
Last Updated: October 20, 2025
Excitability of Muscle Cells
Skeletal muscle cells are voluntary cells, meaning that one can make a muscle cell move in response to a voluntary command initiated within the central nervous system. The peripheral nerves that influence the function of skeletal muscle cells are referred to as motor neurons, specifically somatic alpha (α) and gamma (γ) motor neurons. Alpha motor neurons are the principal motor neurons controlling skeletal muscle (extrafusal muscle fibers) contraction while gamma motor neurons control the contraction of intrafusal skeletal muscle fibers (muscle fibers of specialized sensory organs).
Cardiac and smooth muscle cells are involuntary, meaning that one cannot command these cells to exert an activity. The motor neurons that innervate cardiac and smooth muscle cells are sympathetic and parasympathetic motor neurons of the autonomic nervous system. For more details on the various nerve cell types go to the Biochemistry of Nerve Transmission page.
Skeletal versus Cardiac Muscle Action Potentials
Skeletal Muscle Action Potentials
In order to move a skeletal muscle cell, an action potential must be initiated from a peripheral motor neuron. Cardiac muscle (myocardial) cells on the other hand, can initiate their own electrical activity in the absence of an autonomic nerve-mediated action potential. With respect to skeletal muscle, nerve transmission occurs when an axon of a post-ganglionic nerve terminates on a skeletal muscle fiber, at specialized structures called the neuromuscular junction. An action potential occurring at this site is known as neuromuscular transmission.
At a neuromuscular junction, the axon subdivides and branches into numerous structures, referred to as terminal buttons (pronounced “boo-tawns”) or end bulbs, that can then innervate numerous skeletal muscle fibers. The result is that many muscle fibers can be innervated by a single neuron instead of each fiber having to be dependent upon an individual neuron for contractile activation. The skeletal muscle fibers that are innervated by branches from the same neuron constitute a motor unit. Large muscles in the body (e.g. the gastrocnemius) contain numerous motor units. This arrangement of the motor units in a particular muscle allows for activation of only a specific part of a muscle at any given time. This represents a form of spatial control over muscle fiber contraction within a muscle, a feature not associated with cardiac muscle excitation as discussed below.
The terminal buttons (end bulbs) of the motor neurons reside within depressions formed in the skeletal muscle plasma membrane (sarcolemma). At these locations the skeletal muscle membrane is thickened and is referred to as the motor end plate. The space between the terminal buttons (end bulbs) and the motor end plate is similar to the synaptic cleft that exists where the pre-synaptic and post-synaptic membranes of neurons are in close proximity. The particular neurotransmitter in use at the neuromuscular junction is acetylcholine, ACh. When an action potential reaches the pre-synaptic membrane of a motor neuron the permeability (membrane potential) of the membrane changes. This change in membrane potential activates specific voltage-gated Ca2+ channels.
The resultant influx of Ca2+ ion into the nerve endings triggers mobilization of ACh-containing vesicles to the plasma membrane and resultant exocytosis of the ACh. This vesicle mobilization and ACh exocytosis occurs within a few hundred microseconds of the action potential reaching the pre-synaptic membrane. Vesicle mobilization results from the interaction of Ca2+ with proteins of the synaptotagmin family. The interaction of Ca2+ with synaptotagmins activates these membrane targeted proteins to interact with membrane phospholipids and other proteins of the exocytotic apparatus termed SNARE proteins. SNARE proteins are required for the process of membrane fusion that is a necessary step in the interaction of exocytotic vesicle with the plasma membrane.
The released ACh then binds to nicotinic ACh receptors (nAChR) that are concentrated in the motor end plate membrane. Once released from the motor neuron, the level of active ACh is controlled by its catabolism through the action of acetylcholinesterase. As discussed in the Biochemistry of Nerve Transmission page, nAChR are members of the ionotropic receptor superfamily (ion channel receptors). Activation of nicotinic ACh receptors in the motor end plate results in an increase in Na+ and K+ conductance through the nAChR channel.
The resulting influx of Na+, into the skeletal muscle cell, produces a depolarizing potential. As a result of this depolarization, action potentials are conducted in both directions, away from the motor end plate, along the muscle fiber. These action potentials are the result of the initial membrane depolarization (the resting membrane potential is -70 to -90 mV) to a less negative potential and propagated across the surface membrane (sarcolemma: muscle cell plasma membrane) via the opening of voltage-gated Na+ channels (encoded by members of the SCN gene family; proteins are designated Nav).
Voltage-gated Na+ channels are composed of a large pore-forming α-subunit and a small accessory β-subunit that modulates activity of the α-subunits. Humans express nine α-subunit genes SCN1A–SCN5A, SCN8A–SCN11A and four β-subunit genes SCN1B–SCN4B. The primary skeletal muscle Na+ channel α-subunit is designated Nav1.4 and is encoded by the SCN4A gene. The primary skeletal muscle Na+ channel β-subunit is the Navβ1 protein which is encoded by the SCN1B gene.
Nearly simultaneous with the membrane voltage changes associated with activation is a process of inactivation. The inactivation is triggered as the membrane potential becomes progressively less negative but is a slower process than that of activation. The inactivation results in the closure of the voltage-gated Na+ channels. A second factor that limits action potentials is the voltage activation of inwardly rectifying potassium channels (encoded by genes of the KCNJ family; channels designated: Kir), the result of which is repolarization and a rapid return to the resting membrane potential.
During the initial membrane depolarization event the Kir channels are closed. When inwardly rectifying K+ channels open, K+ enters the cell through these channels against the concentration gradient of potassium. This inward potassium movement returns the membrane potential to the original resting potential which is associated with a much higher concentration of potassium inside the cell than outside (on the order of 40 times).
Inwardly rectifying potassium channels are members of the voltage-gated potassium subfamily J channels, of which there are 16 genes in humans identified as KCNJ1–KCNJ6, KCNJ8–KCNJ16, KCNJ18. Human skeletal and cardiac muscle primarily express the KNCJ2 (Kir2.1) and KCNJ12 (Kir2.2) genes in both the sarcolemma and the T tubule membranes. The activation of the KCNJ2 and KCNJ12 channels is initiated during the onset of the activating action potential but there is a slight delay in channel opening such that repolarization is delayed on the order of 1–1.5 msec in skeletal muscle. The entire time frame of sarcolemma depolarization and repolarization, constituting the skeletal muscle action potential, is on the order of 3 msec.
The action potential is then propagated down the T-tubule system which directly interacts with the sarcoplasmic reticulum, SR (muscle cell endoplasmic reticulum, ER). The action potential results in the opening of voltage-gated Ca2+ channels in the sarcolemma of the T-tubules allowing Ca2+ influx. These T-tubule Ca2+ channels are members of the L-type Ca2+ channels and they are also known as dihydropyridine receptors (DHPR) due to the fact that they were identified through inhibition by dihydropyridine. All Ca2+ channels contain multiple subunits where the α-subunit constitutes the ion pore in the membrane. The α-subunit of the skeletal muscle DHPR is encoded by the CACNA1S gene. This channel is commonly referred to as Cav1.1.
The incoming calcium then binds to ligand-gated Ca2+ channels in the SR membrane activating this membrane. Activation of the SR membrane leads to the release, into the sarcoplasm (cytoplasm of muscle cells), of stored Ca2+ through the opening of Ca2+ release channels. The SR calcium release channels are ligand-gated ion channels where Ca2+ ion is the ligand. The calcium release channels are also known as the ryanodine receptors (RYR) due to the fact that they were originally identified by their high affinity for the plant alkaloid ryanodine. Humans express three ryanodine receptor genes identified as RYR1, RYR2, and RYR3. The end result of the ACh-initiated propagating action potential is muscle contraction. Although there is ubiquitous expression of all three genes, the skeletal muscle the Ca2+ release channels are primarily encoded by the RYR1 gene, in cardiac muscle they are primarily encoded by the RYR2 gene. Expression of the RYR3 gene is predominantly seen in the brain.
Myasthenia Gravis
A particularly devastating disease that results from defects in the overall processes of neuromuscular nerve transmission is myasthenia gravis, MG. MG is a very serious disorder that is often times fatal. The characteristic features of the disease are weakened skeletal muscles that tire with very little exertion. MG is an auto-immune disease associated with antibodies to the nAChR of the neuromuscular junction. Binding of the antibodies to the receptor results in receptor destruction as well as receptor cross-linking. In most patients with MG there is a 70%–90% reduction in motor end plate nicotinic receptor number. Two major forms of MG exist, one in which the extraocular muscles are the ones primarily affected and in the other form there is a generalized skeletal muscle involvement. In the latter form of MG, the muscles of the diaphragm become affected resulting in respiratory failure which contributes to the mortality of MG. Treatment of MG involves numerous approaches including the use of acetylcholinesterase inhibitors. The use of these types of drugs allows for enhanced levels of ACh at the motor end plate during repeated muscle stimulation.
Cardiac Muscle (Cardiomyocyte) Action Potentials
Although cardiac muscle cells (also called cardiac myocytes or cardiomyocytes) are striated like skeletal muscle cells and utilize the same biochemical mechanics for contraction, the physiological characteristics and the electrochemical activation processes of cardiomyocytes are quite distinct. One significant difference between these two striated muscle types is that cardiac muscle cells do not require innervation to be activated to contract even though the heart is innervated by both the sympathetic and parasympathetic nervous systems. In contrast to initiating contractile activity the autonomic nervous system serves to modulate, not initiate, cardiac contractile activity.
In contrast to skeletal muscle cells within a muscle fiber, all cardiomyocytes are electrically coupled and therefore contract simultaneously. The electrical communication between cardiomyocytes occurs through gap junctions at points on the cells referred to as nexi. The major gap junction protein in cardiac myocytes is encoded by the GJA1 gene (gap junction alpha 1; also known as alpha 1 connexin and designated CX43). The initiation of an action potential in one cardiomyocyte is rapidly propagated via the nexi to all cells of the heart. This connectivity, referred to as a functional syncytium, effectively serves to allow the heart muscle to act as one large cell. However, the potential downside to this interconnectivity across the entire heart is that the abnormal (pathophysiological) activation of one or more cardiomyocytes can inadvertently activate the heart as a whole.
Certain cardiac myocytes possess the capacity to initiate their own action potentials, a function referred to as automaticity. In addition, cardiomyocytes exhibit a periodicity to the initiation of action potentials, a process termed rhythmicity. Cardiomyocyte action potentials are characterized as either fast or slow. Ventricular and atrial cardiomyocytes, as well as those of Purkinje fibers, exhibit fast response action potentials. The cardiomyocytes of the sinoatrial (SA) node and atrioventricular (AV) node exhibit slow response action potentials. The primary ions controlling the generation, propagation, and termination of cardiomyocyte action potentials are K+, Na+, and Ca2+.
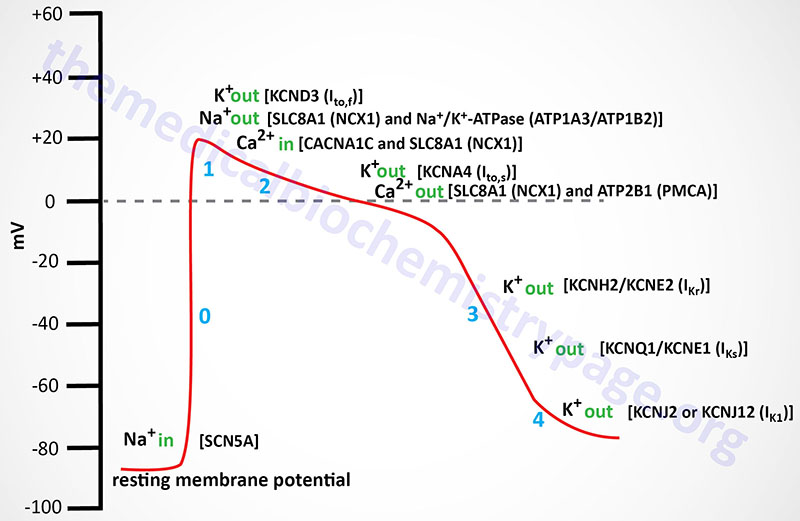
Cardiomyocyte fast response action potentials are unique from those of skeletal muscle in that the depolarization lasts longer and exhibits what is referred to as a plateau phase. The resting membrane potential is primarily a function of the diffusion potential of K+. This makes resting cardiomyocytes extremely sensitive to changes in extracellular K+ concentration. For example, high plasma K+ concentrations (hyperkalemia) depolarize cardiac cells, whereas hypokalemia hyperpolarizes these cells. Depolarization refers to the membrane potential becoming less negative than the resting membrane potential, hyperpolarization refers to the membrane potential becoming more negative than the resting membrane potential. Both of these pathophysiological conditions can adversely affect cardiac function.
A contributor to the resting membrane potential of cardiomyocytes is the low level Na+ influx, not associated with membrane depolarization. When a cardiomyocyte membrane is depolarized (beyond a specific threshold), fast acting voltage-gated Na+ channels open allowing a rapid influx of Na+. The opening of these Na+ channels is the result of voltage-mediated opening of an extracellular activation gate in the channel. The major voltage-gated Na+ channel in cardiomyocytes is the Nav1.5 channel which is composed of the SCN5A encoded α-subunit and one of the four modulatory β-subunit proteins.
The rapid influx of Na+ further depolarizes the membrane, opening more Na+ channels, resulting in a rapid, self-reinforcing depolarization of the cell. However, this Na+ effect is self limiting since the membrane depolarization results in channel inactivation within milliseconds of it opening. The channel inactivation is the result of voltage-mediated closure of the intracellular inactivation gate. This depolarization-mediated Na+ channel closing keeps the channel closed until the membrane repolarizes, thus, limiting the extent of the action potential.
In skeletal muscle cells K+ channels open very quickly after the Na+ channels open, resulting in efflux of K+ from the cell and a consequent rapid membrane repolarization. In cardiomyocytes this does not occur resulting in the plateau phase of the action potential. The plateau phase is physiologically significant because it allows for a single cardiac contraction event to be completed prior to the initiation of a second action potential. The slow refractory (plateau) phase of cardiomyocyte action potentials is mediated by slow acting voltage-gated Ca2+ channels. All cardiomyocytes contain L-type Ca2+ channels with the Cav1.2 channel being the most abundant form in cardiomyocytes. The L-type channels open with the depolarization of the membrane.
Nearly all of the voltage-gated calcium channels are pentameric structures composed of five different protein subunits identified as the α1-, α2-, β-, γ, and δ-subunits. Some voltage-gated calcium channels are composed of only four subunits and lack the δ subunit. These latter four subunit channels are typical of cardiac voltage-gated calcium channels. The α1-subunit is the actual transmembrane channel through which the Ca2+ ions flow. The α2- and the δ-subunits are derived from a single preproprotein that, following cleavage, forms an extracellular disulfide bonded glycoprotein dimer designated α2/δ. The γ-subunit is a transmembrane glycoprotein and the β-subunit is an intracellular protein. It is the distinct α1-subunits that define the various Cav1.x, Cav2.x, and Cav3.x channel types.
While Ca2+ is flowing into the cardiomyocyte the electrochemical gradient is maintained by efflux of K+ through K+-specific voltage-gated outwardly rectifying channels. These depolarization activated K+ channels are termed outward rectifying due to the fact that the movement of K+ ions is from inside the cell to outside. These outwardly rectifying K+ channels are associated with current movement and as such are designated as fast (Ito,f; also termed Ito1,f) or slow (Ito,s; also termed Ito1,s) currents due to their slightly different kinetics. The capital letter “I” designation refers to the movement of current while the subscripts to,f and to,s refer to the “transient outward” fast and slow, respectively, movement of the K+ ions (outward rectifying). The near simultaneous opening of Ca2+ and K+ channels results in a near net neutral charge differential across the membrane since the positive charges entering as Ca2+ and those leaving as K+ balance each other resulting in the plateau phase typical of ventricular cardiomyocyte action potentials.
The Ito,f channels in the human heart are designated Kv4.3 (encoded by the KCND3 gene). In small mammals there is an additional cardiac Ito,f type voltage-gated K+ channel identified as Kv4.2 (encoded by the KCND2 gene). The human KCND2 gene is expressed most prominently in the brain but not at all in the heart. The Ito,s channels are designated Kv1.4 (encoded by the KCNA4 gene). The Ca2+ channels eventually close, thereby preventing further membrane depolarization which aids the eventual repolarization of the cell membrane. As the Ca2+ channels close the complete repolarization of the cardiomyocyte occurs through the opening of two distinct types of delayed rectifier K+ channels defined by the kinetics of current movement as rapid [termed I(Kr)] and slow [termed I(Ks)] channels. The I(Kr) channel is encoded by the KCNH2 gene and the protein is referred to as Kv11.1. The KCNH2 gene is also known as hERG for human Ether-a-go-go Related Gene. The KCNH2 protein forms a heteromeric complex with the regulatory β-subunit encoded by the KCNE2 gene. The proteins encoded by the KCNQ1 (α-subunit) and KCNE1 (regulatory β-subunit) genes compose the I(Ks) channel. The KCNQ1 encoded protein is referred to as Kv7.1. Mutations in the KCNQ1 gene result in the inherited arrhythmia referred to as long-QT syndrome and as such the gene is also referred to as KvLQT1.
If the heart is to function as the efficient blood pump it must, the generation of action potentials and subsequent myocardial contraction must be coordinated into a regular, repetitive, and organized process. Normal cardiac contraction occurs in response to the cardiac electrical activity that is initiated by a specialized group of cardiomyocytes called the sinoatrial (SA) node. The SA node resides on the posterior aspect of the right atrium at the junction of the superior and inferior vena cava. Following the initiation of an action potential by the SA node it travels through both the right and left atria where it then activates another area of specialized cardiomyocytes called the atrioventricular (AV) node. The AV node is localized to the junction between the atria and ventricles in the ventricular septum. The action potential is slowed through the AV node so that its propagation to the ventricles allows time for the ventricles to fill with blood from the atria. Normal cardiac conductance, from the atria to the ventricles, can only occur through the AV node. The action potential that emerges from the AV node enters another specialized group of cells called the bundle of His. The bundle of His divides into left and right bundles that give rise to the Purkinje fibers. Purkinje fibers line the entire surface of both ventricles.
Cardiac Nodal Muscle Action Potential
Action potentials in cardiac nodal (SA and AV) cells do not exhibit the same characteristics as those in the rest of cardiac myocytes. Nodal cells exhibit a slow upward deflecting (to a more positive membrane potential) current associated with phase 4 of the action potential cycle typical of a ventricular cardiomyocyte. This property is referred to as the pacemaker current. The cardiomyocytes of nodal tissues do not contain the fast voltage-gated Na+ channels that are responsible for the rapid depolarization in other cardiomyocytes. The lack of these fast voltage-gated Na+ channels in nodal cells explains the slow depolarization seen in phase 0 of the action potential. At a certain point in the propagation of nodal pacemaker action potentials the cells experience a brief period of hyperpolarization (increase in negative membrane potential) that is associated with a small inward depolarizing Na+ current.
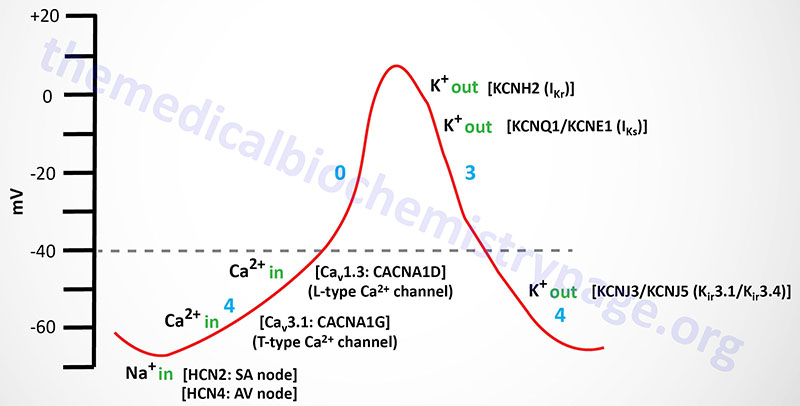
The inward Na+ movement is mediated by the non-selective hyperpolarization-activated cyclic nucleotide-gated channels that are commonly called HCN channels. HCN channels transport both Na+ and K+ ions. Humans express four HCN genes identified as HCN1–HCN4. The HCN1, HCN2, and HCN4 channels are expressed in the heart with HCN2 contributing to action potentials from the SA node and HCN4 contributing to action potentials from the AV node. The HCN channels are referred to as the pacemaker channels, also often referred to as the “funny” current channels. The term funny current was imparted on the pacemaker current since the HCN channels responsible exhibit mixed Na+ and K+ permeability, they are activated by hyperpolarization, and they exhibit slow kinetics.
Nodal cells also do not exhibit the phase 1 (partial repolarization) and phase 2 (Ca2+-mediated plateau) currents seen in other cardiomyocytes. The phase 1 of ventricular cardiomyocyte action potentials results from the closure of the fast-acting voltage gated Na+ channels and since these are not present in nodal cardiomyocytes nodal action potential lack this phase of the action potential. Nodal cells also lack the K+ channels responsible for the Ito,f and Ito,s currents that contribute to the phase 1 and phase 2 portions of ventricular cardiomyocyte action potentials.
Nodal cell mediators of action potential propagation are, initially, low voltage (transient) T-type voltage-gated Ca2+ channels (Cav3.1 whose α-subunits are encoded by CACNA1G gene), followed by L-type Ca2+ channels (Cav1.3 whose α-subunits are encoded by the CACNA1D gene). The L-type Ca2+ channels initiate the phase 0 current of the nodal cells. The rate of spontaneous action potential generation is fastest in the SA node (200-300 msec). This results in continual activation of action potentials from this location before any other cardiac tissue can generate its own action potential. This function of the SA node is why it is referred to as the cardiac pacemaker.
As the action potential propagates in nodal cells to a membrane potential of around -50 mV the T-type Ca2+ channels open allowing inward movement of Ca2+ ions which further depolarizes the cell. Eventually, at a membrane potential of -40 mV, the L-type Ca2+ channels open initiating phase 0 of the action potential. When the membrane potential nears 0 mV the L-type Ca2+ channels close and the outward rectifying K+ channels open to initiate repolarization (phase 3) of the cell. These repolarization K+ channels are the same in nodal cells as they are in other cardiomyocytes, namely IKr and IKs.
Complete repolarization in nodal cells involves inwardly rectifying K+ channels, whose function is the same as the IK1 (encoded by the KCNJ2 gene) channel in ventricular cardiomyocytes. However, in nodal cells these inwardly rectifying K+ channels are identified as IKACh given that they were originally demonstrated to be activated by muscarinic agonists such as acetylcholine and carbachol. The IKACh channels are composed of proteins identified as Kir3.1 (encoded by the KCNJ3 gene) and Kir3.4 (encoded by the KCNJ5 gene). Because the channels formed from the KCNJ3 and KCNJ5 encoded proteins are responsive to G-protein coupled receptor activation they are also referred to as G protein-coupled inwardly-rectifying potassium channels (GIRK). The duration of a typical SA node cell action potential is 200-300 msec.
Calcium: Role in Muscle Contraction and Relaxation
Activation of Striated Muscle Contraction
Events that stimulate muscle activity, by raising sarcoplasmic (cytosolic) calcium, begin with neural excitation at neuromuscular junctions. Excitation induces local depolarization of the sarcolemma (plasma membrane) and the activation of voltage-gated calcium channels allowing Ca2+ ion to enter the skeletal or cardiac muscle cell.
There are three distinct families of voltage-gated calcium channels (also called voltage-dependent calcium channels, VDCC) identified as Cav1, Cav2, and Cav3. The VDCC of striated muscle are of the Cav1 family of calcium channels (also known as L-type calcium channels). The Cav1 channels are composed of five different protein subunits identified as the α1-, α2-, β-, γ, and δ-subunits. The α1-subunit is the actual transmembrane channel through which the Ca2+ ions flow. The α2- and the δ-subunits are derived from a single preproprotein that, following cleavage, forms an extracellular disulfide bonded glycoprotein dimer designated α2/δ. The γ-subunit is a transmembrane glycoprotein and the β-subunit is an intracellular protein. A high degree of variability exists in the Cav1 (L-type) calcium channel subfamily due to multiple genes encoding the various protein subunits as well as due to alternative splicing of the mRNAs derived from these genes.
Humans express four α1-subunit genes that form the various Cav1 calcium channels of striated muscle. These four genes are identified as CACNA1S (forms the core of the Cav1.1 channel), CACNA1C (forms the core of the Cav1.2 channel), CACNA1D (forms the core of the Cav1.3 channel), and CACNA1F (forms the core of the Cav1.4 channel). The Cav1.1 channels are also referred to as the dihydropyridine receptor (DHPR) due to the fact that this drug was shown to block this type of Ca2+ channel. The CACNA1S encoded protein is the binding site for DHP.
There are four different β-subunit encoding genes identified as CACNB1, CACNB2, CACNB3, and CACNB4. The α2/δ preproprotein is encoded by the CACNA2D1 gene. An alternative splice variant mRNA from the CACNA2D1 gene lacks the δ-subunit portion of the preproprotein. There are eight genes encoding γ-subunit proteins which are identified as the CACNG1–CACNG8 genes.
Calcium entry across the sarcolemma causes the initiating neural stimulus to spread the induced action potential of the muscle cell to the associated T-tubule (transverse tubule) system and deep into the interior of the myofiber. T-tubule depolarization results in the opening of the Cav1.1 voltage-gated Ca2+ channels (also called DHPR) leading to an influx of Ca2+ which effectively spreads the action potential to the sarcoplasmic reticulum (SR: muscle form of the endoplasmic reticulum, ER).
The influx of calcium exerts the effect of opening of calcium release channels in the SR membranes. These calcium release channels belong to the family of ligand-gated ion channels, where in this case the ligand is the Ca2+ ion. The SR calcium release channels are also known as the ryanodine receptors (RYR) due to the fact that they were originally identified by their high affinity for the plant alkaloid ryanodine. There are several RYR genes in humans with the RYR1 gene being the predominantly expressed member in skeletal muscle. The RYR2 gene is highly expressed in cardiac myocytes.
The activation of the SR calcium release channel leads to a massive, rapid movement of calcium from the lumen (cisternal compartment) of the SR into the sarcoplasm close to nearby myofilaments. The SR released calcium binds to the TnC subunit of the troponin complex resulting in the production of multiple myosin power strokes, as long as the available calcium concentration remains greater than about 1μM–5μM.
Striated Muscle Relaxation
Normally, cessation of contractile activity and a state of relaxation follow electrical quiescence at the neuromuscular junction. The sarcoplasmic membrane returns to its resting electrical potential (about 60 mV more positive outside), as does the entire T tubule system and the SR membrane. When the initiating signal for muscle contraction is removed, the myocytes need to reverse the localization of the activating calcium from the sarcoplasm back into the SR. Sarcoplasmic calcium is pumped back into the lumen of the SR by an extremely active ATP–driven calcium pump, which comprises one of the main proteins of the SR membrane.
This SR calcium pump is a member of a family of Ca2+-ATPases identified as Sarco/Endoplasmic Reticulum Ca2+–ATPases (SERCA). The human SERCA proteins are encoded by a family of three genes identified as the ATPase sarcoplasmic/endoplasmic reticulum Ca2+ transporting (ATP2A) genes. These three genes are identified as ATP2A1 (SERCA1), ATP2A2 (SERCA2), and ATP2A3 (SERCA3). Within skeletal muscle, the major SERCA transporters are encoded by the SERCA1 (specifically SERCA1A in fast glycolytic fibers) and SERCA2 (specifically SERCA2A in slow twitch oxidative fibers) genes. Within cardiac muscle the major SERCA is encoded by the SERCA2 gene and is identified as SERCA2B.
For each ATP hydrolyzed by the SR Ca2+-ATPase, two calcium ions are moved out of the sarcoplasm into the SR lumen, with sarcoplasmic calcium ultimately falling below 0.1μM, or 50– to 100–fold lower than the KD for calcium binding to the TnC subunit of the troponin complex.
The activity of the SERCA transporters is regulated, in part, via their interactions with two small transmembrane proteins identified as phospholamban (PLB) and sarcolipin (SLN). Phospholamban (encoded by the PLN gene) interacts SERCA transporters where its function is to inhibit the reuptake of Ca2+ into the SR. The primary site of expression of the PLN gene is in cardiac muscle. Phospholamban exists in monomeric and homopentameric conformations where the monomeric form interacts with SERCA to inhibit Ca2+ transport. Phosphorylation of PLB causes it to dissociate from SERCA and form homopentamers. Phosphorylation of PLB by PKA reduces the inhibitory action of PLB on SERCA2A which promotes Ca2+ reuptake. Diastolic relaxation of cardiac myocytes requires Ca2+ reuptake by the SR, thus, the inhibition of PLB allows for an increased rate of myocyte contraction and relaxation resulting in an overall increased force of contraction.
Sarcolipin (encoded by the SLN gene) is abundantly expressed in skeletal muscle. Sarcolipin was originally identified as a proteolipid, a protein with covalently attached lipid, in the SR which contributed to the derivation of the name of the protein. When sarcolipin interacts with SERCA transporters the transport of Ca2+ is uncoupled from ATP hydrolysis. This effect of sarcolipin contributes to the role of skeletal muscle in thermogenesis. Indeed, when experiments were performed in mice in which the SLN gene was knocked out the animals were unable to maintain their body temperatures under conditions of acute cold. Expression of the SLN gene is regulated by a myokine identified as feimin. Production of feimin increases in response to feeding and exercise. During exercise the consumption of skeletal muscle ATP results in the activation of AMPK which phosphorylates feimin. Phosphorylated feimin migrates to the nucleus where it inhibits the transcription factor, FOXC2, which normally activates expression of the SLN gene. The inhibition of SLN expression allows for enhanced exercise performance at the expense of thermogenesis.
The cisternal surface of the SR membrane also contains large quantities of a glycoprotein known as calsequestrin. The skeletal muscle calsequestrin is encoded by the CASQ1 gene and the cardiac muscle form of the protein is encoded by the CASQ2 gene. Calsequestrin avidly binds calcium, decreasing its free concentration within the lumen of the SR, and thus favors calcium accumulation inside the SR.
An additional and physiologically significant repository of sarcoplasmic calcium is the mitochondrial matrix. Calcium uptake by mitochondria, across the outer mitochondrial membrane (OMM), is the function of transporters of the voltage-dependent anion channel (VDAC) family.
Mitochondria have remarkably active calcium transport mechanisms, driven, in part, by the electron transport-generated chemiosmotic potential. Under aerobic conditions this process uses the energy of electron transport to sequester calcium in the mitochondrial matrix, in preference to the synthesis of ATP. Although there are several mitochondrial calcium transporters the major inner mitochondrial membrane (IMM) transporter is called simply the calcium uniporter, encoded by the MCU (mitochondrial calcium uniporter) gene. The mitochondrial uniporter functions to transport calcium across the IMM, into the matrix, in conjunction with the proteins identified as MICU1 and MICU2, mitochondrial calcium uptake 1 and 2, respectively.