oxidation pathway. However, this would not allow distinct regulation of the two pathways to occur even given the fact that the pathways are separated within different cellular compartments. The pathway for fatty acid synthesis occurs in the cytoplasm, whereas, oxidation occurs in the mitochondria. The other major difference is the use of nucleotide co-factors. Oxidation of fats involves the reduction of FAD and NAD+. Synthesis of fats involves the oxidation of NADPH. However, the essential reaction chemistry of the two processes are reversals of each other. Both oxidation and synthesis of fats utilize an activated two carbon intermediate, acetyl-CoA. However, the activated form of acetyl-CoA in fat synthesis exists temporarily bound to the enzyme complex as malonyl-CoA.
Acetyl-CoA Carboxylase (ACC) Reaction
The synthesis of malonyl-CoA is the first committed step of fatty acid synthesis and the enzyme that catalyzes this reaction, acetyl-CoA carboxylase (ACC), is the rate-limited enzyme and major site of regulation of fatty acid synthesis. Like other enzymes that transfer CO2 to substrates, ACC requires a biotin as a co-factor. Acetyl-CoA carboxylase is called an ABC enzyme due to the requirements for ATP, Biotin, and CO2 for the reaction.
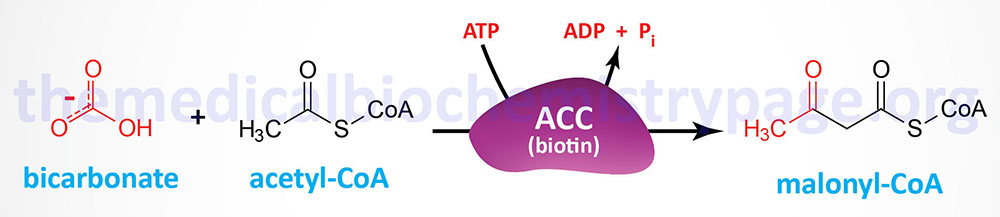
Humans express two major isoforms of ACC. These are identified as ACC1 (also called ACCα) and ACC2 (also called ACCβ).
The ACC1 enzyme is encoded by the ACACA gene. The ACACA gene is located on chromosome 17q12 and is composed of 66 exons that undergo alternative splicing to yield five splice variant mRNAs that generate four different precursor proteins from 2268 to 2383 amino acids in length. Transcriptional regulation of ACACA is effected by three promoters (PI, PII, and PIII), which are located upstream of exons 1, 2, and 5A, respectively. The PI promoter is a constitutive promoter, the PII promoter is regulated by various hormones, and the PIII promoter is expressed in a tissue-specific manner. The presence of the alternatively spliced exons does not alter the translation of the ACC1 protein which starts from an AUG present in exon 5.
The ACC2 enzyme is encoded by the ACACB gene. The ACACB gene is located on chromosome 12q24.11 and is composed of 58 exons that encode a precursor protein of 2,458 amino acids. The highest levels of expression of the ACACB gene are found in adipose tissue followed by heart and skeletal muscle.
Fatty Acid Synthase (FAS) Reactions
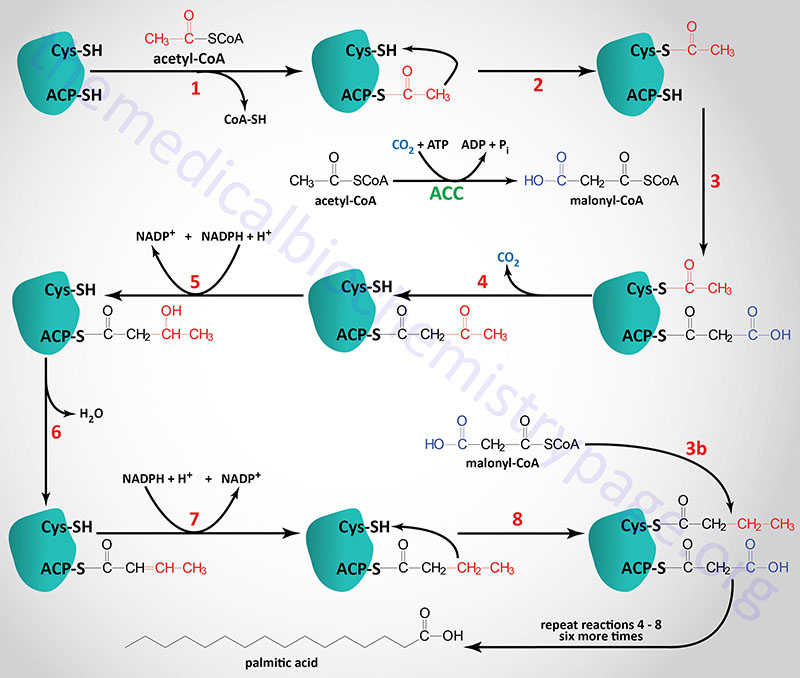
The acetyl groups that are the products of fatty acid and amino acid oxidation are linked to CoASH. As outlined in the Vitamins: Water and Fat Soluble page, CoASH contains a phosphopantetheine group coupled to AMP. The carrier of acetyl groups (and elongating acyl groups) during fatty acid synthesis is also a phosphopantetheine prosthetic group, however, it is attached to a serine hydroxyl in one of the active sites of the fatty acid synthase (FAS) complex.
The acetyl and acyl carrier portion of the FAS complex is called acyl carrier protein, ACP. This is somewhat of a misnomer in eukaryotic fatty acid synthesis since the ACP portion of the synthetic complex is simply one of many catalytic domains of a single polypeptide. As indicated, the ACP of fatty acid synthase, as well as several other lipid biosynthetic enzymes, is 4′-phosphopantetheine, an intermediate in the synthesis of CoASH. The 4′-phosphopantetheine from CoA is covalently attached to a specific Ser residue via a phosphodiester linkage. This reaction is catalyzed by L-aminoadipate-semialdehyde dehydrogenase-phosphopantetheinyltransferase which is encoded by the AASDHPPT gene. During the course of the attachment, 3′-phosphoadenosine 5′-phosphate (PAP) is released.
The acetyl-CoA and malonyl-CoA are transferred to ACP by the action of the malonyl/acetyltransferase activity (also called acetyl-CoA transacylase and malonyl-CoA transacylase). The attachment of these carbon atoms to ACP allows them to enter the fatty acid synthesis cycle.
The synthesis of fatty acids from acetyl-CoA and malonyl-CoA is carried out by fatty acid synthase, FAS. Fatty acid synthase is encoded by the FASN gene. The FASN gene is located on chromosome 17q25.3 and is composed of 43 exons that encode a protein of 2511 amino acids. The highest level of expression of the FASN gene is found in adipose tissue at levels that are several hundred-fold higher than any other tissue.
The active FAS enzyme functions as a head-to-tail homodimer. All of the reactions of fatty acid synthesis are carried out by the multiple enzymatic activities of FAS. Like fat oxidation, fat synthesis involves four primary enzymatic activities. These are (in order of reaction), β-ketoacyl-ACP synthase (contained in a domain composed of amino acids 2–404), β-ketoacyl-ACP reductase (contained in a domain composed of amino acids 1876–2112), 3-hydroxyacyl-ACP dehydratase (contained in a domain composed of amino acids 874–1106) and enoyl-CoA reductase (contained in a domain composed of amino acids 1564–1854). The two reduction reactions require NADPH oxidation to NADP+. The domain that is required for attachment and transfer of acetyl-CoA and malonyl-CoA (acyltransferase domain) is composed of amino acids 493–810. The phosphopantetheine arm of FAS is attached to a domain composed of amino acids 2111-2179.
The primary fatty acid synthesized by FAS is the fully saturated 16 carbon fatty acid, palmitic acid (palmitate). Palmitate is then released from the enzyme via the thioesterase activity of FAS (contained in a domain composed of amino acids 2242–2488). Once released, palmitate can then undergo separate elongation and/or unsaturation to yield other fatty acid molecules.
Metabolic Roles for Palmitate
Given that palmitate is the end-product of de novo fatty acid synthesis, it is not surprising that it represents the most abundant saturated fatty acid in human tissues and blood. In addition, palmitate constitutes 20%–30% of the fatty acid composition of the phospholipid bilayer of cellular membranes.
As detailed in the Elongation and Desaturation section below, palmitate is the primary substrate for the synthesis of numerous longer saturated fatty acids, such stearic acid (18:0), and these saturated fatty acids can undergo desaturation forming numerous mono- (MUFA) and polyunsaturated fatty acids (PUFA). Two of the most significant monounsaturated fatty acids in humans are oleic acid (18:1; a physiologically significant omega-9 fatty acid) and palmitoleic acid (16:1; a physiologically significant omega-7 fatty acid). These two monounsaturated fatty acids represent the majority of monounsaturated fatty acids present in membrane phospholipids, triglycerides, and cholesterol esters.
Palmitic acid functions in the modification of proteins via a process termed protein S-palmitoylation. Protein S-palmitoylation is an important post-translational modification that functions in the regulation of membrane attachment, intracellular trafficking, and membrane subdomain localization. The bulk of S-palmitoylation occurs on the sulfhydryl of internal cysteine residues, however, important examples of N-terminal palmitoylated proteins are known.
Palmitic acid, when activated to palmitoyl-CoA, and serine serve as the precursors for the synthesis of the ceramides and sphingosine.
Palmitic acid has also been shown to exert pro-inflammatory effects in macrophages by triggering an increase in reactive oxygen species (ROS) production and enhancing the secretion of pro-inflammatory cytokines. Pro-inflammatory effects of palmitate are also exerted in adipose tissue and in the brain.
Regulation of Fatty Acid Metabolism
Regulation by Acetyl-CoA Carboxylase: ACC
One must consider the body’s global integrated energy requirements in order to effectively understand how the synthesis and degradation of fats (and also carbohydrates) needs to be exquisitely regulated. The blood is the carrier of triglycerides [contained within very low density lipoproteins (VLDL) and chylomicrons], fatty acids bound to albumin, amino acids, lactate, ketone bodies, and glucose. The pancreas is the primary organ involved in sensing dietary and energy states primarily via glucose concentrations in the blood but also via glutamine levels and to a lesser extent fatty acid levels. In response to low blood glucose, glucagon is secreted, whereas, in response to elevated blood glucose insulin is secreted.
The regulation of fat metabolism occurs via two distinct mechanisms. One is short-term regulation which is regulation effected by events such as substrate availability, allosteric effectors and/or enzyme modification. The other is long-term regulation which involves the regulation of gene expression.
As indicated above, ACC represents the rate-limiting reaction of de novo fatty acid synthesis and humans express two forms of the enzyme identified as ACC1 and ACC2.
ACC1 is strictly cytosolic and is enriched in liver, adipose tissue, and lactating mammary tissue. ACC2 was originally discovered in rat heart but is also expressed in liver and skeletal muscle.
ACC2 has an N-terminal extension that contains a mitochondrial targeting motif and is found associated with carnitine palmitoyltransferase 1 (CPT1) allowing for rapid regulation of CPT1 by the malonyl-CoA produced by ACC2. The localization of ACC2 and the inhibition of CPT1 by its product, malonyl-CoA, indicates that ACC2 is primarily involved in the regulation of fatty acid oxidation as opposed to fatty acid synthesis.
Both isoforms of ACC are allosterically activated by citrate and inhibited by palmitoyl-CoA and other short- and long-chain fatty acyl-CoAs. Citrate triggers the polymerization of ACC1 which leads to significant increases in its activity. Although ACC2 does polymerize in response to citrate, the extent of oligomerization does not appear to be as great as for ACC1, which is presumed to be due to its mitochondrial association. Glutamate and other dicarboxylic acids can also allosterically activate both ACC isoforms.
ACC activity can also be affected by phosphorylation. Both ACC1 and ACC2 contain at least eight sites that undergo phosphorylation. The sites of phosphorylation in ACC2 have not been as extensively studied as those in ACC1. Phosphorylation of ACC1 at three serine residues (S79, S1200, and S1215) by AMPK leads to inhibition of the enzyme. AMPK also phosphorylates and inhibits the activity of ACC2. Glucagon-stimulated increases in cAMP and subsequently to increased PKA activity also lead to phosphorylation of ACC where ACC2 is a better substrate for PKA than is ACC1. Epinephrine activation of β-adrenergic receptors also results in increased PKA activity and consequently phosphorylation and inhibition of ACC1.
The activating effects of insulin on ACC are complex and not completely resolved. It is known that insulin leads to the dephosphorylation of the phosphoserines in ACC1 that are AMPK targets in the heart enzyme. This insulin-mediated effect has not been observed in hepatocytes or adipose tissues cells. At least a portion of the activating effects of insulin are related to changes in cAMP levels. Early evidence had shown that phosphorylation and activation of ACC occurs via the action of an insulin-activated kinase. However, contradicting evidence indicates that although there is insulin-mediated phosphorylation of ACC this does not result in activation of the enzyme.
Regulation of ACC2 by Prolyl Hydroxylation
The activity of ACC2 is also regulated by hydroxylation. As the level of glucose rises the extent of ACC2 hydroxylation increases and the level of malonyl-CoA rises resulting in inhibition of fatty acid oxidation. The primary enzyme responsible for the hydroxylation of ACC2 is a member of the prolyl hydroxylase domain (PHD) family of enzymes. Humans express three PHD family member encoding genes, PHD1, PHD2, and PHD3. The PHD family enzymes are members of the large family of 2-oxoglutarate (α-ketoglutarate) and Fe2+ (iron)-dependent dioxygenases (2OG-oxygenases).
Within cardiac and skeletal muscle the PHD3 encoded enzyme has been shown to hydroxylate ACC2. When AMPK phosphorylates ACC2 during periods of energetic stress, PHD3 can no longer hydroxylate the enzyme. Studies have shown that loss of PHD3 activity (in knock-out mice) results in a significant reduction in ACC2 activity. The reduced ACC2 activity, resulting in reduced levels of malonyl-CoA, is associated with increased fatty acid delivery to the mitochondria for oxidation which promotes exercise capacity.
Regulation by Malonyl-CoA Decarboxylase: MCD
The intracellular levels of malonyl-CoA represent a balance between its synthesis from acetyl-CoA by ACC and its utilization in fatty acid synthesis by FAS as well as by its degradation to acetyl-CoA via the action of malonyl-CoA decarboxylase (MCD). Indeed, MCD is involved in regulating malonyl-CoA levels in multiple tissues. MCD is found in the cytosol, the mitochondria, and in peroxisomes.
The MCD enzyme is encoded by the MLYCD gene. The MLYCD gene is located on chromosome 16q23.3 and is composed of 5 exons encoding a protein of 493 amino acids.
Inhibition of MCD results in reduced rates of fatty acid oxidation in highly oxidative tissues such as the heart. As well, MCD inhibition leads to reduced triglyceride content in lipid synthesizing tissues such as the liver.
When hypothalamic MCD levels are experimentally increased in laboratory animals there is a dramatic increase in food intake, weight gain, and ultimately results in obesity. Conversely, inactivation of the MCD gene in mice protects the animals from high-fat diet-induced insulin resistance. Additionally, chemical inhibition of MCD leads to reduced macrophage-associated inflammation in conditions of insulin resistance. The primary mechanism for this effect appears to be via the accumulation of long-chain fatty acids which in turn activate PPARα.
Interestingly, reduced MCD activity also exerts a beneficial effect in the hypothalamus. Transcriptional regulation of the MCD gene is exerted by PPARα, a major transcription factor involved in the regulation of fatty acid oxidation. Hypothalamic PPARα has been shown to play a role in the regulation of appetite, presumably via enhanced expression of MCD. PPARα-mediated increases in MCD levels results in reduced levels of malonyl-CoA in the hypothalamus. This is associated with increased food intake in the FAS knock-out mice demonstrating that malonyl-CoA levels are indeed responsible for the hypophagic effects observed in the FAS knock-out mice. The potential therapeutic benefits to reduced MCD activity in the treatment of obesity and diabetes are currently undergoing intensive investigation.
Mutations in the MLYCD gene are associated with a very rare disorder referred to as malonic acidemia or malonic aciduria. The symptoms of this disorder are variable but generally appear in early childhood and include developmental delay, hypoglycemia, metabolic acidosis, diarrhea, hypotonia, cardiomyopathy, and seizures.
Additional Processes Regulating Fatty Acid Synthesis
Control of a given pathways’ regulatory enzymes can also occur by alteration of enzyme synthesis and turn-over rates. These changes are long term regulatory effects. Insulin stimulates ACC and FAS synthesis, whereas, starvation leads to decreased synthesis of these enzymes. Adipose tissue lipoprotein lipase levels also are increased by insulin and decreased by starvation. However, in contrast to the effects of insulin and starvation on adipose tissue, their effects on heart lipoprotein lipase are just the inverse. This allows the heart to absorb any available fatty acids in the blood in order to oxidize them for energy production. Starvation also leads to increases in the levels of fatty acid oxidation enzymes in the heart as well as a decrease in FAS and related enzymes of synthesis.
Adipose tissue contains hormone-sensitive lipase (HSL), that is activated by PKA-dependent phosphorylation leading to increased fatty acid release to the blood. The activity of HSL is also affected through the action of AMPK, however, AMPK-mediated phosphorylation of HSL is inhibitory. The phosphorylation and inhibition of HSL by AMPK may seem paradoxical since the release of fatty acids stored in triglycerides would seem necessary to promote the production of ATP via fatty acid oxidation and the major function of AMPK is to shift cells to ATP production from ATP consumption. This paradigm can be explained if one considers that if the fatty acids that are released from triglycerides are not consumed they will be recycled back into triglycerides at the expense of ATP consumption. Thus, it has been proposed that inhibition of HSL by AMPK mediated phosphorylation is a mechanism to ensure that the rate of fatty acid release does not exceed the rate at which they are utilized either by export or oxidation.
In the liver the net result of activation of HSL (due to increased acetyl-CoA levels) is the production of ketone bodies. This would occur under conditions where insufficient carbohydrate stores and gluconeogenic precursors were available in liver for increased glucose production. The increased fatty acid availability in response to glucagon or epinephrine is assured of being completely oxidized since both PKA and AMPK also phosphorylate (and as a result inhibits) ACC, thus inhibiting fatty acid synthesis.
Insulin, on the other hand, has the opposite effect to glucagon and epinephrine leading to increased glycogen and triglyceride synthesis. One of the many effects of insulin is to lower cAMP levels which leads to increased dephosphorylation through the enhanced activity of protein phosphatases such as PP-1. With respect to fatty acid metabolism this yields dephosphorylated and inactive hormone sensitive lipase. Insulin also stimulates certain phosphorylation events. This occurs through activation of several cAMP-independent kinases.
Regulation of fat metabolism also occurs through malonyl-CoA induced inhibition of carnitine acyltransferase 1 (CPT1). This functions to prevent the newly synthesized fatty acids from entering the mitochondria and being oxidized. Indeed, as pointed out above, the localization of ACC2 to the mitochondrial outer membrane allows for rapid inhibition of CPT1 when fatty acid synthesis has been stimulated.
Mitochondrial Fatty Acid Synthesis
Synthesis of fatty acids in the mitochondria plays a critical role in the assembly of the components of the oxidative phosphorylation machinery as well as playing a role in the processing of mitochondrial RNAs. The process of mitochondrial fatty acid synthesis (mtFAS) closely resembles a bacterial type II fatty acid synthesis system (FASII) that involves individual enzymes for each step in the pathway. This is unlike cytosolic fatty acid synthesis which is catalyzed by two enzymes, acetyl-CoA carboxylase 1 (ACC1) and fatty acid synthase (FAS).
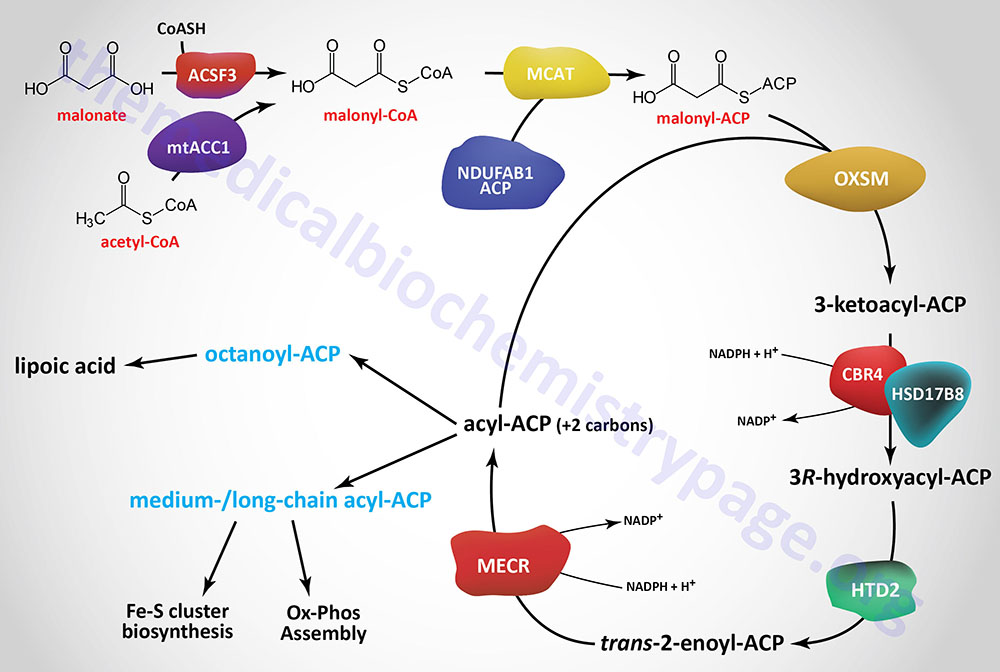
Initiation of mitochondrial fatty acid synthesis involves the use of either malonic acid or acetyl-CoA. Malonic acid is most likely transported into the mitochondria via the carnitine transport process similar to that required for mitochondrial entry of long-chain fatty acids in the process of fatty acid β-oxidation.
Within the mitochondria malonate is converted to malonyl-CoA by the ACSF3 (acyl-CoA synthetase family member 3) encoded enzyme. The ACSF3 encoded enzyme is also referred to as malonyl-CoA synthetase given that it exhibits specificity for malonic acid and methylmalonic acid. The major product of the ACSF3 encoded enzyme is malonyl-CoA.
Mitochondrial acetyl-CoA can be converted to malonyl-CoA by a mitochondria-localized enzyme (mtACC1) originating from an alternatively spliced mRNA derived from the ACACA gene. As described in the Acetyl-CoA Carboxylase (ACC) Reaction section above, the ACACA gene encodes the cytosolic ACC1 enzyme.
Mitochondrial fatty acid synthesis requires an acyl-carrier (ACP) which is the sulfhydryl (–SH) of 4′-phosphopantetheine (4′-PP) which is derived from coenzyme A (CoA) and has been added to a conserved Ser residue. This mitochondrial ACP function is similar to the ACP portion of cytosolic fatty acid synthase (FAS). Similar to fatty acid synthase, the 4′-phosphopantetheine from CoA is covalently attached to a specific Ser residue via a phosphodiester linkage. This reaction is catalyzed by L-aminoadipate-semialdehyde dehydrogenase-phosphopantetheinyltransferase which is encoded by the AASDHPPT gene. During the course of the attachment, 3′-phosphoadenosine 5′-phosphate (PAP) is released.
The mitochondrial ACP is a function of a subunit of mitochondrial oxidative phosphorylation complex I and is encoded by the NDUFAB1 (NADH:ubiquinone oxidoreductase subunit AB1) gene. This modified NDUFAB1 protein is the scaffold upon which mitochondrial fatty acids are synthesized.
The ACSF3 gene is located on chromosome 16q24.3 and is composed of 16 exons that generate four alternatively spliced mRNAs that collectively encode two different precursor proteins of 576 amino acids (isoform 1) and 311 amino acids (isoform 2). Translation of the isoform 2 protein initiates at a downstream start codon relative to isoform 1.
Mutations in the ACSF3 gene result in the disorder called combined malonic and methylmalonic aciduria (CMAMMA). Symptoms of CMAMMA commonly begin in early childhood and include ketoacidosis, hypoglycemia, muscle issues (dystonia and hypotonia), developmental delay, failure to thrive, and potentially coma. In some individuals symptoms don’t appear until adulthood and include seizures, loss of memory, difficulty thinking, or psychiatric abnormalities.
The malonyl-CoA produced in the first reaction of mitochondrial fatty acid synthesis is the substrate for malonyl-CoA-acyl carrier protein transacylase which is encoded by the MCAT gene. The MCAT enzyme is exclusively localized to the mitochondria. MCAT attaches the malonyl-CoA to the mitochondrial ACP, generating malonyl-ACP.
The MCAT gene is located on chromosome 22q13.2 and is composed of 4 exons that generate two alternatively spliced mRNAs encoding two distinct precursor proteins. The isoform a precursor is 390 amino acids and the isoform b precursor is 180 amino acids.
Malonyl-ACP is successively elongated in the next series of reactions that begins through the action of 3-oxoacyl-ACP synthase that is encoded by the OXSM gene. This enzyme catalyzes a condensation between the malonyl-ACP and an acyl-ACP, which would be acetyl-ACP in the first reaction of elongation. The condensation reaction releases CO2 and generates an acyl-ACP of an even number of carbon atoms. The mechanism by which the initial acetyl-ACP is generated is unclear. The OXSM encoded enzymes carries out each subsequent round of elongation where the substrates are the acyl-ACP product and a malonyl-ACP.
The OXSM gene is located on chromosome 3p24.2 and is composed of 3 exons that generate two alternatively spliced mRNAs encoding two distinct precursor proteins of 459 amino acids (isoform 1) and 376 amino acid (isoform 2).
The 3-ketoacyl-ACP product of the OXSM encoded enzyme is the substrate for the NADP(H)-dependent mitochondrial 3-ketoacyl-ACP reductase (KAR1). Mitochondrial 3-ketoacyl-ACP reductase is a heterotetrameric complex (α2β2) where the α-subunits are 17β-hydroxysteroid dehydrogenase type 8 and the β-subunits are carbonyl reductase type 4. The product of the 3-ketoacyl-ACP reductase is a 3R-hydroxyacyl-ACP. The 17β-hydroxysteroid dehydrogenase type 8 is encoded by the HSD17B8 gene and the carbonyl reductase type 4 is encoded by the CBR4 gene. The HSD17B8 encoded enzyme is also involved in the inactivation of estradiol, testosterone, and dihydrotestosterone.
The HSD17B8 gene is located on chromosome 6p21.32 and is composed of 9 exons that encode a 261 amino acid protein.
The CBR4 gene is located on chromosome 4q32.3 and is composed of 9 exons that encode a 237 amino acid protein. The CBR4 encoded enzyme is a member of the large family of short-chain dehydrogenases/reductases (SDR).
The 3R-hydroxyacyl-ACP is then converted to a trans-2-enoyl-ACP via the action of hydroxyacyl-thioester dehydratase type 2 that is encoded by the HTD2 gene. The HTD2 gene is located on chromosome 3p14.3 and is composed of 7 exons that generate four alternatively spliced mRNAs that differ in their 5′-UTR but that encode the same 168 amino acid protein.
The HTD2 gene is located downstream of the gene for ribonuclease P/MRP subunit p14 (RPP14) and these two genes appear to be co-transcribed into a bicistronic mRNA that may contain both open reading frames.
The last step in mitochondrial fatty acid synthesis reduces the trans-2-enoyl-ACP to an acyl-ACP in an NADPH-dependent reaction catalyzed by mitochondrial trans-2-enoyl-CoA reductase encoded by the MECR gene. Mitochondrial trans-2-enoyl-CoA reductase is a member of the medium-chain alcohol dehydrogenase/reductase (MDR) family.
The MECR gene is located on chromosome 1p35.3 and is composed of 18 exons that generate nine alternatively spliced mRNAs that collectively encode five distinct protein isoforms.
Mutations in the MECR gene are associated with hypotonia, dystonia, spasticity, optic atrophy, and abnormalities in brain development, particularly in the basal ganglia.
The acyl-ACP product of the mitochondrial trans-2-enoyl-CoA reductase can then be elongated by serving as the substrate for the OXSM encoded enzyme. When the eight carbon saturated fatty acid octanyl-ACP product is generated there are two fates, one is diversion of the octanoyl moiety into the lipoic acid synthesis pathway, the other is further elongation to either myristoyl-ACP (C14) or palmitoyl-ACP (C16).
The NADPH required by the heterotetrameric mitochondrial 3-ketoacyl-ACP reductase and the mitochondrial trans-2-enoyl-CoA reductase is generated from mitochondrial NAD+ via the activity of the mitochondria-localized NAD kinase encoded by the NADK2 gene. Loss of activity of NADK2 significantly impairs mitochondrial fatty acid synthesis.
The mitochondrial acyl carrier protein, although being a subunit of complex I of the oxidative phosphorylation pathway, is also critical for the assembly of other complexes of oxidative phosphorylation. Mitochondrial ACP interacts with proteins of the LYRM (Leucine-tYrosine-aRginine Motif) family. Individual LYRM proteins not only serve as accessory subunits or assembly factors of complexes I, II, III, and IV, they serve as assemble or accessory subunits of mitochondrial ribosomes, they are essential for the biogenesis of Fe-S cluster proteins/complexes, and they are also involved in the metabolism of acetate.
Humans express ten genes that encode LYRM family proteins. These genes are LYRM1, LYRM2, LYRM4, LYRM7, LYRM9, NDUFB8 (LYRM3), ETFRF1 (LYRM5), NDUFA6 (LYRM6), SDHAF1 (LYRM8), and SDHAF3 (LYRM10). NDUFB8 encodes NADH:ubiquinone oxidoreductase subunit B9. ETFRF1 encodes electron transfer flavoprotein regulatory factor 1. NDUFA6 encodes NADH:ubiquinone oxidoreductase subunit A6. SDHAF1 and SDHAF3 encode succinate dehydrogenase complex assembly factor 1 and 3, respectively.
Mitochondrial Synthesis of Lipoic Acid
Alpha-lipoic acid, LA, (chemical name: 1,2-dithiolane-3-pentanoic acid; also known as thioctic acid), is a naturally occurring dithiol compound synthesized by intestinal bacteria or derived enzymatically in the mitochondria. For mitochondrial synthesis, the initial substrate for lipoic acid synthesis is the octanoic acid intermediate of mitochondrial fatty acid synthesis that is attached to the ACP sulfhydryl of the mitochondrial fatty acid synthesis protein encoded by the NDUFAB1 (NDUFAB1 (NADH:ubiquinone oxidoreductase subunit AB1) gene as described in the Mitochondrial Fatty Acid Synthesis section above.
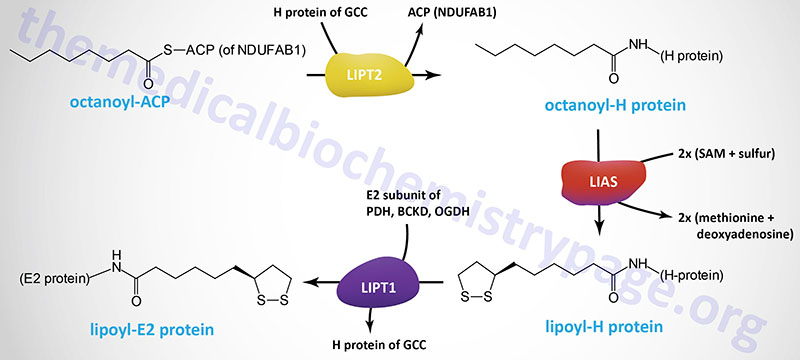
The octanoyl group is transferred to the H-protein (encoded by the GCSH gene) of the glycine cleavage complex (GCC) via the action of lipoyl (octanoyl) transferase 2 (encoded by the LIPT2 gene). Lipoic acid synthase (encoded by the LIAS gene) then inserts sulfur atoms, from S-adenosylmethionine (SAM or AdoMet) into the octanoyl group on the H-protein. Finally, lipoyltransferase 1 (encoded by the LIPT1 gene) transfers the lipoyl group from the H-protein to E2 subunits of several enzyme complexes. This important post-translational modification is termed protein lipoylation.
In addition to the GCSH encoded H-protein, the lipoylated E2 subunit proteins includes the DLAT encoded protein of the pyruvate dehydrogenase complex (PDHc), the DLST encoded protein of the 2-oxoglutarate (α-ketoglutarate) dehydrogenase complex (OGDH), and the DBT encoded protein of the branched-chain keto acid dehydrogenase complex (BCKD). In addition to the E2 subunit of the PDHc, the PDHX encoded protein of this complex is also lipoylated.
The LIPT2 gene is located on chromosome 11q13.4 and is composed of 2 exons that as a result of the use of alternative splice sites generates three mRNAs, each of which encode distinct protein isoforms.
The LIAS gene is located on chromosome 4p14 and is composed of 11 exons that generate six alternatively spliced mRNAs, each of which encodes a distinct protein isoform.
The LIPT1 gene is located on chromosome 2q11.2 and is composed of 7 exons that generate five alternatively spliced mRNAs, all of which encode the same 373 amino acid precursor protein.
Mutations in the LIPT2, LIAS, and LIPT1 genes have been identified in humans and they result in defects in lipoic acid synthesis. Defective lipoic acid synthesis is associated with neonatal-onset epilepsy, defective mitochondrial energy metabolism, and hyperglycinemia.
Additional clinical significance is associated with protein lipoylation, in particular under conditions of excess copper. Copper binds to lipoylated proteins causing them to aggregate which results in inhibition of enzymatic activity. The loss of PDHc and TCA cycle activity results in loss of energy production and mitochondrial stress leading to cell death. This copper-mediated programmed cell death process is termed cuproptosis.
ChREBP: Master Lipid Regulator in the Liver
When glycogen stores are maximal in the liver, excess glucose is diverted into the lipid synthesis pathway as a direct result of the mechanisms activated by insulin in this organ. Glucose is catabolized to acetyl-CoA and the acetyl-CoA is used for de novo fatty acid synthesis. The fatty acids are then incorporated into triglycerides and exported from hepatocytes as very-low-density lipoproteins (see the Lipoproteins, Blood Lipids, and Lipoprotein Metabolism page for more details) and ultimately stored as triglycerides in adipose tissue.
A diet rich in carbohydrates leads to stimulation of both the glycolytic and lipogenic pathways. The transcription rates of genes encoding glucokinase (GCK) and liver pyruvate kinase (PKLR) of glycolysis and ATP-citrate lyase (ACLY), ACC1 (ACACA), and FAS (FASN) of lipogenesis are modulated in response to increased carbohydrate intake. In addition, the enzymes encoded by these genes are subject to post-translational and allosteric regulation. These genes contain glucose- or carbohydrate-response elements (ChoRE) that are responsible for their transcriptional regulation.
One transcription factor that exerts control over glucose and lipid homeostasis is sterol-response element-binding protein (SREBP), in particular SREBP-1c. The regulation and actions of SREBP are discussed in the Cholesterol: Synthesis, Metabolism, and Regulation page. SREBP controls the expression of a number of genes involved in lipogenesis and its own transcription is increased by insulin and repressed by glucagon. However, SREBP activity alone cannot account for the stimulation of glycolytic and lipogenic gene expression in response to a carbohydrate rich diet.
A search for potential additional regulatory factors revealed a basic helix-loop-helix/leucine zipper (bHLH/LZ) transcription factor which was identified as carbohydrate-responsive element-binding protein (ChREBP). ChREBP is most highly expressed in the liver but is also abundantly expressed in white adipose tissue (WAT), brown adipose tissue (BAT), pancreatic β-cells, small intestine, kidney, adrenal glands, and brain. ChREBP was identified as a major glucose-responsive transcription factor and it is required for glucose-induced expression of the PKLR gene (encoding the liver pyruvate kinase, L-PK) and the lipogenic genes ACACA (encoding acetyl-CoA carboxylase 1, ACC1) and FASN (encoding fatty acid synthase, FAS). Subsequent to the identification of glucose as an enhancer of ChREBP activity it was found that fructose is a more potent activator than glucose. Fructose-mediated activation of ChREBP involves acetylation of the protein and increased DNA binding to ChREBP target genes.
ChREBP is encoded by the MLXIPL (MLX interacting protein like) gene. The MLXIPL gene is located on chromosome 7q11.23 and is composed of 19 exons that generate four alternatively spliced mRNAs that encode isoforms alpha (α), beta (β), gamma (γ), and delta (δ). Deletion of several genes, including MLXIPL, is the cause of Williams-Beuren syndrome. In skeletal muscle, the ChREBP homolog is called MondoA and it is encoded by the MLXIP gene.
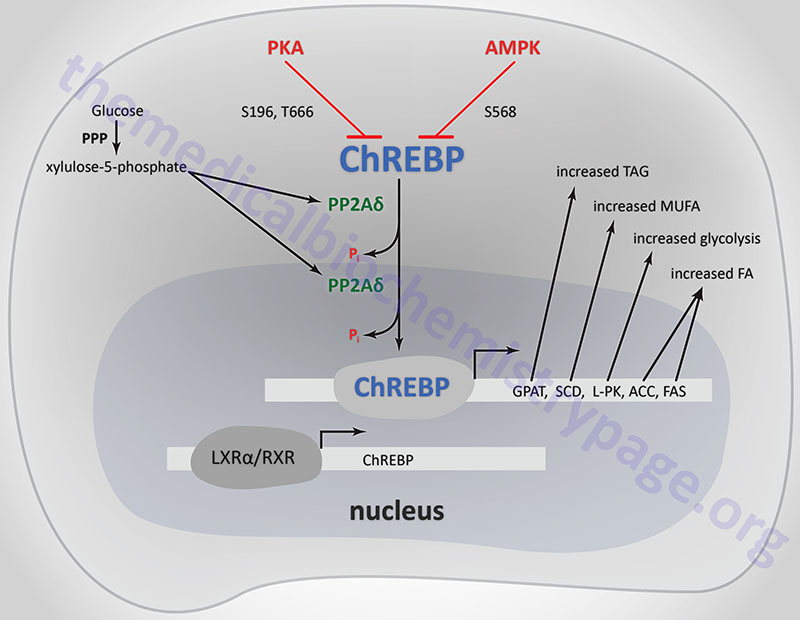
ChREBP does not bind to ChoREs as a typical homodimeric bHLH transcription factor. ChREBP interacts with another bHLH protein identified as MAX-like protein X (MLX). MLX is a member of the MYC/MAX/MAD family of transcription factors that serve as interacting partners in transcription factor networks. The binding of MLX to ChREBP occurs within a domain located in the C-terminal portion of ChREBP. This interaction between ChREBP and MLX is essential for DNA-binding.
Regulation of ChREBP (MLXIPL) Expression
The liver X receptors (LXR) are members of the steroid/thyroid hormone superfamily of nuclear receptors that regulate gene expression by binding to specific target DNA sequences. There are two forms of the LXRs, LXRα and LXRβ. The LXR form heterodimers with the retinoid X receptors (RXR) and as such can regulate gene expression either upon binding oxysterols (e.g. 22R-hydroxycholesterol) or 9-cis-retinoic acid. LXR are also important regulators of the lipogenic pathway.
Recent evidence has demonstrated that the MLXIPL gene is a direct target of LXR and that glucose itself can bind and activate LXRs. There are two LXR binding sites in the promoter region of the MLXIPL gene. Within the liver the thyroid hormone receptor beta (TRβ), a member of the nuclear receptor family, also binds to and activates expression of the MLXIPL gene.
Regulation of ChREBP Activity
When glucose levels rise, protein phosphatase 2A delta (PP2Aδ) is activated by xylulose 5-phosphate which is a product of glucose metabolism in the pentose phosphate pathway. PP2Aδ dephosphorylates S196 resulting in translocation of ChREBP into the nucleus. In the nucleus PP2Aδ dephosphorylates T666 which allows ChREBP to bind to specific sequence elements (ChoRE) in target genes. Other glucose-derived metabolites, including glucose 6-phosphate and acetyl-CoA, are potential activators of ChREBP translocation into the nucleus.
The activity of ChREBP is regulated by post-translational modifications as well as sub-cellular localization. The kinases PKA and AMPK both phosphorylate ChREBP rendering it inactive as a transcriptional activator. PKA is known to phosphorylate ChREBP on serine 196 (S196) and threonine 666 (T666), whereas, AMPK phosphorylates ChREBP at serine 568 (S568). Under conditions of low (basal) glucose concentration, ChREBP is phosphorylated and resides in the cytosol.
Subsequent to the identification of glucose as an enhancer of ChREBP activity it was found that fructose is a more potent activator than glucose. Fructose-mediated activation of ChREBP involves acetylation of the protein and increased DNA binding to ChREBP target genes.
Additional mechanisms of glucose-mediated regulation of ChREBP activity were made apparent when it was shown that mutations in the PKA phosphorylation sites (S196 and T666) did not completely abolish glucose-responsiveness. Further analysis of ChREBP regulation in response to glucose administration was shown to be due to domains present in the amino terminal portion of ChREBP. The glucose sensing domain (GSM) is actually composed of two distinct sub-domains identified as the low-glucose inhibitory domain (LID) and the glucose-responsive activation conserved element (GRACE). Under conditions of low glucose the LID inhibits transcriptional transactivation by the GRACE domain. This inhibition is reversed by glucose or a glucose metabolite such as glucose-6-phosphate or acetyl-CoA.
Recently a newly discovered mechanism of regulated ChREBP activity involves the tissue-specific transcription of an alternatively spliced form of ChREBP. This alternative splice variant contains a novel upstream exon (identified as exon 1b) and bypasses the originally identified exon 1 (now identified as exon 1a). This novel mechanism of ChREBP activity has been shown to occur in adipose tissue and represents a potent mechanism for glucose-mediated modulation of adipose tissue fatty acid synthesis and insulin sensitivity.
The original ChREBP is now referred to as ChREBP-α and the novel alternative splice form is called ChREBP-β. The means by which glucose plays a role in adipose tissue ChREBP activity is that glucose, or a metabolite, activates the transcriptional activity of ChREBP-α by the mechanisms described earlier. One of the important adipose tissue targets for glucose-activated ChREBP-α is the upstream transcriptional activation site that regulates the transcription of ChREBP-β. Following adipose tissue activation of ChREBP-β expression, both ChREBP-α and ChREBP-β work in concert to dramatically alter lipogenic gene expression.
Regulation of ChREBP Activity by O-GlcNAcylation
In addition to phosphorylation by PKA and AMPK and fructose-mediated acetylation, ChREBP activity is regulated by O-GlcNAcylation. The O-GlcNAcylation of ChREBP results in increased stabilization of the ChREBP protein and increased transcriptional activity.
Additional mechanisms of ChREBP activity are the result of its O-GlcNAcylation. Upon O-GlcNAcylation, ChREBP interacts with host cell factor 1 (encoded by the HCFC1 gene) which increases the recruitment of O-GlcNAc transferase (OGT) to ChREBP. In addition, the HCF1–ChREBP complex is involved in the recruitment of the histone demethylase, PHF2 (PHD finger protein 2 where PHD refers to plant homeodomain), resulting in the epigenetic modification of the promoter region of several lipogenic genes. PHF2 is a member of the Jumonji C domain-containing (JmjC) histone demethylase family of enzymes.
ChREBP Target Genes
Genes whose patterns of expression are under the control of ChREBP activity are involved in glycolysis, the pentose phosphate pathway, gluconeogenesis, and lipogenesis.
Genes that are involved in glucose metabolism that are regulated by ChREBP include those encoding liver pyruvate kinase (L-PK; encoded by the PKLR gene), glucokinase regulatory protein (encoded by the GCKR gene), and GLUT2 (encoded by the SLC2A2 gene).
Genes encoding enzymes in the gluconeogenesis pathway that are subject to regulation by ChREBP such as G6PC1, which encodes the predominant form of glucose-6-phosphatase, and FBP1 which encodes the liver form of fructose-1,6-bisphosphatase.
Genes encoding enzymes involved in the pentose phosphate pathway that are regulated by ChREBP include those encoding glucose-6-phosphate dehydrogenase (G6PD) and transketolase (TKT).
ChREBP also regulates the expression of several genes involved in the metabolism of fructose in the small intestines and the liver. These genes include SLC2A2 and SLC2A5 which encode GLUT2 and GLUT5, respectively, KHK which encodes ketohexokinase (commonly referred to as fructokinase), ALDOB which encodes aldolase B, and TKFC which encodes triokinase and FMN cyclase.
Within adipose tissue ChREBP-α and ChREBP-β function together to dramatically increase the transcription of lipogenic genes in this tissue such as FASN (fatty acid synthase) and ACACA (acetyl-CoA carboxylase 1, ACC1). In addition, it has been shown that when expression of MLXIPL is reduced, the expression levels of the genes encoding glycerol-3-phosphate acyltransferases (GPAM, GPAT2, GPAT3, GPAT4) and Δ9-stearoyl-CoA desaturase (SCD) are also reduced.
The glycerol-3-phosphate acyltransferases esterify glycerol-3-phospate generating lysophosphatidic acid (LPA) which is the first step in the synthesis of triglycerides (TG; or triacylglycerides, TAG) as described in detail in the Synthesis of Triglycerides page.
SCD is the rate-limiting enzyme involved in the synthesis of the major monounsaturated fatty acids oleic acid (18:1) and palmitoleic acid (16:1) as described below in the section on Elongation and Desaturation.
Elongation and Desaturation
Elongation
The fatty acid product released from FAS is palmitate (via the action of palmitoyl thioesterase) which is a 16:0 fatty acid (i.e. 16 carbons and no sites of unsaturation). Elongation of fatty acids occurs in the cytosol, the mitochondria, and the endoplasmic reticulum (microsomal membranes). The predominant site of fatty acid elongation is in the ER membranes.
Elongation involves condensation of fatty acyl-CoA groups with either acetyl-CoA or malonyl-CoA. The resultant product is two carbons longer (CO2 is released from malonyl-CoA as in the FAS reaction) which undergoes reduction, dehydration and reduction yielding a saturated fatty acid extended in length by two carbon atoms. The reduction reactions of elongation require NADPH as co-factor.
Cytoplasmic fatty acid elongation is essentially an extension of the normal fatty acid synthesis reaction. Acetyl-CoA carboxylase (ACC) produces malonyl-CoA and then FAS utilizes acetyl-CoA and the malonyl-CoA to elongate the primary end-product of normal fatty acid synthesis, palmitate. Mitochondrial fatty acid elongation involves a fatty acyl-CoA and acetyl-CoA units and the enzyme trans-2-enoyl-CoA reductase (encoded by the MERC gene). The mitochondrial fatty acid elongation pathway is a minor pathway in humans.
The microsomal fatty acid elongation pathway represents the major pathway for the elongation of both endogenous and dietary fatty acids. The overall pathway involves four enzymes with the substrates being fatty acyl-CoAs, malonyl-CoA, and NADPH.
In the initial step in the pathway a 3-keto acyl-CoA synthase catalyzes the condensation of malonyl-CoA with a fatty acyl-CoA precursor. Humans express seven distinct 3-keto acyl-CoA synthase encoding genes. These seven genes are designated ELOVL1–ELOVL7 which stands for ELOngation of Very Long fatty acids. Fatty acyl-CoA substrate specificity and the rate of fatty acid elongation in the microsomes is determined by the ELOVL enzymes and not the reductases nor the dehydratase.
- The ELOVL1 encoded enzyme exhibits preference for saturated fatty acids in the 18-carbon (C18) to C26 length with highest activity towards the C22:0 saturated fatty acid called behenic acid (docosanoic acid).
- The ELOVL2 encoded enzyme elongates polyunsaturated fatty acids (PUFA) of C20 and C22 length with highest activity towards the C20:4 PUFA, arachidonic acid.
- The ELOVL3 encoded enzyme elongates saturated fatty acids with highest preference for stearic acid (C18:0).
- The ELOVL4 encoded enzyme preferentially elongates very long-chain saturated fatty acids such as lignoceric (C24:0; tetracosanoic acid) and cerotic (C26:0; hexacosanoic acid) acids. Critical enzyme in the synthesis of very long-chain PUFA (C26 to C38) and very long-chain saturated fatty acids. Mutations in the ELOVL4 gene are associated with inherited forms of macular degeneration.
- The ELOVL5 encoded enzyme elongates PUFA with highest preference for the C18:3 fatty acids, α-linolenic acid (ALA) and gamma(γ)-linolenoyl-CoA (GLA). ALA has sites of unsaturation at carbons 9, 12, and 15 and is the precursor fatty acid in the pathway of omega-3 PUFA synthesis. GLA has sites of unsaturation at carbons 6, 9, and 12 and is an intermediate in the pathway of arachidonic acid synthesis. ELOVL5 can also elongate some monounsaturated fatty acids, such as palmitoleic acid (C16:1).
- The ELOVL6 encoded enzyme elongates saturated fatty acids with 12, 14, and 16 carbon atoms with highest activity towards palmitic acid (C16:0).
- The ELOVL7 encoded enzyme has been shown to elongate a range of both saturated and unsaturated fatty acids such as the C16:0, C18:0, C18:1, C18:2, C18:3, and C20:4 fatty acids. The C18:3 substrates of ELOVL7 are the same as those used by the ELOVL5 enzyme. ELOVL7 shows no elongation activity towards fatty acids longer than 20 carbons.
The ELOVL genes are expressed in tissue specific patterns as well as being subjected to dietary and hormonal regulation. ELOVL1 expression is highest in skin, stomach, lung, and intestine, with moderate levels of expression in the liver, kidney, brain, heart, and muscle. ELOVL2 expression is highest in liver, testes, and retina. ELOVL3 expression is highest in liver, adipose tissue, and skin. ELOVL4 expression is highest in brain, retina, skin, and testes. ELOVL5 expression is highest in liver, adrenal glands, testes, and mammary glands. ELOVL6 expression is highest in liver, adipose tissue, brain, and mammary glands. Expression of ELOVL7 is highest in kidney, colon, pancreas, and prostate gland.
In the second reaction of fatty acid elongation a 3-ketoacyl-CoA reductase activity (also called 3-oxoacyl-CoA reductase) reduces the resulting 3-ketoacyl-CoA intermediate to a 3-hydroxyacyl-CoA. The 3-ketoacyl-CoA reductase activity of mammalian fatty acid elongation is associated with the enzyme hydroxysteroid 17-β dehydrogenase 12 encoded by the HSD17B12 gene.
The HSD17B12 gene is located on chromosome 11p11.2 and is composed of 18 exons that encode a 312 amino acid protein. Elongation of fatty acids in the mitochondria utilizes a related enzyme called 3-oxoacyl-ACP synthase that is encoded by the OXSM gene.
The third step in fatty acid elongation involves the dehydration of the 3-hydroxy species and it is catalyzed by 3-hydroxyacyl-CoA dehydratase 2 (encoded by the HACD2 gene). Humans express four genes of the HACD family (HACD1, DACD2, DACD3, and HACD4) but only the protein encode by HACD2 is involved in fatty acid elongation.
The HACD2 gene is located on chromosome 3q21.1 and is composed of 11 exons that generate five alternatively spliced mRNAs that collectively encode four protein isoforms.
In the fourth step of fatty acid elongation the product of HACD2 catalyzed reaction is reduced by trans-2,3-enoyl-CoA reductase, an member of the steroid 5-alpha reductase family. The trans-2,3-enoyl-CoA reductase is encoded by the TECR gene.
The TECR gene is located on chromosome 19p13.12 and is composed of 15 exons that generate two alternatively spliced mRNAs, both of which encode distinct protein isoforms.
The fatty acid elongases function in concert with the fatty acid desaturases (next section) to generate many of the long chain monounsaturated fatty acids (MUFA) and polyunsaturated fatty acids (PUFA) that are found in cellular lipids. For example ELOVL6 and stearoyl-CoA desaturase (encoded by the SCD gene) generate the 18-carbon monounsaturated fatty acid (C18:1), oleic acid. ELOVL2 and ELOVL5 function along with the delta-5 (D5D) and delta-6 (D6D) desaturases to generate the omega-6 PUFA, arachidonic acid (C20:4) and the omega-3 PUFA, docosahexaenoic acid, DHA (C22:6).
Desaturation
Like the elongation of fatty acids, the desaturation of fatty acids occurs in the ER membranes. In mammalian cells fatty acid desaturation involves three broad specificity fatty acyl-CoA desaturases (non-heme iron containing enzymes). These enzymes introduce unsaturation at C5, C6, or C9 of various fatty acid substrates. The names of these enzymes are Δ5-eicosatrienoyl-CoA desaturase (D5D), Δ6-oleoyl(linolenoyl)-CoA desaturase (D6D), and Δ9-stearoyl-CoA desaturase (SCD).
The D5D enzyme is encoded by the FADS1 gene. The FADS1 gene is located on chromosome 11q12.2 and is composed of 13 exons that encode a 501 amino acid protein. The FADS1 encoded enzyme has near exclusive activity towards PUFA with 20 carbon atoms.
The D6D enzyme is encoded by the FADS2 gene. The FADS2 gene is located on the same chromosome as the FADS1 gene, 11q12.2. The FADS2 gene is composed of 13 exons that generates three alternatively spliced mRNAs that encode three distinct isoforms of the enzyme. This chromosomal region is referred to as the FADS cluster and harbors an additional desaturase gene identified as FADS3.
The FADS2 encoded enzyme desaturates linoleic acid (omega-6 18:2; also written 18:2n-6) generating γ-linolenic acid (GLA; 18:3n-6). FADS2 also desaturates α-linolenic acid (omega-3 18:3; also written 18:3n-3) generating stearidonic acid (18:4n-3).
FADS2 also functions as a Δ4 and a Δ8 desaturase. The Δ4 desaturase activity is functional on 22:4n-6 (adrenic acid) generating docosapentaenoic acid (22:5n-6; DPAn-6; trivial name: osbond acid) and on the omega-3 form of docosapentaenoic acid (22:5n-3; DPAn-3; trivial name clupanodonic acid) generating docosahexaenoic acid (22:6n-3). The Δ8 desaturase activity acts on 20:2n-6 (eicosadienoic acid) and 20:3n-3 (eicosatrienoic acid) generating 20:3n-6 (dihomo-γ-linolenic acid, DGLA) and 20:4n-3 (eicosatetraenoic acid; specifically all-cis-8,11,14, 17-eicosatetraenoic acid), respectively.
Eicosatetraenoic acid can exist in eight different isomeric forms where the omega-6 form (arachidonic acid) is all-cis-5,8,11,14-eicosatetraenoic acid while the omega-3 form is all-cis-8,11,14,17-eicosatetraenoic acid.
The SCD enzyme is encoded by the SCD gene (also known as FADS5). The SCD gene is located on chromosome 10q24.31 and is composed of 6 exons that generate at least two mRNAs that differ by alternative polyadenylation signal sequence utilization. Both SCD mRNAs encode the same 359 amino acid protein.
The electrons transferred from the oxidized fatty acids during desaturation are transferred from the desaturases to cytochrome b5 and then NADH-cytochrome b5 reductase. These electrons are uncoupled from mitochondrial oxidative-phosphorylation and, therefore, do not yield ATP.
Since the mammalian desaturase enzymes cannot introduce sites of unsaturation beyond C9 they cannot synthesize either linoleic acid (18:2Δ9,12) or α-linolenic acid (ALA: 18:3Δ9,12,15). These two fatty acids must be acquired from the diet and are, therefore, referred to as essential fatty acids.
Linoleic is especially important in that it required for the synthesis of arachidonic acid. Arachindonate is a precursor for the eicosanoids (the prostaglandins, thromboxanes, and leukotrienes). It is this role of fatty acids in eicosanoid synthesis that leads to poor growth, wound healing, and dermatitis in persons on fat free diets. Also, linoleic acid is a constituent of epidermal cell sphingolipids that function as the skins’ water permeability barrier. The significance of ALA is that it serves as the precursor fatty acid for the synthesis of several clinically relevant omega-3 fatty acids, namely eicosapentaenoic acid (EPA) and docosahexaenoic acid (DHA).
The FADS3 encoded enzyme functions as a ceramide desaturase in the synthesis of the ceramide derivatives of the sphingadiene (SD) class. Sphingadienes are long chain sphingoid bases that contain an additional carbon-carbon double bond in the long-chain acyl moiety when compared to sphingosine. Sphingosine is the most common mammalian sphingoid base. FADS3 has been shown to be required for the synthesis of 4,14-sphingadiene (specifically: 4E,14Z-sphingadiene). The sphingadienes constitute a class of lipid that has been shown to exert cytotoxic effects on certain types of cancers through their ability to inhibit the activity of the AKT/PKB signaling pathway.
Humans express eight genes that encode enzymes of the fatty acid desaturase family that includes the FADS1, FADS2, FADS3, and SCD genes. Additional members of this family, that are involved in the synthesis of the ceramides, include the DEGS1 (also identified as FADS7) and DEGS2 (also identified as FADS8) genes. The other members of this family are the FADS6 and SCD5 (also identified as FADS4) genes. These latter two genes encode enzymes that function similar to the FADS2 and SCD encoded enzymes, respectively.
Clinical Significance of SCD-Mediated Desaturation
Stearoyl-CoA desaturase (SCD) is the rate-limiting enzyme catalyzing the synthesis of monounsaturated fatty acids (MUFAs). The expression of SCD is under the control of the transcription factor ChREBP as discussed above. The major products of SCD are oleic acid (18:1; a physiologically significant omega-9 fatty acid) and palmitoleic acid (16:1; a physiologically significant omega-7 fatty acid). These two monounsaturated fatty acids represent the majority of monounsaturated fatty acids present in membrane phospholipids, triglycerides, and cholesterol esters. Palmitoleic acid is the most abundant fatty acid in the blood and in adipose tissue. Palmitoleic acid possesses numerous biological activities and is referred to as a lipokine.
The ratio of saturated to monounsaturated fatty acids in membrane phospholipids is critical to normal cellular function and alterations in this ratio have been correlated with diabetes, obesity, cardiovascular disease, and cancer. Thus, regulation of the expression and activity of SCD has important physiological significance.
SCD activity has been shown to play a critical role in the prevention of cytotoxic stress. Cellular stress activates stress-activated kinases that in turn initiate a series of adaptive mechanisms that includes the unfolded protein response (UPR), autophagy, and apoptosis.
The effects of SCD in the regulation of cytotoxic stress are due to its role in the production of the MUFA oleic acid (18:1) and the subsequent generation of the phosphatidylinositol (PI) that contains oleic acid at both the sn1 and sn2 positions [1,2-dioleoyl-sn-glycero-3-phospho-(1’-myo-inositol)] designated PI[(18:1/18:1)]. This PI has been shown to inhibit p38 MAPK activation, counteract activation of the UPR and apoptosis, as well as regulating activation of autophagy. Given the role of PI[(18:1/18:1)] is maintenance of cellular integrity it has been define as a lipokine. The level of expression of the SCD gene has also been shown to be decreased during the onset of apoptosis and other forms of cell death. In addition, depletion of PI[(18:1/18:1)] has been linked to activation of cellular stress response pathways.
Synthesis and Biological Activities of Palmitoleic Acid
Palmitoleic acid can be acquired in the diet as well as synthesized from palmitic acid via the actions of SCD. Significant dietary sources of palmitoleic acid include salmon, cod liver oil, olive oil, chocolate, eggs, macadamia nut oil, and sea buckthorn.
The major site of synthesis of palmitic acid is adipose tissue. Another source of palmitoleic acid is hepatic synthesis of trans-palmitoleic acid from trans-vaccenic acid (trans-C18:1; an omega-7 MUFA) via peroxisomal β-oxidation. It was originally believed that trans-palmitoleic acid could be obtained only from the diet. This novel hepatic synthesis pathway represents a mechanism by which a dietary fatty acid (trans-vaccenic acid) can contribute to the accumulation of palmitoleic acid in numerous tissues.
Increased synthesis of palmitoleic acid is associated with enhanced peripheral insulin sensitivity and increased glucose uptake by the liver, adipose tissue, and skeletal muscle. Both the cis– and trans-palmitoleic acid forms are associated with enhanced insulin sensitivity and glucose homeostasis. Palmitoleic acid, produced by adipose tissue, acts in an autocrine manner to stimulate adipocyte lipolysis via increased PPARα activity resulting in increased adipose tissue triglyceride lipase (ATGL) and hormone-sensitive lipase (HSL) phosphorylation. Palmitoleic acid also stimulates insulin sensitivity of adipose tissue resulting in increased GLUT4 translocation to the plasma membranes of adipocytes and increased glucose uptake.
In mouse models of obesity and type 2 diabetes the administration of palmitoleic acid protects against hyperglycemia and hypertriglyceridemia, decreases the expression of hepatic lipogenic genes (SREBP1, FAS, and SCD), and results in reduced accumulation of triglycerides in the liver. Within the pancreas palmitoleic acid has been shown to stimulate pancreatic lipogenesis, phospholipid remodeling, and triglyceride synthesis. These effects of palmitoleic acid in the pancreas result in increased cell viability and endocrine functions of the organ.
Palmitoleic acid is an important regulator of cellular stress and inflammation, particularly in skeletal muscle and adipose tissue. Within skeletal muscle palmitoleic acid delays the activation of pro-oxidative and pro-inflammatory enzyme, COX2, and blocks the activation of p38 MAPK-mediated insulin resistance. Within adipocytes palmitoleic acid also exerts negative effects on the activation of inflammatory processes by inducing downregulation of genes encoding chemokines and cytokine receptors. Within the liver palmitoleic acid inhibits the expression of liver ER-stress genes and decreases expression of apoptotic genes. Palmitoleic acid also reduces apoptosis in pancreatic β-cells.
Exercise plays an important role in the level of circulating palmitoleic acid by activating adipose tissue lipolysis and release of stored palmitoleic acid. In addition, exercise increases de novo synthesis of palmitoleic acid by adipocytes. In mice in which the ATGL gene has been knocked out there is a failure of exercise-induced lipolysis, as well as in the normal process of cardiac hypertrophy and non-proliferative cardiomyocyte growth that normally occurs with exercise. When these mice are supplemented with palmitoleic acid there is restoration of left ventricular mass to the level of that in exercising wild-type mice. Exercise also leads to the accumulation of palmitoleic acid in triglycerides in skeletal muscle which has been shown to result in increased insulin sensitivity n skeletal muscle.
Clinical Significance of D5D and D6D Activities
Physiological and pathophysiological significance is also associated with the activities of D5D and D6D. The activity of D6D requires pyridoxal phosphate (PLP, derived from vitamin B6), Mg2+, and Zn2+ ions and its activity is, therefore, affected by nutritional status. Both of D5D and D6D exhibit reduced and inhibited activity in conditions associated with hyperglycemia such as is typical of type 2 diabetes. Additional factors impairing D5D and D6D activity include alcohol consumption, hypercholesterolemia, and the processes of aging. Conversely, normal insulin responsiveness results in increased D5D and D6D activity as does a caloric restriction diet.