
Last Updated: April 8, 2026
Introduction to Processes of Protein, Organelle, and Cell Turnover
The concepts of protein, organelle, and cell turnover relate to the fact that during the normal course of cellular life, macromolecules serve their intended purposes and must be degraded or turned over to ensure their function(s) remain tightly regulated. In addition, cellular components such as proteins and even organelles can become damaged and the cell requires mechanisms to remove the damaged entities in order to ensure viability.
The processes by which proteins and organelles are degraded, removed, and even recycled are vital for cellular survival. These processes are regulated in highly specific ways that include protein modification, protein degradation and amino acid recycling, organelle tagging, engulfment, and lysosomal degradation with component parts being recycled. Ultimately, if these controlled and regulated processes become overwhelmed a program of cell death, called apoptosis, can be initiated in order to destroy aberrant cells as a means to ensure overall tissue survival.
This page is not intended to be an exhaustive overview of the details of all of these macromolecule and cell turn-over processes. However, certain details of protein turnover, involving the ubiquitin and ubiquitin-like modifier pathways, and the pathways of organelle turnover, and the programmed cell death pathways will be highlighted.
Ubiquitin and Protein Ubiquitylation (Ubiquitination)
The protein ubiquitin is the founding member of a family of proteins that includes at least 20 members, all of which are involved in post-translational modifications of numerous substrate proteins. The originally characterized function of ubiquitin was its attachment to proteins to target them for degradation in the protein degradation apparatus termed the proteasome. Humans express four different genes that produce the protein ubiquitin, UBB, UBC, UBA52, and RPS27A (also known as UBA80).
The UBB gene generates multiple alternatively spliced mRNAs that encode proteins that are composed of three direct repeats of the ubiquitin coding sequence and, therefore, produce a polyubiquitin precursor protein.
Like the UBB gene, the UBC gene generates an mRNA that encodes a polyubiquitin precursor protein.
The UBA52 gene generates multiple alternatively spliced mRNAs that encode precursor ubiquitin fusion proteins. The UBA52 mRNAs encode proteins that contain ubiquitin at the N-terminal part of the fusion protein and large ribosomal subunit protein L40 at the C-terminal part.
The RPS27A gene encodes a ubiquitin fusion protein like that encoded by the UBA52 gene. The RPS27A gene generates multiple alternatively spliced mRNAs that encode precursor proteins containing ubiquitin in the N-terminal part fused to small ribosomal subunit protein S27a in the C-terminal part.
The other members of the ubiquitin protein family are referred to as ubiquitin-like proteins or ubiquitin-like modifiers (or ubiquitin-like molecules) and given the designation UBL and also often designated ULM. The UBL/ULM family of proteins represents one of two classes of the ubiquitin-like molecules while the second class is composed of proteins that contain a ubiquitin domain and are, therefore, referred to as ubiquitin-domain proteins (UDP). The UBL/ULM family of proteins are used to modify protein substrates in a manner similar to that of ubiquitin as described below. The UDP family all contain a domain similar to ubiquitin but they are not conjugated to protein targets. The UDP function as adapter proteins that noncovalently interact with ubiquitin or other UBL/ULM proteins.
The UBL/ULM family includes the small ubiquitin-related modifier (SUMO) proteins and NEDD8 (originally isolated in a screen for Neural precursor cell Expressed, Developmentally Down-regulated genes) which are the most well characterized of the UBL family proteins.
Additional ubiquitin-like modifiers include, but are not limited to, members of the human ATG8 (autophagy related gene 8) family of proteins, ATG12 (autophagy related gene 12), ISG15 (interferon-stimulated gene 15), URM1 (ubiquitin-related modifier 1), UFM1 (ubiquitin-fold modifier 1), UBD (ubiquitin-like modifier D, originally identified as FAT10 for HLA-F adjacent transcript 10), and FUB1 (also known as FUBI for fusion ubiquitin-like and also known as MNSFβ for monoclonal nonspecific suppressor factor β).
Like the UBA52 and RPS27A genes, which encode fusion proteins containing ubiquitin, the FAU gene encodes the FUB1 protein sequence at the N-terminal end of a fusion protein where the small ribosomal subunit protein S30 corresponds to the C-terminal portion of the fusion protein.
Although originally characterized for its role in protein degradation, ubiquitin, and the other UBL proteins, are also involved in diverse metabolic processes that include regulation of subcellular localization, nuclear transport, protein synthesis, DNA repair, regulation of cellular responses to oxidative stress, regulation of inflammatory responses, and autophagy.
Protein Ubiquitylation
Proteins are in a continual state of flux, being synthesized and degraded. In addition, when proteins become damaged they must be degraded to prevent aberrant activities of the defective proteins and/or other proteins associated with those that have been damaged. One of the major mechanisms for the destruction of cellular proteins involves a complex structure referred to as the proteasome.
In eukaryotic cells the proteasome is found in the cytosol and the nucleus and has a large mass such that it has a sedimentation coefficient of 26S. The 26S proteasome comprises a 20S barrel-shaped catalytic core as well as 19S regulatory complexes at both ends. Degradation of proteins in the proteasome occurs via an ATP-dependent mechanism.
The other major orderly protein (and other cellular constituents) degradation pathway is referred to as autophagy, from the Greek for “self” and “to eat”. The process of autophagy involves the sequestration of targeted cytoplasmic constituents into a double membrane vesicle termed the autophagosome. Following its formation, the autophagosome fuses with the lysosomal machinery of the cell and the contents are degraded.
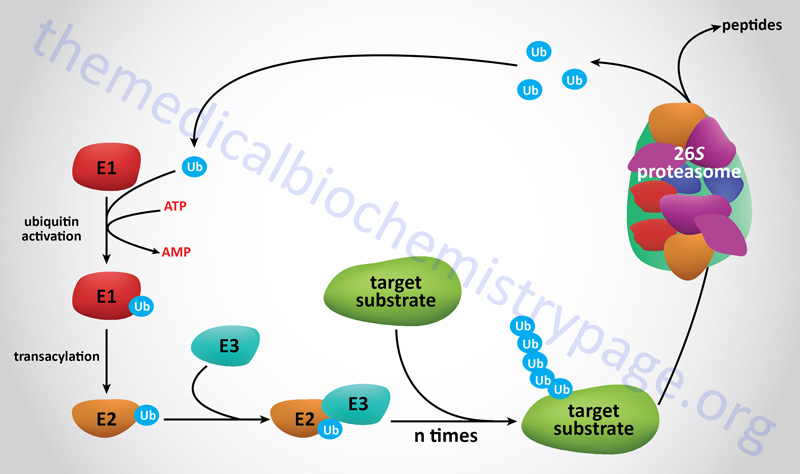
Proteins that are to be degraded by the proteasome are first tagged by attachment of multimers of the 76 amino acid protein ubiquitin, a process termed ubiquitylation or ubiquitination. Many proteins involved in cell cycle regulation, control of proliferation and differentiation, programmed cell death (apoptosis), DNA repair, immune and inflammatory processes, and organelle biogenesis have been discovered to undergo regulated degradation via the 26S proteasome. This proteolytic system is referred to as the ubiquitin-proteasome system, UPS.
The degradation of proteins via the 26S proteasome involves a two-step process starting with ubiquitylation of the target protein followed by entry into, and degradation by, the proteasome complex with release of ubiquitin monomers that can be re-used to tag additional proteins.
The process of ubiquitin addition to the substrate protein involves multiple ubiquitin additions such that the targeted protein is polyubiquitylated. Of clinical significance are the observations that deregulation of the functions of the proteasome can contribute to the pathogenesis of various human diseases such as cancer, myeloproliferative diseases, and neurodegenerative diseases.
Prior to target protein ubiquitylation, the precursor ubiquitin proteins need to be cleaved at their C-terminal end to expose a di-glycine motif. The enzymes that cleave the C-terminus of ubiquitin are called ubiquitin C-terminal hydrolases, UCH. The UCH enzymes are also members of the large family of deubiquitylating enzymes described below. There are four human UCH encoding genes, UCHL1, UCHL3, UCHL5, and BAP1 (BRCA1 associated protein 1). Attachment of the C-terminal glycine of ubiquitin to lysine residues (via an isopeptide linkage) in target proteins involves a series of three enzyme activities.
E1 Ubiquitin-Activating Enzymes
The first activity, identified as E1 (also called ubiquitin-activating enzymes and also ubiquitin like modifier activating enzymes), activates ubiquitin in an ATP-dependent manner such that the ubiquitin is bound to the E1 enzyme via a high-energy thiol ester. Humans express a family of ten genes encoding E1 class ubiquitin-activating enzymes.
E2 Ubiquitin-Conjugating Enzymes
The next class of enzyme, referred to as E2 (also called ubiquitin-conjugating enzymes, UBE; or ubiquitin-carrier proteins), transfers the ubiquitin via an E2 thiol ester intermediate to the substrate protein. The substrate proteins are recognizable by E2 because they are bound by the third class of enzyme called E3 ligases or ubiquitin protein ligases. Humans express a family of 43 genes encoding E2 class ubiquitin conjugating enzymes.
E3 Ubiquitin Ligases
The E3 enzymes carry out the final step in the process which is the covalent attachment of ubiquitin to the targeted substrate protein. The ubiquitin is generally transferred to the ε-amino group of an internal lysine residue in the substrate protein. There are examples where the ubiquitin is attached to the N-terminal amino group in a substrate protein following acetylation of the target protein. More than 600 (prediction of more than 1000) genes encoding E3 class ubiquitin ligases have been identified in humans.
The E3 ligases are divided into three major families that includes the RING finger (RNF) domain containing family that acquired the term RING from Really Interesting New Gene. The RING E3 ligases represent the largest family of ubiquitin ligases. The RING finger domain is a zinc finger-like domain which is a protein domain involved in DNA-binding, RNA-binding, protein-binding, and lipid-binding. There are at least 340 genes in humans that express RING finger domain containing proteins most of which are E3 ubiquitin ligases. The RING E3 ligases directly transfer ubiquitin from E2 proteins to target substrates.
RING domain E3 ubiquitin ligases have been classified into four distinct categories defined by their functional structure. These four categories are monomeric, homodimeric, heterodimeric, and multi-subunit complexes.
The multi-subunit RING finger domain E3 ligases associate in complexes with proteins of the cullin family forming the cullin-RING E3 ligase family. Cullin proteins are a family of proteins that serve as molecular scaffolds. Humans express eight cullin genes identified as CUL1, CUL2, CUL3, CUL4a, CUL4b, CUL5, and CUL7 and PARC (p53-associated parkin-like protein). Each of these eight proteins contains a highly homologous domain termed the cullin domain. The proteins encoded by the CUL genes all assemble into multi-subunit complexes forming the cullin-RING E3 ubiquitin ligase (CRL) family.
In addition to a cullin and a RING finger domain protein, the CRL also contain substrate recognition proteins that represent the E2 ubiquitin-conjugating enzymes. The archetypal CRL (CRL1) is also commonly called the SCF complex where SCF stands for SKP1 (S-phase kinase associated protein 1), CUL1, and F-box containing complex.
The second E3 ligase family is the HECT domain family. The term HECT is derived from Homologous to the papilloma virus E6 protein associated protein (E6AP) Carboxy Terminus. Humans express 28 genes that encode HECT domain containing E3 ligases. HECT domain E3 ligases transfer ubiquitin from E2 proteins to target substrates in a 2-step process.
The third family of E3 ligases is the RBR family where RBR stands for Ring Between Ring. Humans express 14 genes that encode RBR family E3 ligases. Like the HECT domain E3 ligases, the RBR E3 ligases transfer ubiquitin from E2 proteins to target substrates in a 2-step process.
Protein Deubiquitylation
Whereas, ubiquitylation targets proteins for degradation in the proteasome there needs to be a mechanism to ensure that inappropriately tagged proteins, i.e. those that are not destined for degradation, can be untagged, as well as deubiquitylating proteins that have been modified for the purposes of regulation not degradation.
There is a large family of isopeptidases called deubiquitylating enzymes (DUBs) that carry out this vital function of removing ubiquitin from proteins to which it is either added for temporary regulation or has been mistakenly attached. The DUB enzymes also cleave monoubiquitin from polyubiquitin chains that have been removed from proteins, effectively recycling active ubiquitin.
The DUB enzymes are all members of either the large family of cysteine proteases or the large family of metalloproteases. Humans express over 90 genes whose encoded proteins have been identified as DUBs or are putative DUB enzymes with the largest group of DUBs belonging to the cysteine protease family of enzymes.
The DUB enzymes are divided into seven subfamilies identified as the ubiquitin-specific proteases (USP; 56 genes), the ubiquitin C-terminal hydrolases (UCH; 4 genes), the Machado-Josephin domain proteases (MJD; 4 genes), the ovarian tumor proteases (OTU; 17 genes), the JAMM/MPN+ zinc metallopeptidase family (12 genes), the ZUP1 deubiquitinase (1 gene), and the MINDY deubiquitinases (5 genes).
The Machado-Josephin domain was originally identified in the protein that is encoded by the gene that is mutated in Machado-Josephin disease, MJD. MJD is also known as spinocerebellar ataxia type 3 (SCA3).
The JAMM/MPN+ metallopeptidases are so-called from the amalgam of three domains JAB1, MPN, and Mov34 that are found in the proteins of this subfamily. JAB1 is Jun Activation domain-Binding protein 1. MPN refers to Mpr1 and Pad1 N-terminus. Mpr1 is Metalloprotease promoting regression 1. Pad1 is Peptidylarginine deiminase 1. The Mov34 protein was originally identified in mice and refers to Mouse ovary morphogenesis 34.
The ZUP1 protein refers to Zinc finger containing Ubiquitin Peptidase 1.
The MINDY deubiquitinases represent the most recently identified DUB subfamily of enzymes. The acronym MINDY refers to Motif Interacting with Ubiquitin-containing Novel DUB familY. There are currently five identified human genes encoding MINDY family deubiquitinases where MINDY1 was the originally characterized member. The MINDY1 gene is identified as MINDY lysine 48 deubiquitinase 1. The MINDY family proteins are also known as FAM63 (Family with sequence similarity 63) where MINDY1 has also been identified as FAM63A.
Recent work has shown that the MINDY1 protein is a target of the AKT/PKB kinase and that when MINDY1 is phosphorylated it interferes with insulin receptor-mediated signaling.
Ubiquilin Family Proteins in Protein Degradation
The ubiquilin (UBQLN) family of proteins comprises five members, UBQLN1-UBQLN4 and UBQLNL, that contain a ubiquitin-like (UbL) domain at the N-terminus and a ubiquitin-associated (UbA) domain at the C-terminus. The UBQLN proteins function as adaptor proteins to facilitate the degradation of proteins via both the ubiquitin-proteasome pathway and the autophagy pathway. The UBQLN family proteins also participate in the endoplasmic reticulum (ER)-associated protein degradation pathway (ERAD).
Clinical Significance of Abnormal Ubiquitylation
Inactivation of a critical activity such as that catalyzed by the E1 enzymes results in lethality. However, there are numerous pathological states that can be attributed, in part, to mutations in recognition motifs in ubiquitylation substrates and enzymes in the ubiquitylation process. Disease states associated with the ubiquitin modification system can be classified into two groups.
One group results from a loss of function mutation in a ubiquitin system enzyme or target protein that results in stabilization of the protein that should normally be degraded. The other group results from gain of function mutations that result in abnormal or accelerated degradation of target proteins.
The most obvious disease state that could be expected to arise as a result of defective ubiquitylation processes is cancer. In fact, many malignancies are known to result from defective ubiquitin-mediated degradation of growth promoting proteins such as FOS, MYC, and SRC. Likewise, inappropriate degradation of key regulators of cell cycle progression such as the tumor suppressor p53 and p27KIP1 (CDKN1B), which is an inhibitor of cyclin-dependent kinases (CDKs which control progression through the cell cycle) is also associated with various types of cancer. In addition to cancers, defective ubiquitylation is found associated with neurodegenerative diseases such as Parkinson disease, Alzheimer disease, and amyotrophic lateral sclerosis.
Protein Neddylation
The NEDD8 gene was originally isolated in a screen for abundantly expressed genes in the embryonic mouse brain. There were ten genes isolated in this screen and the were all designated as Neural precursor cell-Expressed, Developmentally Downregulated genes. These ten genes all have distinct functions in a variety of disparate biochemical pathways. The NEDD8 gene encodes a ubiquitin-like protein and of all the UBLs it is the most closely related to ubiquitin exhibiting 59% sequence identity between human NEDD8 and ubiquitin. Like ubiquitin, the NEDD8 protein is attached to target proteins and this modification is accomplished by the largest subfamily of E3 ligases, the cullin-RING E3 ubiquitin ligases described earlier. The attachment of NEDD8 to target proteins is a process referred to as neddylation.
Functional NEDD8 protein is localized primarily to the nucleus. Following its synthesis, the precursor NEDD8 protein is proteolytically processed to expose the necessary C-terminal glycine residue that is the amino acid attached to substrate proteins. There are two primary NEDD8 processing enzymes, one that functions on both ubiquitin and NEDD8 (C-terminal hydrolase isozyme 3, UCHL3) and one that is specific for NEDD8 (deneddylase 1, DEN1).
Similar to the mechanism of ubiquitylation, neddylation involves E1 activating enzymes, E2 conjugating enzymes, and E3 ligases. The NEDD8 E1 activating enzyme is termed NAE. NAE is a heterodimeric complex composed of ubiquitin activating enzyme 3 (UBA3) and amyloid-β precursor protein binding protein 1 (APPBP1). There are at least two NEDD8-specific E2 conjugating enzymes identified as ubiquitin conjugating enzyme E2 M (UBE2M, also known as UBC12) and UBE2F. All of the NEDD8 E3 ligases also function to attach ubiquitin to target substrates. The cullin-RING E3 ligases, which represent the largest subfamily of ubiquitin E3 ligases, all function in the neddylation reaction.
Similar to the process by which ubiquitin can be removed from a target proteins through the action of the deubiquitylation (DUB) enzymes, neddylated proteins can also be deneddylated. The deneddylation reaction is catalyzed by the protein encoded by the CSN5 (COP9 signalosome complex subunit 5) gene when this protein is engaged in the eight-subunit COP9 signalosome complex. The COP9 signalosome is the sole regulator (deactivator) of cullin-RING ligase (CRL) complexes. The COP9 signalosome is highly similar to the 19S complexes of the 26S proteasome.
Important substrates for neddylation are the eight human cullin gene encoded proteins. All eight of these proteins contain a highly conserved C-terminal neddylation site (IVRIMKMR) where the bolded lysine residue is the NEDD8 attachment site. As discussed above, the cullin proteins serve as molecular scaffolds for the RING finger domain family of E3 ubiquitin ligases which together form the CRL family of E3 ligase complexes.
In addition to the cullin proteins, at least 50 other proteins, including transcription factors, have also been identified as being substrates for neddylation. Some important examples of NEDDylated proteins are p53, HIF-1α, pVHL, and PEPCK.
In the case of transcription factors the consequences of neddylation is, in general, a suppression of their activity as a result of altered protein-protein or protein-DNA interactions as well as altered protein stability and subcellular localization. One very clinically relevant transcription factor that is neddylated is the tumor suppressor, p53. In normal cells the p53 protein is trapped in the cytosol and targeted for degradation through interaction with the E3 ubiquitin ligase MDM2. When cells experience DNA damage or other forms of cellular stress p53 is released from MDM2 and enters the nucleus to initiate a program of gene expression designed to allow the cell to respond to the stress event. The MDM protein also serves as the E3 ligase for NEDD8 allowing p53 to neddylated. The effect of neddylation of p53 is not degradation but inhibited transcriptional activity. Other important neddylated transcription factors are members of the E2F family. Neddylation of members of this transcription factor family, like the effect of p53 neddylation, results in down-regulation of their transcriptional activity.
SUMO Family of Ubiquitin-Like Proteins
As discussed earlier, many proteins are post-translationally modified via the addition of ubiquitin. Over the past several years numerous ubiquitin-like (UBL) proteins have been identified. Like ubiquitin, UBLs are added to other proteins via post-translational reactions. However, unlike ubiquitylation which primarily targets proteins for degradation in the proteasome, UBL modifications are not for protein degradation. Although there are several types of UBLs (e.g. NEDD8 as discussed in the section above), those with the broadest range of functions and the largest number of known substrates are members of the SUMO (small ubiquitin-related modifier) proteins. Proteomics analysis has identified over 3600 different proteins (representing almost 18% of the human proteome), that function in a variety of different capacities within cells, to be modified by SUMOylation. Modification of proteins by SUMO addition has been shown to occur in all tissues and at all developmental stages. However, in contrast ubiquitylation which occurs in almost all cell compartments, SUMOylation predominates in the nucleus with roughly 65% of SUMOylated proteins being associated with the nucleus.
There are four SUMO proteins in mammalian cells designated SUMO-1 to SUMO-4. Although it has been reported that a fifth SUMO (SUMO-5) protein in mammals it may actually be a pseudogene.
SUMO-1 is also identified as UBL1, PIC1 [promyelocytic leukemia (PML) interacting clone 1], sentrin, GMP1 [GTPase activating protein (GAP) modifying protein 1], and Smt3c (suppressor of mif two 3 homolog 1c).
SUMO-2 is also identified as sentrin 2, Smt3b, and GMP1-related protein.
SUMO-3 is also identified as sentrin 3 and Smt3a. SUMO-2 and SUMO-3 differ only at three N-terminal amino acid residues and are, therefore, often referred to collectively as SUMO-2/3. SUMO-2/3 share only 50% amino acid similarity to SUMO-1.
SUMO-4 is based upon DNA sequence analysis and exhibits 87% amino acid similarity to SUMO-2. It is believed that the SUMO-4 gene is actually a pseudogene since it does not contain any introns. Although SUMO-4 mRNA has been detected in tissues such as spleen and lymph nodes, no protein product has been detected.
Following de novo synthesis, SUMO proteins must undergo a C-terminal cleavage processing event in order to render the proteins biologically active. The C-terminal cleavage reactions are catalyzed by a family of proteins called SENP (sentrin/SUMO-specific protease). There are at least seven SENP proteins (encoded by the SENP1–SENP3 and SENP5–SENP8 genes) in mammals. The removal of the C-terminal amino acids reveals a di-glycine (–G–G) motif that allows the SUMO protein to subsequently conjugate to lysine (K) residues in target proteins. The SENP family of enzymes are required for both the maturation of SUMOylated proteins as well as for the removal (deSUMOylation) of SUMO from protein substrates.
SUMO conjugation to target proteins requires a series of enzymatic steps that is similar in mechanism to the conjugation of ubiquitin to target proteins. SUMO proteins are initially activated via an ATP-dependent reaction catalyzed by the E1 activating complex. This complex is a heterodimer composed of SUMO-activating enzyme E1 (SAE1) and SAE2. This activating reaction forms a covalent bond between an active site cysteine in SAE2 and the C-terminal glycine of the SUMO protein.
The next step involves the transfer of the SUMO protein to an active site cysteine of the protein identified as Ubc9 (ubiquitin-conjugating 9). As yet Ubc9 is the only known SUMO-conjugating (E2) enzyme in mammalian tissues. Ubc9 brings the SUMO protein to the target protein by recognizing, and binding to, the consensus SUMOylation motif in target proteins.
The consensus motif for SUMOylation is the following: ΨKxD/E where Ψ is a large hydrophobic amino acid and x is any amino acid. Approximately 75% of all SUMOylated proteins contain this target motif, however, not all proteins that contain this motif are SUMOylated and some proteins are SUMOylated on lysine residues that do not lie in this motif.
As described above for ubiquitylation, there are three classes of enzyme involved in the SUMOylation process: E1, E2, and E3. Since Ubc9 can directly conjugate SUMO proteins to their targets it was thought that no E3-like activity was required. However, SUMO E3 ligases have been identified and although they do not function directly in the enzymatic process of SUMO attachment to target proteins they do act as a scaffold.
SUMO E3 ligases bring Ubc9-SUMO complexes into contact with target proteins. The mammalian SUMO E3 proteins are members of the PIAS [protein inhibitor of activated STAT (where STAT refers to signal transducer and activator of transcription)] family of proteins. There are currently five members of the mammalian PIAS protein family encoded by the PIAS1, PIAS2 (encodes the PIASxα and PIASxβ proteins), PIAS3, PIAS4 (encodes the PIASγ protein) genes. At least ten additional human genes have been identified that encode SUMO E3 ligase proteins.
Whereas, ubiquitylation of a protein results in is destruction in the proteasome, SUMOylation is a dynamic process and once attached the SUMO residue can be removed. Removal of SUMO from a target protein is accomplished by the same SENP enzymes that are required for the activation step of SUMO processing. SENP1 and SENP2 have broad specificity for SUMO-1 and SUMO-2/3 and are involved in their processing and deconjugation. SENP3 and SENP5 exhibit a preference for SUMO-2/3. Although SENP6 and SENP7 exhibit the same preference for SUMO-2/3 they are only minimally involved in deconjugation reactions. The primary functions of SENP6 and SENP7 are in editing the length of poly-SUMO-2/3 chains on target proteins. Therefore, it seems clear that SENP1 and SENP2 are responsible for maturation of SUMO proteins and deconjugation of SUMO-1 and SUMO-2/3 conjugated targets. SENP3 and SENP5 function in the removal of monomeric SUMO-2/3 from target proteins. SENP6 and SENP7 function as editors of SUMO-2/3 chains in target proteins.
The exact functional consequences of SUMOylation of a particular target protein is difficult to predict. However, modification of target proteins by SUMO addition is likely to lead to at least three non-mutually exclusive effects. The attachment of SUMO can result in the masking of a site in the target protein that is required for binding or interaction with a substrate protein. The addition of SUMO may alternatively result in the formation of an attachment site allowing for the recruitment of proteins that can now interact with the SUMOylated protein. The third consequence could be that the conformation of the SUMOylated protein is altered such that activity is regulated in some way.
Table of Several Mammalian Substrates for SUMO
The following Table is not intended to represent a complete list of SUMO target proteins it is just a representative list.
| Gene Symbol: Protein | Protein Function | Role of SUMOylation |
| AR androgen receptor | transcriptional activation | reduces the transcriptional activation activity of the receptor |
| GLUT1 glucose transporter 1 | glucose transport | exact consequence not fully defined but GLUT1 protein levels are down-regulated by Ubc9 |
| GLUT4 glucose transporter 4 | glucose transport | exact consequence not fully defined but GLUT4 protein levels are up-regulated by Ubc9 |
| HIPK2 homeodomain-interacting protein kinase 2 | transcriptional co-repression | mediates the localization of HIPK2 nuclear bodies, also called promyelocytic leukemia (PML) bodies |
| NFKBIA NFκB inhibitor α (IκBα) | inhibitor of NF-κB (nuclear factor κB) signal transduction | inhibits ubiquitylation of IκBα thereby blocking NF-κB activity |
| MDM2 Mdm2 proto-oncogene (originally isolated from mouse tumorigenic cell line 3T3DM) | E3 ubiquitin ligase for tumor suppressor p53 | inhibits ubiquitylation of Mdm2 resulting in activation of the E3 function of Mdm2 |
| TP53 p53 | tumor suppressor, is a transcription factor activated in response to DNA damage | activates p53 transactivation leading to increased apoptosis |
| PML promyelocytic leukemia | tumor suppressor | allows for the formation of nuclear bodies and the recruitment p53 |
| TOP1 topoisomerase I | topoisomerase involved in DNA replication and repair | exact consequence not fully defined but SUMOylation is induced after DNA damage with camptothecin |
| TOP2 topoisomerase II | topoisomerase involved in DNA replication and repair | exact consequence not fully defined but SUMOylation is induced after DNA damage with teniposide |
The Integrated Stress Response, ISR
Cells are continually exposed to varying levels of stress such as oxidative stress, endoplasmic reticulum (ER) stress (also referred to as the unfolded protein response, UPR), mitochondrial stress (ISRmt), nutrient stress (deprivation or restriction), hypoxia, and viral infection, to name the most common forms of stress. Each of these stressors affects the normal processes of translational homeostasis (proteostasis). Disturbances in proteostasis will ultimately exacerbate oxidative stress and trigger cell death. The primary organelles that manifest the effects of stress and disturbed proteostasis are the ER, the mitochondria, and the Golgi apparatus. The collective interplay between various stressors and organelles defines the cellular responses as the Integrated Stress Response, ISR. With respect to mitochondrial stress, and its role in the ISR, the designation ISRmt is commonly utilized.
Disturbances in proteostasis result in the activation of an eIF-2α kinase that phosphorylate the α-subunit of the translational initiation factor, eIF-2. There are four of these related kinases known to exist in mammalian cells. Each eIF-2α kinase contains unique regulatory domains that interact with various inducing agents in response to different stress-related conditions. All four eIF-2α kinase phosphorylates the eIF-2α protein on Ser 51 (S51).
Because these four eIF-2α kinases are involved in the responses of cells to a variety of stressful conditions, their activation, and subsequent effects, are collectively referred to as the integrated stress response (ISR). Indeed, the core component of the ISR is eIF-2. A distinguishing feature of the ISR, that is not associated with the UPR, is that there is regulation of both translation and transcription as a result of the activation of the ISR. The function of the ISR is to regulate complex downstream events that direct stressed cells to metabolic reprogramming or, under severe stress conditions, lead to activation of apoptosis, autophagy, or senescence.
During the acute phase of stress, activation of the ISR results in inhibited protein synthesis so as to maintain cellular homeostasis under conditions of impaired protein folding (the UPR) and impaired protein transport. Chronic stress causes the ISR to enhance the translation of the mRNA encoding the transcription factor, ATF4 (activating transcription factor 4; also known as CREB2: cAMP response element-binding protein 2).
The ATF4 mRNA contains two upstream open reading frames (uORFs) identified as uORF1 and uORF2. uORF1 encodes only three amino acids while uORF2 encodes 59 amino acids and overlaps the first 83 amino acids of the functional ATF4 protein. uORF1 serves a positive role in translational control allowing ribosomes to overcome the inhibitory role of uORF2 in response to eIF-2α phosphorylation. Under non-stressed conditions when eIF-2α phosphorylation is low the ribosomes rapidly scan to uORF2 and translate a non-functional protein thus, reducing the overall level of functional ATF4 protein. However, in the stressed condition there is less eIF-2-GTP which slows translational initiation allowing ribosomes that have engaged the limited eIF-2-GTP to scan to the correct ATF translational initiation site allowing for increases in functional ATF4 protein.
The function of ATF4 is to induce the expression of genes that allow the cell to respond to the stress conditions such as the transcription factors FOS, JUN, and CHOP (C/EBP homologous protein, where C/EBP is CCAAT/enhancer binding protein). The primary activity of CHOP is activation of apoptosis.
The activation of ATF4 leads to increased transcription of target genes that includes FGF21, GDF15, MTHFD2, and ATF5. Increased expression of the FGF21 gene leads to enhanced systemic glucose uptake and increased hepatic lipid metabolism. GDF15 is involved in central control of feeding behaviors. Increased expression of the MTHFD2 gene (encoding the mitochondrial NAD+/NADP+-dependent enzyme that possesses 5,10-methylene-THF dehydrogenase and 5,10-methenyl-THF cyclohydrolase activities) enhances the generation of the mitochondrial pool of tetrahydrofolate (THF) derivatives. Enhancement of the THF pool allows for increased nucleotide biosynthesis, a process that is required in the context of DNA damage repair.
ATF4 translation also activates a feedback regulatory mechanism to control the level of eIF-2α phosphorylation via increased expression of the GADD34 gene (growth arrest and DNA damage-inducible protein 34). GADD34 is a regulatory subunit of protein phosphatase-1 (PP-1) and when the GADD34 gene is activated by ATF4 there is a resultant increase in PP-1-mediated phosphate removal from eIF-2α due to increased GADD34 protein.
Forms of Regulated Cell Death
There are many forms of cell death and they can be divided into two broad categories, regulated cell death (RCD) and accidental cell death (ACD). Within the context of RCD there is the commonly utilized description of programmed cell death (PCD). The more classical forms of PCD are of three distinct types, apoptosis, autophagy, and necroptosis. Apoptosis is also referred to as type I PCD, autophagy is also referred to as type II PCD, and necroptosis is also referred to type III PCD.
The many forms of regulated cell death (RCD), in addition to apoptosis, autophagy, and necroptosis, include ferroptosis, pyroptosis, cuproptosis, oxeiptosis, entotic cell death (entosis), netotic cell death (netosis), parthanatos, lysosome-dependent cell death (LCD), alkaliptosis, mitoptosis, paraptosis, methuosis, erebosis, and autosis.
Pyroptosis represents a form of lytic cell death that involves the activation of the inflammasome which is a cytosolic multiprotein complex of the innate immune system. The inflammasome is responsible for the release of interleukin-1 (IL-1) family member proteins. Initiation of pyroptosis involves proteins of the gasdermin (GSDM) family which includes GSDMA, GSDMB, GSDMC, GSDMD, GSDME, and pejvakin (PJVK). All of the gasdermin family proteins, excluding PJVK, contain two conserved domains, an N-terminal pore-forming (PFD) and a C-terminal repressor domain (RD). The PFD is the primary inducer of pyroptosis. Stimulation by the appropriate exogenous or endogenous factors results in cleavage of the gasdermins. Cleavage is executed by some of the caspases as well as by granzymes which releases the PFD. The PFD oligomerizes and forms pores in the plasma membrane resulting in the release of inflammatory molecules and the initiation of pyroptotic cell death.
As the name implies, cuproptosis is a copper-dependent form of regulated cell death.
The term oxeiptosis was coined to describe a reactive oxygen species (ROS)-sensitive, caspase independent, and non-inflammatory cell-death pathway.
Entosis represents a form of cell death whereby one cell engulfs and kills another cell and is characterized by the formation of cell-in-cell structures. Entosis has been primarily associated with epithelial tumor cells in the context of aberrant proliferation, glucose starvation, and loss of cell adhesion.
Netosis (also written NETosis) is a form of cell death that is associated with the release of NETs which are extracellular net-like DNA-protein structures. Netosis was originally observed to occur in neutrophils. NETs are released by cells in response to infection or injury. At the level of chromatin the process of netosis involves the activation of peptidyl arginine deiminase 4 (encoded by the PADI4 gene), an enzyme that catalyzes the conversion of arginine to citrulline residues in histones.
Parthanatos is a poly(ADP-ribose) polymerase 1 (PARP1)-dependent RCD that is activated by oxidative stress-induced DNA damage and lysis of chromatin. PARP1 is a nuclear-localized protein that is associated with chromatin and plays a role in the repair of DNA single-strand or double-strand breaks. PARP1 utilizes NAD+ and ATP to trigger poly(ADP)-ribosylation. Hyperactivated PARP1 is the mediator of parthanatos through depletion of NAD+ and ATP and by dissipation of the mitochondrial inner transmembrane (MIM) potential. Depletion of NAD+ and ATP is also associated with necroptosis whereas dissipation of MIM potential is also associated with apoptosis. Similar to apoptosis and oxeiptosis, activation of apoptosis inducing factor, mitochondria associated 1 (AIFM1; also known simply as apoptosis inducing factor, AIF) is required for the activation of parthanatos.
Lysosome-dependent cell death (LCD) is a form of cell death that is mediated by the release of lysosomal hydrolases following lysosomal membrane permeabilization. The permeabilization of lysosomal membranes may also amplify or initiate cell death signaling in the context of apoptosis, autophagy, and ferroptosis. The cathepsins are the major lysosomal hydrolases responsible for the initiation of LCD and they also constitute the most abundant lysosomal hydrolases. Cathepsin is derived from Ancient Greek kata– “down” and hepsein “boil”. Humans express 15 genes that encode enzymes of the cathepsin family. The cathepsin proteins are divided into three subfamilies based upon their mechanism of catalysis. Cathepsins A and G are serine proteases, cathepsins D and E are aspartate proteases, while the remaining cathepsins (B, C, F, H, K, L, O, S, V, X, and W) are cysteine proteases. Not all members of the cathepsin family are found within the lysosomes as cathepsin K is excreted from osteoclasts during the process of bone resorption.
As the name implies, alkaliptosis represents a form of cell death that is initiated in response the the alkalization of cells. The process of alkaliptosis was originally characterized with studies on the opioid receptor-like 1 (ORL1) antagonist JCT-801. This compound kills numerous types of cancer by a cytotoxic process that was shown not to be the result of apoptosis, necroptosis, autophagy, or ferroptosis. The inhibition of intracellular alkalinization with the use of N-acetylcysteine, N-acetylalanine, and acidic culture media blocked JTC-801-induced cell death resulting in the definition of the process as alkaliptosis.
Autosis is a form of cell death that is dependent on autophagy and as such involves autophagy-inducing peptides as well as a requirement for Na+/K+-ATPase activity. Cellular stress signals such as hypoxia-ischemia and nutrient deprivation play roles in the activation of autosis.
Autophagy
Autophagy (from Greek for “self-eating”) represents the highly regulated pathways by which cellular components can be degraded and recycled as a means to support cell survival during times of energy stress and nutrient starvation. Three primary pathways of autophagy have been characterized. Microautophgy represents the pathway whereby the lysosome directly engulfs a small portion of the cytosol. Chaperone-mediated autophagy (CMA) represents the pathway by which chaperones target selective substrates for degradation in the lysosome. Macroautophagy (most commonly just referred to as autophagy) is the pathway by which cytoplasmic organelles are sequestered within membrane compartments referred to as pre-autophagosomes or phagophores (also referred to as isolation membranes, IM). In yeast the initial autophagy-associated structure is referred to as the pre-autophagosomal structure, PAS. The sources of the membrane material from which the phagophore forms is quite varied. Evidence demonstrates that membrane from the endosomes, the endoplasmic reticulum (ER), the Golgi, the ER-Golgi intermediate complex (ERGIC), the mitochondria, and ER-mitochondrial contact sites are sources of the phagophore membranes.
The newly formed membrane compartments fuse at their edges to form a double membrane vesicular structure called the autophagosome. The autophagosome is, therefore, the membrane enclosed structure which contains cytosolic constituents and subcellular organelles destined for degradation.
The autophagosome ultimately fuses with the lysosome allowing its contents to be degraded. Although the induction of the various pathways of autophagy include both physiological and pathological processes, the response of cells to nutrient starvation represents the canonical form of (macro)autophagy. Dysfunction in the processes of autophagy can have severe clinical consequences. The etiology of numerous diseases and disorders such as Crohn disease, many cancers, diabetes, and several neurodegenerative disorders is associated with defects in components of autophagy such as members of the autophagy related family of proteins.
Transcription Factor EB: Master Regulator of Autophagy
Primary control over the process of autophagosome formation and subsequent fusion with the lysosomes is the transcription factor identified as TFEB (transcription factor EB, where the EB refers to enhancer box binding site). TFEB is considered a master regulator of the biogenesis of lysosomes through its ability to regulate the expression of genes encoding lysosomal hydrolases and lysosomal membrane proteins. With respect to the regulation of lysosomal hydrolase gene expression, TFEB binds to a motif in the promoter region of theses genes that is referred to as the CLEAR motif (Coordinated Lysosomal Expression and Regulation). The CLEAR motif is the 10-base-pair sequence, GTCACGTGAC. The regulation, by TFEB, of genes involved in autophagy involves its binding to E-box (enhancer box) sequences in the promoter region of these genes.
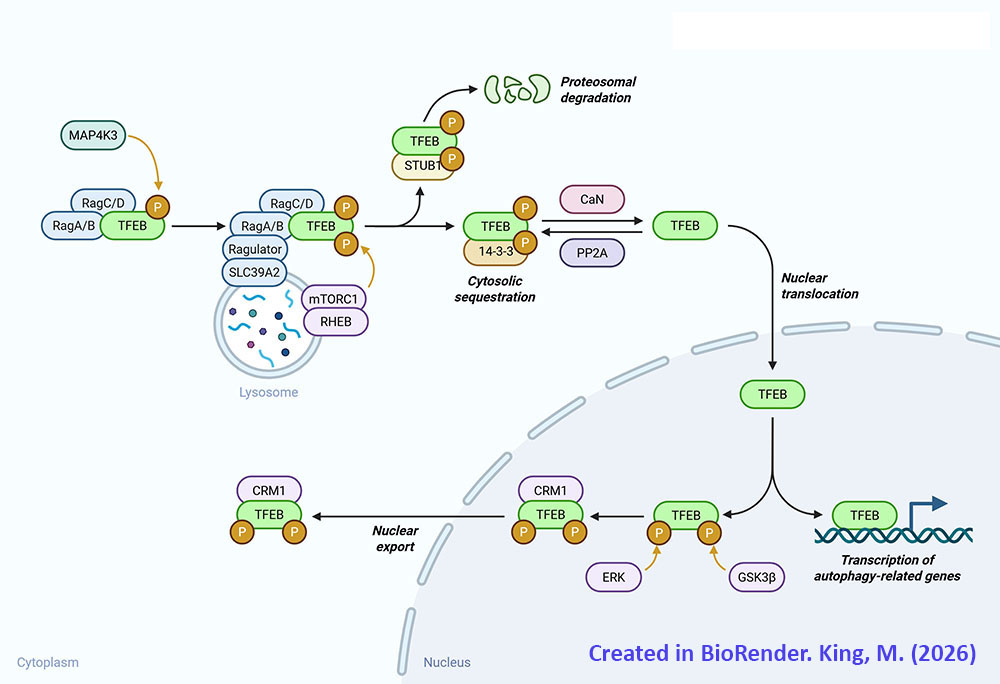
Transcription factor EB is encoded by the TFEB gene. The TFEB gene is located on chromosome 6p21.1 and is composed of 15 exons that generate five alternatively spliced mRNA that collectively encode three distinct protein isoforms. Expression of the TFEB gene is ubiquitous with the highest levels of expression being in tissues responsible for immune function such as the spleen and the lymph nodes.
During normal cellular function TFEB is localized to the cytosol due to its state of phosphorylation. During periods of nutrient deprivation, oxidative stress, or with DNA damage, TFEB is dephosphorylated and migrates to the nucleus where it can activate expression of target genes. In addition to phosphorylation, TFEB is known to be modified by acetylation, SUMOylation, poly-ADP-ribosylation, glycosylation, and cysteine oxidation.
The primary kinase that phosphorylates TFEB is mTOR of the mTOR complex 1 (mTORC1) multiprotein complex. The mTORC1 phosphorylates TFEB on at least three serine residues, S122, S142, and S211. In addition to mTOR the kinases, glycogen synthase kinase 3 beta (GSK3β) and AKT/PKB are known to phosphorylate TFEB. GSK3β phosphorylates TFEB on S134 and S138 which promotes TFEB localization to the surface of the lysosomes. AKT/PKB phosphorylates TFEB on S467.
In order to migrate to the nucleus where it can carry out its transcription factor functions TFEB must be dephosphorylated. There are two primary phosphatases responsible for dephosphorylation of TFEB, calcineurin and protein phosphatase 2A (PP2A). Calcineurin is also identified as protein phosphatase 2B (PP2B) and protein phosphatase 3 (PP3). During periods of nutrient deprivation the calcium (Ca2+) that is stored in lysosomes is effluxed to the cytosol where it activates calcineurin. Calcineurin dephosphorylates S142 and S211 in TFEB which stimulates nuclear translocation. Although PP2A is primarily involved in the regulation of cell cycle progression, during periods of oxidative stress it is known to dephosphorylate S109, S114, S122, and S211 in TFEB which promotes nuclear translocation.
In addition to its role in the regulation of lysosomal functions and autophagosome formation, TFEB plays a role in the regulation of carbohydrate and lipid metabolism and in the signaling cascade of the insulin receptor. The CLEAR motif, to which TFEB interacts, is present in the promoter regions of the genes encoding proteins involved in glucose metavbolism such as GLUT1 (SLC2A1) and GLUT4 (SLC2A4), as well as those encoding hexokinase 1 (HK1), hexokinase 2 (HK2), pyruvate kinase (PKLR), and the muscle form of PFK1 (PFKM). TFEB also regulates the expression of several genes in the liver and adipose tissue that are involved in lipid metabolism. These genes include, but is not limited to, CD36 which encodes the fatty acid transporter, FAT/CD36, ACADS which encodes short-chain acyl-CoA dehydrogenase (SCAD), CPT1A which encodes the liver form of carnitine palmitoyltransferase 1, CPT1B which encodes the muscle form of carnitine palmitoyltransferase 1.
Table of Membrane Trafficking Proteins in Autophagosome Formation
List is not intended to be a complete catalog of all membrane trafficking proteins involved in autophagy
| Protein Name | Gene(s) | Functions / Comment |
| Rab1 family | RAB1A, RAB1B, RAB1C | member of the RAB family of monomeric G-proteins; belongs to RAS superfamily; involved in phagophore biogenesis from ER exit sites (ERES) which are membrane domains involved in the protein secretory pathway |
| Rab5 family | RAB5A, RAB5B, RAB5C | member of the RAB family of monomeric G-proteins; belongs to RAS superfamily; associated with early endosomes; involved in the process of induction of autophagy |
| Rab7 family | RAB7A, RAB7B | member of the RAB family of monomeric G-proteins; belongs to RAS superfamily; associated with lysosomes and late endosomes; involved in the fusion of autophagosomes with the lysosomes |
| Rab11 family | RAB11A, RAB11B | member of the RAB family of monomeric G-proteins; belongs to RAS superfamily; associated with recycling endosomes; involved in the transport of endosomal membrane to the phagophore |
| Rab25 | RAB25 | member of the RAB family of monomeric G-proteins; belongs to RAS superfamily; associated with recycling endosomes; involved in the regulation of autophagosome maturation |
| Rab32 | RAB32 | member of the RAB family of monomeric G-proteins; belongs to RAS superfamily; associated with the ER; involved in autophagosome formation |
| synaptosome associated protein 29 | SNAP29 | involved in the fusion between autophagosome membranes and lysosomal membranes |
| sorting nexin 18 | SNX18 | associated with recycling endosomes; participant in endocytosis and intracellular vesicle trafficking; involved in formation of the autophagosome |
| syntaxin 7 | STX7 | involved in trafficking membranes from the plasma membrane to early endosomes along with STX8; involved in formation of the autophagosome |
| syntaxin 8 | STX8 | involved in trafficking membranes from the plasma membrane to early endosomes along with STX7; involved in formation of the autophagosome |
| syntaxin 17 | STX17 | a member of the SNARE (SNAP REceptor, where SNAP is Soluble N-ethylmaleimide-sensitive factor Attachment Protein) family of membrane fusion proteins; the SNARE family proteins are required for membrane fusion processes of the exocytotic and endocytotic pathways; involved in the control of autophagosome membrane fusion with the lysosome membrane |
| vesicle associated membrane protein 3 | VAMP3 | a member of the SNARE family; involved in vesicular transport from the late endosomes to the trans-Golgi network (TGN); associated with the ATG12-ATG5-ATG16L1 conjugate fusion with the phagophore membranes |
| vesicle associated membrane protein 7 | VAMP7 | involved in the targeting/fusion of transport vesicles to their target membrane during protein transport to endosomes and lysosomes; role in ATG16L1 association with membranes forming the phagophore |
| vesicle associated membrane protein 8 | VAMP8 | a member of the SNARE family; interacts with STX17; involved in the control of autophagosome membrane fusion with the lysosome membrane |
In addition to the more global processes of autophagy there are several selective processes that include autophagic removal of mitochondria (called mitophagy and described in more detail below), peroxisomes (termed pexophagy), endoplasmic reticulum (called reticulophagy), and ribosomes (called ribophagy). Part of the nucleus can also be removed via specialized autophagic processes referred to as nucleophagy. There are two stages of nucleophagy termed piecemeal microautophagy of the nucleus (PMN) which is the early stage process, and late nucleophagy (LN) which occurs following prolonged nutrient deprivation. The process referred to as xenophagy involves the autophagic elimination of invading pathogens such as bacteria and viruses.
The complete process of autophagy induction, organelle turnover, and recycling of the required proteins of autophagy is exceedingly complex and beyond the scope of this discussion. However, an overview of the basics of the regulation of induction of autophagy and the major protein activities of (macro)autophagy will be carried out. Numerous genes that encode proteins that regulate the processes of autophagy are identified by the ATG acronym which refers to AuTophaGy related gene. Not all proteins in the large family of proteins referred to as the autophagy related family are designated by the ATG nomenclature. Humans express a total of 33 genes in the autophagy related protein family, several of which are described in the following Table. In addition to these 33 proteins of the autophagy related family, there are numerous other proteins required to activate and regulate the overall processes of autophagy. Indeed, several proteins that regulate autophagy also play important roles in the regulation of apoptosis.
Table of Mammalian Autophagy Related Protein Family
| Protein Name | Gene | Functions / Comment |
| autophagy related 2A | ATG2A | required for the formation of the autophagosome via its role in the regulation of lipid droplet formation and dispersion; functions along with the ATG2A, WIPI1, and WIPI2 encoded proteins to forma critical functional complexes of autophagy |
| autophagy related 2B | ATG2B | required for the formation of the autophagosome via its role in the regulation of lipid droplet formation and dispersion; functions along with the ATG2B, WIPI1, and WIPI2 encoded proteins to form a critical functional complexes of autophagy |
| autophagy related 3 | ATG3 | functions as a ubiquitin conjugating E2-like activity; covalently attaches phosphatidylethanolamine (PE) to the C-terminus of the human ATG8 family member proteins: GABARAP, GABARAPL1, GABARAPL2, and MAP1LC3A |
| autophagy related 4A | ATG4A | functions as a cysteine protease; cleaves the C-terminus of ATG8 family member ubiquitin-like modifier proteins exposing a terminal glycine residue |
| autophagy related 4B | ATG4B | functions as a cysteine protease; cleaves the C-terminus of ATG8 family member ubiquitin-like modifier proteins exposing a terminal glycine residue |
| autophagy related 4C | ATG4C | functions as a cysteine protease; cleaves the C-terminus of ATG8 family member ubiquitin-like modifier proteins exposing a terminal glycine residue |
| autophagy related 4D | ATG4D | functions as a cysteine protease; cleaves the C-terminus of ATG8 family member ubiquitin-like modifier proteins exposing a terminal glycine residue |
| autophagy related 5 | ATG5 | is conjugated with ATG12 via the ubiquitin conjugating E2-like activity of ATG10 |
| autophagy related 7 | ATG7 | functions as a ubiquitin conjugating E1-like activity to activate ATG12 |
| autophagy related 9A | ATG9A | multipass membrane protein that is thought to assist in brining membrane to the developing phagophore |
| autophagy related 9B | ATG9B | multipass membrane protein that is thought to assist in brining membrane to the developing phagophore |
| autophagy related 10 | ATG10 | functions as a ubiquitin conjugating E2-like activity to transfer ATG12 to ATG5 forming a complex |
| autophagy related 12 | ATG12 | activated by ATG7 then transferred to ATG5 via the ubiquitin conjugating E2-like activity of ATG10 |
| autophagy related 13 | ATG13 | involved in the complex formed from the ULK1, ULK2, RB1CC1, and ATG101 encoded proteins; required to stabilize ATG101 |
| autophagy related 14 | ATG14 | also known as ATG14 like (ATG14L); component of the beclin 1 complex; involved in the association of the ATG12-ATG5-ATG16L1 complex and the ATG8-PE conjugate to the phagophore |
| autophagy related 16 like 1 | ATG16L1 | forms a ubiquitin conjugating E3-like activity when conjugated with ATG12 and ATG5 |
| autophagy related 16 like 2 | ATG16L2 | forms a ubiquitin conjugating E3-like activity when conjugated with ATG12 and ATG5 |
| autophagy related 101 | ATG101 | interacts with and stabilizes, in a reciprocal manner, ATG13; involved in the formation of the ULK1/ULK2 autophagy initiating complex |
| beclin 1 | BECN1 | mammalian homolog of yeast ATG6 (which is also known as Vps30); functions in a complex with several proteins that includes ATG14; activity regulated by ULK1/ULK2; activity also regulated by AMPK; activity inhibited by the pro-survival proteins BCL-2 and BCL-XL; |
| RB1 induced coiled-coil 1 | RB1CC1 | gene name derived from fact that the encoded protein enhances retinoblastoma 1 (RB1) gene expression in cancer cells; commonly referred to as FIP200 (focal adhesion kinase family interacting protein of 200 kDa); interacts with the ULK1 kinase; is the human ortholog of the yeast ATG17 gene |
| UNC-51-like autophagy activating kinase 1 | ULK1 | represents one of two human homologs of the yeast ATG1 gene (often identified as ATG1A); critical regulator of autophagy induction; activity regulated by the mTOR complex 1 (mTORC1) |
| UNC-51-like autophagy activating kinase 2 | ULK2 | represents one of two human homologs of the yeast ATG1 gene (often identified as ATG1B); critical regulator of autophagy induction; activity regulated by the mTOR complex 1 (mTORC1) |
| WD repeat domain, phosphoinositide interacting 1 | WIPI1 | represents one of two human homologs of the yeast ATG18 gene; often designated ATG18A; functions along with the ATG2A, ATG2B, and WIPI2 encoded proteins to form a critical functional complex of autophagy |
| WD repeat domain, phosphoinositide interacting 2 | WIPI2 | represents one of two human homologs of the yeast ATG18 gene; often designated ATG21 or ATG18B; functions along with the ATG2A, ATG2B, and WIPI1 encoded proteins to form a critical functional complex of autophagy |
Transcriptional Regulation of Autophagy
Although autophagy can be stimulated by several intracellular and extracellular cues, the induction of autophagy as a result of nutrient deprivation represents the most well-characterized. Changes in expression of several genes, many of which encode transcription factors, occurs in response to nutrient deprivation, as well as in response to endoplasmic reticulum (ER) and mitochondrial stress, results in enhanced levels of autophagy. Although the majority of the genes described in the following Table promote autophagy, there are some genes whose encoded proteins function to suppress autophagy.
Table of Transcription Factors Regulating Autophagy in Response to Nutrient Deprivation
| Protein Name | Gene | Functions / Comment |
| activating transcription factor 4 | ATF4 | activates expression of ATG5 and ULK1 genes thereby promoting autophagy |
| activating transcription factor 5 | ATF5 | activates expression of MTOR gene; the mTOR kinase is the kinase of the mTORC1 which represses the activity of the ULK1 complex; repression of the ULK1, which normally enhances autophagy, will therefore, results in inhibition of autophagy |
| CCAAT enhancer binding protein beta | CEBPB | is a member of the CCAAT/enhancer-binding protein (C/EBP) family of transcription factors which are also identified by the CHOP acronym |
| DNA damage inducible transcript 3 | DDIT3 | is a member of the CCAAT/enhancer-binding protein (C/EBP) family of transcription factors which are also identified by the CHOP acronym; also known as C/EBP zeta and also as CHOP10 |
| E2F transcription factor 1 | E2F1 | is a member of the E2F family of transcription factors; regulates expression of several ATG family member genes thus promotes autophagy |
| forkhead box O1 | FOXO1 | is a member of the forkhead family of transcription factors; so-called by the presence of a distinct forkhead domain named for a domain originally identified in the Drosophila fork head protein; regulates expression of ATG5 and BECN1 genes; also enhances autophagy outside of the nucleus by interacting with XBP1 and ATG7 |
| forkhead box O3 | FOXO3 | is a member of the forkhead family of transcription factors; so-called by the presence of a distinct forkhead domain named for a domain originally identified in the Drosophila fork head protein; regulates expression of PIK3CA (phosphatidylinositol-4,5-bisphosphate 3-kinase, catalytic sub- unit alpha) gene; leads to activation of AKT1 which phosphorylates FOXO1 leading to enhanced autophagy |
| GATA binding protein 1 | GATA1 | belongs to the GATA family of transcription factors defined by the presence of the consensus sequence WGATAR (W = T or A; R = G or A) |
| Jun proto-oncogene | JUN | JUN is so-called for the Japanese word for the number 5; is subunit of the AP1 transcription factor |
| nuclear factor kappa B (NFκB) | NFKB | major immune response modulating transcription factor; enhanced cytokine production can promote or inhibit autophagy |
| p53 | TP53 | tumor suppressor identified by its molecular size of 53 kDa; transcription factor that regulates expression of numerous genes controlling cell cycle progression; enhanced activity of p53 promotes autophagy; also activates expression of DRAM1 (DNA-damage regulated autophagy modulator 1) which promotes autophagy |
| SRY-box transcription factor 2 | SOX2 | transcriptional repressor of MTOR; reduced mTOR kinase leads to enhancement of autophagy |
| sterol regulatory element binding (SREBP) transcription factor 2 | SREBF2 | activates expression of several autophagy genes including ATG4B and ATG4D |
| signal transducer and activator of transcription 1 | STAT1 | represses expression of several autophagy genes resulting in reduced autophagy |
| signal transducer and activator of transcription 3 | STAT3 | represses expression of several autophagy genes resulting in reduced autophagy |
| transcription factor EB | TFEB | master regulator of autophagy |
| zinc finger with KRAB and SCAN domains 3 | ZKSCAN3 | specifically represses expression of genes involved in autophagy such as ULK1 thus inhibiting autophagy |
Role of mTOR and Beclin 1 in Autophagy
A central regulator of the signaling pathways that trigger autophagy is the Ser/Thr kinase mTOR (mechanistic target of rapamycin) which is the principal component of both the mTOR complex 1 (mTORC1) and mTORC2 protein complexes. The mTORC1 is the major mTOR containing complex that regulates cellular responses to growth factor signaling, nutrient deprivation, and various forms of cellular stress.
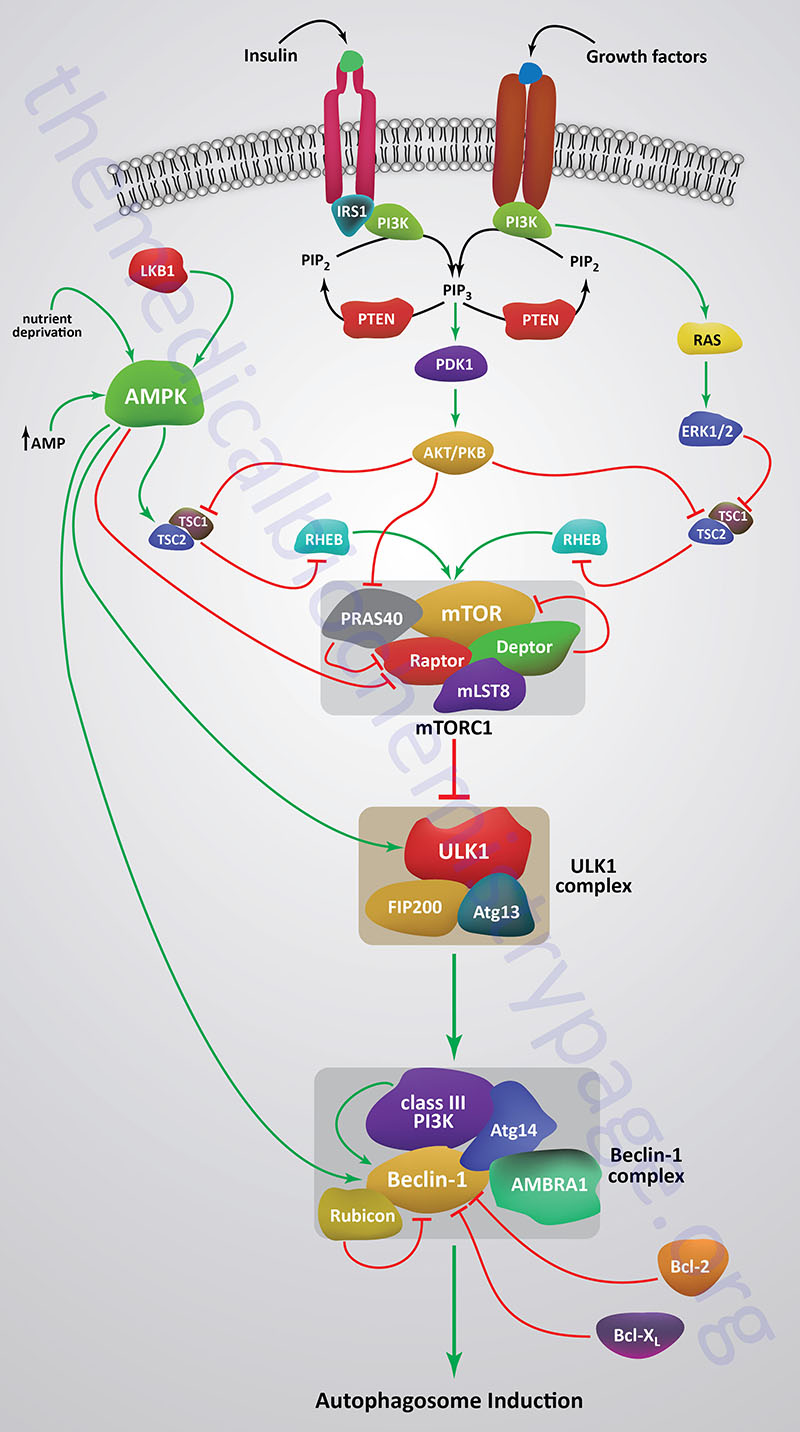
When mTOR is activated by a variety of different growth factor receptors autophagy is suppressed. Upstream signaling proteins that stimulate the activity of mTOR include the Ser/Thr kinases, protein kinase B/AK strain transforming (PKB/AKT), phosphatidylinositol-3-kinase (PI3K), and several members of the MAPK family of kinases. PKB/AKT was originally identified as the tumor inducing gene in the AKT8 retrovirus found in the AKR strain of mice. Humans express three genes in the AKT family identified as AKT1 (PKBα), AKT2 (PKBβ), and AKT3 (PKBγ). Under conditions of energy depletion, such as nutrient starvation, the master metabolic regulatory kinase, AMPK, is activated which phosphorylates and inhibits mTOR, thereby promoting autophagy induction. The tumor suppressor p53 can also activate autophagy under conditions of genotoxic stress through repression of mTOR activity.
Within the context of autophagy there are two related downstream kinases whose activities are regulated by activated mTORC1. These Ser/Thr kinases are known as UNC-51-like autophagy activating kinases -1, and -2, which are encoded by the ULK1 and ULK2 genes, respectively. Humans express a third related gene, UKL3, whose encoded kinase is primarily involved in the regulation of the activity of the sonic hedgehog (SHH) protein. However, evidence does indicate that ULK3 can function in some capacity in the regulation of autophagy.
The UNC genes represent a large family of genes identified in the round worm C. elegans that, when mutated, result in uncoordinated motor activity. The human ULK1 protein functions similarly to the yeast ATG1 protein. ULK1 (or its close family member ULK2) interacts with several other proteins to form a large complex that is required for the formation of an autophagosome. The other proteins that complex with ULK1/ULK2 include ATG13, ATG101, and RB1CC1 (also known as FIP200/ATG17). RB1CC1 is RB1-inducible coiled-coil 1 where RB1 is the gene encoding the retinoblastoma protein (pRB). FIP200 is FAK (focal adhesion kinase) family kinase-interacting protein of 200 kD.
As indicated, under conditions of nutrient deprivation AMPK is activated, thereby phosphorylating and inhibiting the mTORC1. When the mTORC1 is active it phosphorylates ULK1 on a regulatory Ser residue (S757) thereby, inhibiting the activation of ULK1. The mTORC1 also phosphorylates the ATG13 protein of the ULK1 complex. Thus, AMPK-mediated inhibition of mTORC1 activity allows ULK1 to induce autophagy. AMPK also directly phosphorylates ULK1 on two different Ser residues (S317 and S777) which results in activation of the autophagy induction properties of ULK1.
In addition to the role of AMPK in the regulation of ULK1/ULK2 activity via its effects on mTORC1 activity, this master metabolic regulatory kinase also regulates the autophagy complex downstream of ULK1/ULK2 at the level of the beclin 1 complex, also called the class III PI3K complex.
The beclin 1 complex consists of several proteins including, but not limited to beclin 1 (the mammalian homolog of yeast ATG6), ATG14 (specifically the ATG14 long isoform, ATG14L), UVRAG (ultraviolet irradiation resistance-associated gene), AMBRA1 (autophagy and beclin 1 regulator 1), RUBICON (RUN and cysteine-rich domain containing beclin 1 interacting protein), and SH3 domain containing GRB2 like endophilin B1 (encoded by the SH3GLB1 gene; also known as BIF-1).
The beclin 1 complex is also inhibited by activated mTORC1 a result of the phosphorylation of AMBRA1 and ATG14L. Thus, when AMPK is activated, and mTOR is phosphorylated and inhibited, the beclin 1 complex is activated to initiate autophagy.
The primary function of the activation of the beclin 1 complex is to activate the class III PI3K encoded by the PIK3C3 gene. PIK3C3 encodes a p110 catalytic subunit which forms a heterodimer with the p150 regulatory subunit encoded by the PIK3R4 gene. Activation of the class III PI3K results in the production of phosphatidylinositol 3-phosphate (PI3P) at the site of phagophore formation. One important role of PI3P is the recruitment of the WIPI proteins (human homologs of the yeast ATG18 protein) to the phagophore membrane. In addition, PI3P may recruit other downstream effector proteins of the autophagy process such as the ATG5-ATG12-ATG16L1 complex. The function of the beclin 1 protein within the complex can be inhibited via interaction with the pro-survival (anti-apoptotic) proteins BCL-2 and BCL-XL (see Mitochondrial Apoptosis section below).
BCL-2 and BCL-XL bind to the BH3 domain of beclin 1 thereby inhibiting its activity. The interaction of beclin 1 with either of the two pro-survival proteins can be inhibited by phosphorylation of beclin 1 or BCL-2 or by the ubiquitylation of beclin 1. Therefore, it is clear that the beclin 1 complex plays an important role in promoting mitochondrial apoptosis as well. Mitochondria localized beclin 1 induces the translocation of the BAX protein which forms a mitochondrial outer membrane (MOM) pore complex allowing pro-apoptotic factors, such as cytochrome c to be released setting off the intrinsic apoptotic cascade.
ATG Proteins in Autophagosome Formation
The nucleation of membrane into the initial pre-autophagosome (phagophore) requires several ATG proteins and lipids in what is referred to as the conjugation step of autophagosome formation. Phosphatidylethanolamine (PE) is known to be a critical lipid component of the conjugation process and it is believed that the PI3P derived via the action of the class III PI3K is also an important lipid.
Phagophore formation begins when ATG12 is activated via the ubiquitin conjugating E1-like activity of ATG7. Activated ATG12 is then transferred to ATG5 via the ubiquitin conjugating E2-like activity of ATG10. The ATG12-ATG5 conjugate then interacts with ATG16L1 forming an ATG12-ATG5-ATG16L1 complex that possesses ubiquitin conjugating E3-like activity. The ATG12-ATG5-ATG16L1 complex is functionally coupled to proteins of the human ATG8 family. The ATG8 proteins are considered members of the ubiquitin-like modifier family of proteins.
In yeast there is but a single ATG8 protein involved in autophagy, however, in humans there are at least seven ATG8 family members. The human ATG8 family proteins are GABA type A receptor-associated protein (encoded by the GABARAP gene; considered ATG8A), GABA type A receptor-associated protein like 1 (encoded by the GABARAPL1 gene; considered ATG8B), GABA type A receptor-associated protein like 2 (encoded by the GABARAPL2 gene; considered ATG8C), microtubule associated protein 1 light chain 3 alpha (encoded by the MAP1LC3A gene; considered ATG8E; also called simply LC3), microtubule associated protein 1 light chain 3 beta (encoded by the MAP1LC3B gene; considered ATG8F), microtubule associated protein 1 light chain 3 beta2 (encoded by the MAP1LC3B2 gene; considered ATG8G), and microtubule associated protein 1 light chain 3 gamma (encoded by the MAP1LC3C gene; considered ATG8J).
The ATG8 family proteins are cleaved at their C-termini via the action of the several human ATG4 family cysteine proteases. The cleavage results in the exposure of a C-terminal glycine residue. In the process of protein ubiquitylation described in detail above, the exposed C-terminal glycine of ubiquitin would be the site of attachment to protein substrate. However, in the case of the ATG8 family proteins they are attached to phosphatidylethanolamine, PE. The attachment of ATG8 family proteins to PE is catalyzed by the E3-like activity of the ATG12-ATG5-ATG16L1 conjugate in concert with the E2-like activity of ATG3. The ATG12-ATG5-ATG16L1 conjugate, along with the associated ATG8 family protein covalently attached to PE, are recruited to the membranes of the forming phagophore via the function of the ATG14 protein which is in a large complex with the beclin 1 protein.
The recognition of the “cargo” that is to be engulfed by the autophagosome is carried out be specific proteins termed autophagy receptors. All autophagy receptors characterized to date possess both an ATG8E (LC3) interacting region (more commonly referred to as an LC3-interacting region, LIR) and a ubiquitin-binding domain, UBD. The selective autophagy receptor called sequestosome 1 (encoded by the SQSTM1 gene) was the first member of the family characterized. The SQSTM1 encoded protein is also known as nucleoporin 62 (p62). There are five well characterized autophagy receptors which includes SQSTM1, NBR1 (Neighbor of BRCA1 gene 1; encoded by the NBR1 gene), NDP52 [Nuclear Domain 10 Protein 52; encoded by the calcium binding and coiled-coil domain 2 (CALCOCO2) gene], OPTN (optineurin; encoded by the OPTN gene), and BNIP3-like [BCL-2 interacting protein 3 (BNIP3) like; encoded by the BNIP3L gene; BNIP3 and BNIP3L are members of the pro-apoptotic BH3-domain only BCL-2 related protein family). BNIP3L is also called NIX (NIP3-like protein X).
When the formation of the autophagosome is complete it fuses with endosomes which then fuses with lysosomes to form autolysosomes. When the fusion with the lysosomes takes place, the contents of the autophagosome are degraded by the resident hydrolases. The resulting amino acids and other basic cellular constituents are reused by the cell. When intracellular amino acids rise high enough through the actions of the autophagosome and subsequent lysosomal protein degradation they will reactivate the mTORC1 resulting in suppression of autophagy.
Mitochondrial Autophagy (Mitophagy)
Mitochondrial function is critical for metabolic homeostasis and the production of energy. As mitochondria age they become progressively damaged primarily from the accumulated effects of the reactive oxygen species (ROS) that are inherently generated in this organelle during the process of electron transport. The progressive ROS damage leads to release of mitochondrial components, some of which, such as cytochrome c, can trigger the intrinsic apoptotic program described in detail below.
Another mitochondrial damage response pathway is the autophagic pathway termed mitophagy. Mild oxidative stress triggers mitophagy and equally significant is that ROS accumulation is required for hypoxia-induced mitophagy as well as for nutrient deprivation-induced autophagy. Thus, ROS limits its own production via stimulation of mitophagy, thereby, preventing cellular damage. Although mitophagy involves several proteins of the primary (macro)autophagy pathway, the induction of mitophagy occurs via a pathway that is distinct in its protein effectors.
The ultimate role of mitophagy is to remove damaged or surplus mitochondria from the cell so as to ensure cell survival. However, if the damage is significant the intrinsic apoptotic program may be initiated. Defects in the mechanisms that ensure mitochondrial viability, such as mitophagy, are associated with numerous disease states, in particular neurodegenerative diseases such as Parkinson disease (PD).
Parkinson disease is characterized by death of dopaminergic neurons in the substantia nigra which is located in the mesencephalon. Although substantia nigra neuron degeneration represents the primary pathological feature of PD, there are multiple neuron types and neuronal circuits that become disrupted in the course of the disease. The dopaminergic neurons in the substantia nigra possess dense axonal arborizations and because of their continuous pacemaking activity they are under high mitochondrial stress. It is this intrinsic property of dopaminergic neurons that makes then exceptionally sensitive to defects in mitochondrial quality control mechanisms and the resultant mitochondrial dysfunction.
Familiar (inherited) forms of PD do not represent a major proportion of cases, however, study of patients with these forms has been instrumental in identifying proteins involved in the pathways of mitophagy. In patients with early-onset autosomal recessive forms of PD, two genes, whose encoded proteins are critical for mitochondrial quality control, were identified. One gene, identified as PINK1 (PTEN INduced putative Kinase 1; where PTEN stands for Phosphatase and TENsin homolog) is a Ser/Thr kinase and the other gene, identified as PARK2 (parkin RBR E3 ubiquitin protein ligase) is a ubiquitin conjugating E3 ligase. The PARK2 encoded protein is commonly called Parkin. The Parkin protein is a member of one of the three E3 ligase families referred to as RBR (Ring-Between-Ring) family as described above.
During mitochondrial stress the PINK1 protein is localized to the mitochondrial outer membrane (MOM), whereas the Parkin protein is normally localized to the cytosol. Using genetic studies in fruit flies has been determined that PINK1 functions to recruit Parkin from the cytosol to the damaged mitochondrial membrane and to activate its E3 ligase activity. The PINK1 and Parkin mediated pathway represents the most well characterized inducer of mitophagy.
PINK1, which a nuclear encoded protein, is imported into healthy mitochondria by the translocase of outer membrane (TOM) and translocase of inner membrane (TIM) complexes which are described in detail in the Protein Targeting page. The N-terminal transmembrane domain of PINK1 is inserted into the mitochondrial inner membrane (MIM) and subsequently cleaved by the serine protease encoded by the PARL (presenilin associated rhomboid like) gene. The cleaved C-terminal fragment of PINK1 is transported back to the cytosol and degraded via the N-end rule proteasomal pathway (described in the section on Protein Acetylation in the Protein Modifications page).
Within the cytosol the C-terminal fragment of PINK1 can also bind to Parkin, thereby, preventing its recruitment to the MOM. This process of PINK1 synthesis, mitochondrial import, proteolysis, release from the mitochondria, and subsequent N-end rule proteasomal degradation keeps the steady state level of PARK1 in the mitochondria low which is a signal of mitochondrial health. Under conditions where PINK1 activity accumulates mitophagy is stimulated. Mitochondrial stabilization and accumulation of PINK1 can occur due to the over expression of misfolded proteins in the mitochondrial matrix. This most likely occurs due to impaired protein import at the TIM complex.
When PARK1 import into the MIM, or when the MIM-associated proteases that hydrolyze the N-terminus of PARK1 are impaired, the protein autophosphorylates and dimerizes in the MOM at the TOM complex. Under these conditions the kinase domain of PARK1 faces the cytosol. In the unstimulated state, Parkin remains in a closed conformation unable to attach ubiquitin to an internal Cys residue (C431) which is the typical intermediate step in the activity of ubiquitin conjugating E3 ligases. When PINK1 dimerizes in the MOM, and is activated by autophosphorylation, it can phosphorylate Parkin (on S65), as well as ubiquitin, which is required for the full E3 ligase activity of Parkin.
The phosphorylated ubiquitin serves as a Parkin receptor on the mitochondria. When activated Parkin ubiquitylates numerous substrates such as the mitochondrial voltage-dependent anion channels (encoded by the VDAC1, VDAC2, and VDAC3 genes), the mitochondrial RHO family protein call Miro, and the mitochondrial fission protein dynamin related protein-1 (encoded by the DRP1 gene).
As described above, autophagosome cargo selection is determined, in part, by recognition of ubiquitylated proteins via the ubiquitin-binding domains (UBD) of autophagy receptors. The autophagy receptors, NDP52 and OPTN are critical activators of mitophagy. These two autophagy receptors recruit components of the autophagosome, such as the ULK1 complex and the Pi3P-binding proteins WIPI1 and WIPI2, to the mitochondrial membrane, thereby, stimulating mitophagy.
Control of mitophagy can be exerted by the removal of ubiquitin from Parkin substrates. As for the deubiquitylation process described above in the Ubiquitin section, removal of ubiquitin from Parkin substrates is catalyzed by several deubiquitylating enzymes, DUBs. The DUBs encoded by the USP15, USP30, and USP35 (ubiquitin specific peptidase) genes are known to limit the extent of mitophagy by removal of ubiquitin from Parkin substrates. USP30 is likely to be the major DUB controlling mitophagy since it is tethered to the MOM with its catalytic domain facing the cytoplasm.
Apoptosis
Apoptosis refers to the highly regulated and coordinated processes of PCD. The control of apoptosis represents a balance between the activity of pro-survival genes and pro-apoptotic genes.
In general, cell survival pathways predominate provided cells have adequate access to the appropriate growth (trophic) promoting signals. Under certain conditions some cells will actually release specific hormones that triggers the apoptotic program of adjacent or nearby cells. Two primary pathways exist for the initiation of the apoptotic process in a wide variety of different cell types. These two pathways are termed the intrinsic and extrinsic pathways of apoptosis.
The intrinsic apoptotic pathway is predominantly centered around and/or regulated by the mitochondria. An additional intrinsic pathway involves the activation of apoptosis in response to endoplasmic reticulum (ER) stress. The major extrinsic pathway is mediated by receptors that represent a subfamily of the tumor necrosis factor receptor (TNFR) superfamily that includes TNFR1 and FAS. These receptors are activated by an appropriate ligand, such as TNFα and the FAS ligand (FASL), respectively, and initiate a specific apoptotic program. Since these ligands and receptors activate a pathway of PCD these pathways are collectively referred to as the death receptor pathways of apoptosis.
A related extrinsic pathway of apoptosis is the perforin/granzyme pathway that is exclusive to immune cells.
Caspases: Inducers of the Apoptotic Cascades
Within the context of the various apoptotic programs there are similar features and overlapping protein functions. One family of proteins, the caspases, are central to both the intrinsic and extrinsic apoptotic programs. Caspases are so-called because they are cysteine aspartate proteases. Caspases are synthesized as precursor inactive proteins (zymogens) that are proteolytically processed, often by another member of the caspase family. The proteolysis of a caspase results in the production of a large and a small subunit that dimerize to form the catalytically active caspase. Within the large caspase family several members serve as initiators of the apoptotic program and others serve as effectors of the upstream inducing signals.
Table of Human Caspases
| Caspase | Gene | Functions / Comments |
| 1 | CASP1 | referred to as an inflammatory initiator caspase; was originally identified as interleukin-1 converting enzyme (ICE); critical central enzyme acting as an inflammatory response initiator; cleaves interleukin 1β (IL-1β) and IL-8 resulting in the active forms of these cytokines being released from the cell; contains a protein interaction domain called the CARD (Caspase Activation and Recruitment Domain); 30 genes in humans express proteins that contain the CARD including several caspases |
| 2 | CASP2 | referred to as an initiator caspase; translated as a proenzyme (zymogen) with a long pro-domain; contains a CARD; associates with numerous apoptotic proteins, via the CARD, such as caspase 8 |
| 3 | CASP3 | referred to as an executioner (effector) caspase; major central caspase of both the mitochondrial and the death receptor apoptotic pathways; referred to as an executioner caspase; interacts with caspases 8 and 9 which cleave and activate the zymogen form; activated caspase 3 then cleaves and activates caspases 6 and 7; in the mitochondrial apoptotic pathway cytochrome c, caspase 9, and apoptosis activating factor 1 (APAF1) utilize the energy of ATP hydrolysis to activate procaspase 3; activation of procaspase 3 is inhibited by several proteins of the inhibitor of apoptosis (IAP) family of proteins such as X-linked inhibitor of apoptosis protein, XIAP |
| 4 | CASP4 | referred to as an inflammatory initiator caspase; primarily involved in the inflammatory apoptosis pathway initiated by death receptor activation; contains a CARD |
| 5 | CASP5 | referred to as an inflammatory initiator caspase; primarily involved in the inflammatory apoptosis pathway initiated by death receptor activation; contains a CARD |
| 6 | CASP6 | referred to as an executioner (effector) caspase; procaspase 6 activated by caspases 7, 8, and 10; interacts with caspase 8 |
| 7 | CASP7 | referred to as an executioner (effector) caspase; procaspase 7 is activated by caspases 3, 9, and 10; interacts several proteins such as caspase 8 and XIAP |
| 8 | CASP8 | referred to as an initiator caspase; central caspase in the death receptor pathway of apoptosis; activates procaspase 3; contains an N-terminal FADD-like (Fas-Associated protein with Death Domain) domain; known to interact with numerous proteins including both pro-survival proteins and pro-apoptotic proteins |
| 9 | CASP9 | referred to as an initiator caspase; major mediator of the mitochondrial apoptotic pathway; interacts with the APAF1-cytochrome c complex which results in cleavage of procaspase 9 to active caspase 9; active caspase 9 then cleaves procaspase 3; contains a CARD |
| 10 | CASP10 | referred to as an initiator caspase; activated by caspase 8 and granzyme B cleavage; active caspase 10 cleaves caspases 3 and 7; major caspase in the perforin/granzyme pathway of apoptosis; also interacts with proteins in the death receptor pathway of apoptosis |
| 12 | CASP12 | human gene contains a polymorphism, that in the vast majority of individuals, results in a premature stop codon in the encoded protein; the read-through codon is found almost exclusively in individuals of African descent and is associated with bacterial endotoxin hypo-responsiveness resulting in an increased susceptibility to sepsis |
| 14 | CASP14 | predominant caspase in epidermal stratum corneum; plays a role in keratinocyte differentiation |
Inhibitors of Apoptosis, IAP
Apoptosis must not only be efficiently and correctly activated in response to the appropriate signals but the process must also be tightly controlled at the level of inhibition. Inhibition of apoptosis can occur through the action of anti-apoptotic proteins, also referred to as pro-survival factors. Several pro-survival proteins are encoded by genes of the BCL-2 superfamily of apoptosis regulators that are discussed in detail below in the Mitochondrial Apoptosis section. Another class of proteins that serve to prevent apoptosis are collectively referred to as inhibitors of apoptosis, IAP. Numerous IAP proteins have been characterized with several described in the Table below. Proteins of the IAP family contain a homologous domain referred to as the IAP repeat domain. This domain was also identified in the baculovirus inhibitor of apoptosis protein and therefore the domain is also referred to as the baculovirus inhibitor of apoptosis protein repeat (BIR) domain. Humans express eight genes that encode proteins containing the baculovirus IAP repeat with six being involved in the inhibition of apoptosis.
Table of Inhibitors of Apoptosis Proteins, IAP
| Protein | Common/Other Name | Gene | Functions / Comments |
| NLR family apoptosis inhibitory protein | NAIP | NAIP | also known as BIRC1; inhibits the activities of caspase 3, 7, and 9; prevents motor neuron apoptosis |
| baculoviral IAP repeat containing 2 | cIAP1 | BIRC2 | contains a caspase recruitment domain (CARD); multifunctional protein involved in inhibition of apoptosis and modulation of inflammation, mitogenic kinase signaling, and cell proliferation; functions as an E3 ubiquitin ligase with numerous apoptosis proteins as substrates such as caspases 3, 7, and 8, and DIABLO |
| baculoviral IAP repeat containing 3 | cIAP2 | BIRC3 | contains a caspase recruitment domain (CARD); multifunctional protein involved in inhibition of apoptosis and modulation of inflammation, mitogenic kinase signaling, and cell proliferation; functions as an E3 ubiquitin ligase with numerous apoptosis proteins as substrates such as caspases 3, 7, and 8 |
| baculoviral IAP repeat containing 5 | survivin | BIRC5 | inhibits caspase 3 and 7; serves as a component of the complex involved in chromosome alignment and segregation during mitosis and cytokinesis (chromosome passage protein complex, CPC) |
| baculoviral IAP repeat containing 6 | bruce | BIRC6 | functions as an E3 ubiquitin ligase with numerous apoptosis proteins as substrates such as caspases 3, 7, and 9, and DIABLO |
| X-linked inhibitor of apoptosis | XIAP | XIAP | also known as BIRC4; directly binds to and inhibits caspase 3, 7, and 9; activity of XIAP blocked by the mitochondrial proteins DIABLO and HTRA2 that are released in response to pro-apoptotic signals; functions as an E3 ubiquitin ligase with numerous apoptosis proteins as substrates |
Mitochondrial Apoptosis
The mitochondria-mediated pathway of apoptosis represents the major intrinsic apoptotic pathway. One of the first major proteins to be identified as being critical for the regulation of the mitochondrial apoptotic pathway was BCL-2. As a result of the role of BCL-2 in this intrinsic apoptotic pathway it is also referred to as the BCL-2-regulated pathway or also as the mitochondrial stress pathway to cell death. The BCL-2 protein (encoded by the BCL2 gene) was originally identified from the breakpoint region of a recurrent chromosomal translocation found in human B cell lymphomas (hence the BCL designation). The gene within the translocation region, and the encoded protein was called BCL-2. The BCL-2 gene was subsequently shown to be able to replace the function of the C. elegans CED-9 gene which prevents developmental cell death in this organism. Because the function of BCL-2 prevents programmed cell death it is referred to as a cell survival (anti-apoptotic) gene.
The overall threshold that regulates the mitochondrial apoptosis pathway is determined by interactions that take place between numerous proteins at the mitochondrial outer membrane. These protein interactions involve functionally and structurally distinct protein subfamilies of the larger BCL-2-related protein superfamily. The proteins of the BCL-2 superfamily all possess one or more regions of homology referred to as BCL-2 homology (BH) domains. There are four BH domains identified as BH1, BH2, BH3, and BH4. Proteins of the BCL-2 superfamily are classified as either initiators, pro-survival proteins, or effector proteins. The pro-survival proteins are commonly referred to as guardians or as anti-apoptotic proteins and are exemplified by BCL-2 itself. The effector proteins are commonly referred to as pro-apoptotic proteins. The initiator BCL-2 family members only possess a BH3 domain and are, therefore, referred to as the BH3-only subfamily.
Table of Pro-Survival BCL-2 Family Proteins
| Protein | Common/Other Name | Gene | Functions / Comments |
| BCL-2 | Bcl-2 | BCL2 | associated with the mitochondrial outer membrane (MOM); interferes with the release of cytochrome c induced by activated BAK and/or BAX; also inhibits activation of caspase cascades thereby preventing apoptosis |
| BCL-2 like 1 | Bcl-X | BCL2L1 | two isoforms result from alternative mRNA splicing; the long isoform (identified as BCL-XL) is a pro-survival (anti-apoptotic) protein while the short isoform (identified as BCL-XS) is a pro-apoptotic protein; BCL-XL is associated with the mitochondrial outer membrane (MOM); interferes with the release of cytochrome c induced by activated BAK and/or BAX; also inhibits activation of caspase cascades thereby preventing apoptosis; BCL-XS allows cytochrome c to be released |
| BCL-2 like 2 | Bcl-W | BCL2L2 | over-expression prevents myeloid and lymphoid cell apoptosis; involved in normal spermatogenesis |
| BCL-2 related protein A1 | Bfl-1 | BCL2A1 | common name derived from BCL-2-related protein expressed in fetal liver |
| BCL-2 like 10 | Bcl-B | BCL2L10 | selectively interacts with Bax; cannot interact with Bak, Bad, and Bid |
| BCL-2 family apoptosis regulator | Mcl-1 | MCL1 | originally identified as induced myeloid leukemia cell differentiation protein 1 (Mcl-1); multiple isoforms result from alternatively spliced mRNAs; the longest isoform (isoform 1) is a pro-survival (anti-apoptotic) protein while the two shorter isoforms (isoform 2 and 3) are pro-apoptotic |
Table of Pro-Apoptosis BCL-2 Family Proteins
| Protein | Gene | Functions / Comments |
| BCL-2 antagonist/killer 1 (BAK) | BAK1 | pro-apoptotic effector protein; contains BH1, BH2, BH3, and BH4 domains; localized to the mitochondrial outer membrane (MOM); forms an oligomeric pore in the outer mitochondrial membrane allowing release of cytochrome c when stimulated by an apoptotic signaling cascade |
| BCL-2 associated X, apoptosis regulator (BAX) | BAX | pro-apoptotic effector protein; contains BH1, BH2, BH3, and BH4 domains; localized to the cytosol; activation of an apoptotic signaling cascade induces mitochondrial outer membrane (MOM) association; when associated with mitochondria the protein induces the opening of the mitochondrial voltage dependent anion channels (VDAC1, VDAC2, VDAC3); also forms an oligomeric pore in the MOM allowing release of cytochrome c |
| BCL-2-related ovarian killer protein (BOK) | BOK | pro-apoptotic effector protein; at least three protein isoforms identified via alternative mRNA splicing |
| BCL-2-associated agonist of cell death (BAD) | BAD | member of the BH3-only subfamily; when in the dephosphorylated state BAD forms heterodimers with BCL-2 and BCL-XL which inactivates these pro-survival proteins; in the absence of BCL-2 or BCL-XL the BAK and BAX proteins can promote cytochrome c release from the mitochondria; when BAD is phosphorylated it cannot associated with BCL-2 or BCL-XL which promotes cell survival; BAD phosphorylated by the kinase PKB/AKT and is dephosphorylated by Ca2+-stimulated calcineurin |
| BCL-2-interacting mediator of cell death (BIM) | BCL2L11 | member of the BH3-only subfamily |
| BH3-interacting domain death agonist (BID) | BID | member of the BH3-only subfamily; interacts with BAX promoting the insertion of BAX into the mitochondrial outer membrane (MOM) |
| BCL-2 interacting killer (BIK) | BIK | member of the BH3-only subfamily |
| BCL-2 modifying factor (BMF) | BMF | member of the BH3-only subfamily; associates with dynein light chain 2 |
| BCL-2 interacting protein 3 | BNIP3 | member of the BH3-only subfamily; localized to the mitochondrial outer membrane, MOM; also involved in induction of mitochondrial autophagy (mitophagy) by recruiting the autophagic machinery to mitochondria |
| BCL-2 interacting protein 3 like | BNIP3L | member of the BH3-only subfamily; localized to the mitochondrial outer membrane, MOM; also involved in induction of mitochondrial autophagy (mitophagy) by recruiting the autophagic machinery to mitochondria |
| phorbol-12-myristate-13-acetate-induced protein 1 | PMAIP1 | member of the BH3-only subfamily; commonly referred to as NOXA which means damage in Latin; involved in p53-mediated apoptosis pathway |
| BCL-2-binding component 3 | BBC3 | member of the BH3-only subfamily; originally identified as p53 up-regulated modulator of apoptosis (PUMA); |
| harakiri, BCL-2-interacting protein | HRK | member of the BH3-only subfamily; interacts with both BCL-2 and BCL-XL |
The mitochondrial apoptosis pathway is initiated by the stress-mediated release of cytochrome c. Mitochondrial stress can be initiated internally due to excess reactive oxygen species (ROS) generation or triggered by external factors such as growth factor deprivation or DNA damage. In addition to cytochrome c, mitochondria contain several other pro-apoptotic proteins such as apoptosis inducing factor, mitochondria associated 1 (AIFM1; also known simply as apoptosis inducing factor, AIF) and DIABLO (also known as SMAC: second mitochondria-derived activator of caspase). Both AIF and DIABLO are released from the mitochondria in response to stress and/or apoptosis induced signaling. The release of these mitochondrial pro-apoptotic proteins is carried out through pores in the mitochondrial outer membrane (MOM) created by oligomeric BCL-2 family member proteins BAK and BAX.
When cytochrome c is released, it interacts in the cytosol with apoptotic protease activating factor 1 (APAF1) and procaspase 9, along with ATP, to form what is referred to as the apoptosome. The formation of the apoptosome results in cleavage and activation of procaspase 9. Active caspase 9 then cleaves procaspase 3 to its active form setting off the full intrinsic apoptotic program.
The normal control of the release of cytochrome c through BAK and/or BAX is exerted by the pro-survival proteins, BCL-2 and BCL-XL. In order to mount an appropriate apoptotic response the inhibition of BAK and BAX by BCL-2 (or BCL-XL) must be countered. The inhibition of BCL-2 and BCL-XL function is exerted by several proteins of the large BCL-2 homology family, specifically the BH3 domain-only family members. Several BH3-only proteins that interfere with BCL-2 and BCL-XL function are BAD, BID, and BCL2L11 (commonly called BIM). The activity of the BAD protein is controlled by its state of phosphorylation. When dephosphorylated BAD interacts with BCL-2 and BCL-XL to inhibit their ability to inhibit cytochrome c release through BAK and/or BAX oligomeric complexes. Growth factor receptor activation results in the activation of the kinase PKB/AKT which can then phosphorylate BAD. When phosphorylated, BAD is trapped in the cytosol via interaction with proteins of the 14-3-3 family. The cytosolic sequestration of BAD allows BCL-2 and BCL-XL to interact with and inhibit BAK and BAX complexes in the MOM.
Additional regulation of the mitochondrial apoptotic program is exerted upon the apoptosome by members of the inhibitor of apoptosis protein (IAP) family. The most well studied member of the human IAP family is X-linked inhibitor of apoptosis, XIAP. The XIAP protein exerts numerous effects to inhibit apoptosis including directly binding to and inhibiting caspases 3, 7, and 9. XIAP also functions as an E3 ubiquitin ligase to ubiquitylate several proteins in the apoptotic cascade resulting in their proteasomal degradation. The inhibitory activity of XIAP is blocked by the mitochondrial protein DIABLO that, like cytochrome c, is released in response to pro-apoptotic signals.
Cell stress caused by DNA damage also ultimately impacts activation of the mitochondrial apoptotic pathway. DNA damage results in the activation of the tumor suppressor, p53 which in turn transcriptionally activates numerous genes whose encoded proteins participate in activation of apoptosis. Expression of the PMAIP1 (phorbol-12-myristate-13-acetate-induced protein 1; commonly called NOXA), the BBC3 (BCL-2 binding component 2; commonly referred to as PUMA), and the BAX genes are all induced by activated p53. The PUMA and NOXA proteins are BH3-only BCL-2 homology family proteins termed apoptosis initiators.
Endoplasmic Reticulum Stress-Mediated Apoptosis
The endoplasmic reticulum (ER) is the central intracellular organelle responsible for synthesis and processing of almost all proteins that are destined for insertion into membranes, to be carbohydrate modified, and to be secreted. In addition to its critical role in protein synthesis, lipids and membrane components from the ER contribute to the production of exocytotic vesicles and to the formation of the peroxisomes. Any number of events that lead to disturbances in normal ER function result in what is collectively termed ER stress. The primary response to ER stress is the activation of the unfolded protein response, UPR. The UPR represents a tightly regulated series of intracellular signal transduction reactions that are designed to restore normal protein homeostasis.
The functions of three key signaling proteins distinguish the characteristics of the UPR. These three transmembrane proteins are called IRE1 (inositol-requiring protein-1; also identified as IRE1α), PERK [RNA-dependent protein kinase (PKR)-like ER Kinase], and ATF6 (activating transcription factor 6). The human IRE1 protein is encoded by the ERN1 (endoplasmic reticulum to nucleus signaling 1) gene. PERK is one of four enzymes of the translation initiation factor, eIF-2 kinase family. All three proteins contain luminal stress-sensing domains and cytoplasmic activity domains. IRE1 possesses a kinase domain and an RNase domain. PERK possesses a kinase domain. ATF6 possesses as transcription activation domain.
In addition to ER stress, numerous other conditions of cellular stress, such as mitochondrial stress, lead to the activation of one of more of the eIF-2α kinases. Because these eIF-2α kinases are involved in the responses of cells to a variety of stressful conditions, their activation, and subsequent effects, are collectively referred to as the integrated stress response (ISR). A distinguishing feature of the ISR, that is not associated with the UPR, is that there is regulation of both translation and transcription as a result of the activation of the ISR.
As ER-associated protein synthesis proceeds the proteins are extruded into the lumen of the ER in the unfolded state. Protein folding is a process that is facilitated, not only in the ER, but in every cellular compartment where protein synthesis or post-translational protein import occurs through the actions of molecular chaperones. The molecular chaperones of protein folding belong to the large superfamily of heat shock proteins (Hsp). The Hsp superfamily is composed of multiple protein subfamilies that are loosely designated by molecular weights. These Hsp families include the Hsp40, Hsp60, Hsp70, Hsp90, and Hsp100 families.
Several folding chaperones also possess enzymatic activity and are required for the correct formation of disulfide bonds in proteins. Protein disulfide isomerases (PDI) catalyze the formation of disulfide bonds between cysteine residues in a substrate protein while peptidyl-prolyl cis-trans isomerases (PPI) catalyze isomerization of peptidyl-prolyl bonds.
Protein folding chaperones in the ER belong to the Hsp40, Hsp70, Hsp90, and Hsp100 families. One critically important member of the Hsp70 family of molecular chaperones is the protein commonly identified as glucose-regulated protein 78-kDa, GRP78. GRP78 is also known as BiP (Binding immunoglobulin heavy chain Protein). The human GRP78 protein is encoded by the HSPA5 [heat shock protein family A (Hsp70) member 5] gene. GRP78 is referred to as the master regulator of the ER. The function of GRP78 is not only to participate in protein folding but to also maintain the permeability barrier of the ER by sealing the luminal side of inactive translocons (protein complexes through which nascent polypeptides are extruded), to facilitate the translocation of growing polypeptide chains into the ER lumen, to regulate the aggregation of nonnative polypeptides, and to contribute to calcium homeostasis in the ER. Of significance to the role of ER function in apoptosis, GRP78 plays an important role in the regulation of the UPR.
Under normal physiological conditions, the ER luminal domains of the PERK and ATF6 proteins are bound to BiP. The interaction of PERK and ATF6 with BiP keeps them in the inactive state. When unfolded proteins accumulate in the ER, BiP is released from these complexes to assist with the folding of accumulated proteins. In contrast to PERK and ATF6, IRE1 becomes activated when unfolded proteins bind directly to it. Upon activation, PERK, IRE1 and ATF6 induce signal transduction events that are designed to alleviate the accumulation of misfolded proteins in the ER and to facilitate a protective response.
PERK functions in the UPR by phosphorylating the α-subunit of the translation initiation factor, eIF-2. The phosphorylation of eIF-2α prevents further general protein synthesis but allows for the translation of selective mRNAs such as that encoding the transcription factor ATF4 (activating transcription factor 4). ATF4 transcriptionally activates UPR target genes facilitating the responses to ER stress. Important ATF4 target genes include the transcription factor ATF3 gene and the DNA damage inducible transcript 3 (DDIT3) gene. The protein encoded by the DDIT3 gene is the transcription factor commonly called CHOP [CCAAT-box/enhancer-binding protein (C/EBP) homologous protein] and is also known as growth arrest and DNA damage-inducible protein 153 (GADD153). Another ATF4 target is the protein phosphatase 1 regulatory subunit 15A (PPP1R15A) gene which encodes a protein of the growth arrest and DNA damage-inducible (GADD) family commonly referred to as GADD34.
When BiP releases ATF6 it is allowed to migrate to the Golgi where it undergoes proteolytic cleavage by site 1 protease (S1P) and site 2 protease (S2P) releasing the transcription factor domain which then enters the nucleus and activates additional UPR target genes. The proteases S1P and S2P are the same proteases involved in the release of the transcription factor, SREBP from the Golgi membrane. ATF6 target genes contain ATF/cAMP response elements (CRE) and ER-stress response elements (ERSE-1). Important ATF6 target genes include the HSP5A (BiP) gene, HSP90B1 (also identified as Grp94 and endoplasmin) gene, and DDIT3. Another important ATF6 target is the protein disulfide isomerase associated 6 (PDIA6) gene whose encoded protein assists with protein folding in the lumen of the ER.
Activated IRE1 oligomerizes which activates its RNase domain via autophosphorylation leading to unconventional splicing of the mRNAs. A critical target mRNA is that encoding the ubiquitously expressed XBP1 (X-box binding protein 1) transcription factor. IRE1 action on the XBP1 mRNA results the production of a smaller protein (XBP1s) that transcriptionally activates UPR target genes.
In addition to altering transcription in response to ER stress, IRE1, via its kinase activity, stimulates retrograde transport of misfolded proteins from the ER into the cytosol where they are ubiquitylated and degraded in the proteasome. This latter process is referred to as regulated IRE1-dependent decay (RIDD) which represent a specialized form of ER-assisted degradation (ERAD).
Activation of the RNase activity of IRE1 not only modifies the processing of the XBP1 mRNA but it also results in the cleavage of microRNAs (miRNAs) that control the level of caspases, thereby, inducing the process of apoptosis. Induction of apoptosis is further activated by IRE1 through its ability to stimulate the activity of mitogen-activated protein kinase kinase kinase 5 (MAP3K5; also known as apoptosis signal-regulating kinase-1, ASK1). MAP3K5 phosphorylates and activates the downstream stress kinases, Jun-N-terminal kinase (JNK) and the p38 MAPK proteins both of which promote apoptosis.
The JNK protein is a member of the MAPK superfamily of Ser/Thr kinases and it is encoded by the MAPK8 gene. JNK activation promotes apoptosis by phosphorylating and inhibiting BCL-2 and phosphorylating and activating BIM. There are multiple members of the p38 MAPK family with the one encoded by the MAPK14 gene being the founding member. Activation of MAPK14 leads to phosphorylation and activation of the transcription factor CHOP. CHOP in turn activates the expression of several pro-apoptotic genes such as BIM and the TNF receptor superfamily (TNFRSF) member TRAILR2 (see the next section).
Both JNK and MAPK14 phosphorylate and activate the BAX protein which, as discussed earlier, forms an oligomeric pore in the mitochondrial outer membrane (MOM) that facilitates the release of cytochrome c which, in turn, activates the intrinsic apoptosis pathway. Activation of MAPK14 also results in decreased expression of the pro-survival BCL2 gene.
Death Receptor Pathway of Apoptosis
The death receptor pathway of apoptosis consists of numerous type I transmembrane proteins and their associated ligands many of which are themselves transmembrane proteins (type II transmembrane). Upon ligand-receptor interaction there is activation of signal transduction pathways that involve numerous additional effector proteins. All of the receptors that participate in the death receptor pathway of apoptosis possess an approximately 80 amino acid domain in their cytoplasmic sequence which is termed the death domain (DD).
Death receptors all belong to the large superfamily of tumor necrosis factor (TNF) receptor-related proteins identified as TNF receptor superfamily members (TNFRSF). Death receptors are activated by their cognate ligands which constitute a family of complementary cytokines that belong to the TNF-related protein superfamily identified as TNF superfamily members (TNFSF). TNFSF proteins are commonly referred to as death ligands. With the exception of the soluble, lymphocyte-derived cytokine identified as lymphotoxin alpha (LTα, also known as TNFβ), these TNFSF proteins are mainly expressed as type II transmembrane proteins that contain an intracellular N-terminal domain, a transmembrane region, and a C-terminal extracellular tail. In some cases, the membrane-associated death ligands can be released as soluble cytokines by proteolytic cleavage. However, the apoptosis-inducing capacity of soluble death ligands is generally significantly lower when compared to the corresponding membrane-bound forms.
Following death ligand binding, an activated death receptor stimulates the recruitment of intracellular signaling proteins via interactions with the DD of the receptor and homologous DD in the adapter molecules. Characteristic adapter molecules include the proteins encoded by the TRADD (in the TNFRSF pathway of apoptosis) and FADD (in the FAS pathway of apoptosis) genes described in the Table below.
Following the docking of death effector proteins to the death receptor the initiator caspases 8 and 10 are recruited. The activation of a death receptor by an appropriate death ligand and the subsequent recruitment of death effector proteins and a procaspase 8 or 10 forms what is referred to as the death-inducing signaling complex, DISC. Conversion of procaspase 8 to its active form then sets of the apoptotic cascade via the caspase 8-mediated activation of caspases 3, 6, and 7. The overall activity of the DISC can be regulated by various forms of the proteins encoded by the CFLAR gene. These protein isoforms are commonly called cFLIP and at least three different isoforms have been identified resulting from alternative mRNA splicing. The full-length long form (cFLIPL) is pro-apoptotic via its participation in the conversion of procaspase 8 to active caspase 8 by the DISC. One of the short cFLIP isoforms (cFLIPS) acts as an anti-apoptotic protein by interfering with procaspase 8 activation.
Although the TNFR1 activation pathway is primarily involved in mediating inflammation it is also known to activate apoptotic programs under appropriate conditions. Both TNFR1 and TNFR2 bind three distinct but related ligands. These ligands are the membrane-bound and soluble forms of TNFα (mTNFα and sTNFα) and the soluble lymphocyte-derived cytokine identified as lymphotoxin-α (LTα) but also known as TNFβ. In the apoptosis-inducing pathway activated by TNFR1 and TNFR2 the ligand-receptor complexes are internalized, whereas, the growth promoting complexes stay associated with lipid rafts in the plasma membrane. During the process of TNFR-ligand endocytosis, RIPK1, FADD, and procaspase 8 and 10 are recruited to form a cytosolic DISC, often referred to as the TNF receptosome. This intracellular complex then activates the procaspases resulting in the activation of apoptosis.
A link between the extrinsic (death receptor) and intrinsic (mitochondrial) pathways of apoptosis exists via the ability of activated caspase 8 to proteolyze the BID encoded protein releasing a C-terminal 15 kDa fragment termed tBID (tiny BID). Following cleavage the tBID peptide migrates to the mitochondria where it inhibits the activity of BCL-2 and BCL-XL, thereby allowing cytochrome c release through the BAK and BAX complexes in the mitochondrial outer membrane (MOM).
All of the major death receptor pathways of apoptosis, the TNFR1 (and TNFR2)-, TNFRSF- and the FAS-induced pathways, not only activate the extrinsic pathways of apoptosis but they can also function in non-cytotoxic pathways. In certain cell types, under appropriate conditions, the binding of FASL to FAS actually promotes cell survival and proliferation.
FAS activation can lead to the activation of the MAPK cascade, specifically the extracellular signal-regulated kinase (ERK) cascade. In addition to the proliferation inducing ERK cascade activation, FAS stimulation has also been shown to activate an anti-apoptotic cascade via the activation of NF-κB (nuclear factor kappa-light-chain-enhancer of activated B cells) pathway. NF-κB is a transcription factor that plays a major role in regulating cytokine production and cell survival, particularly in response to numerous cell stress stimuli.
Of significance to the regulation of apoptosis, NF-κB activates the expression if the XIAP (X-linked inhibitor of apoptosis) gene. The TNFRSF receptors can also trigger cell survival pathways, such as the MAPK, NF-κB, and PKB/AKT pathways, under the appropriate conditions. Both the TNFRSF10A (TRAILR1) and the TNFRSF10B (TRAILR2) encoded receptors have been shown to activate NF-κB via a TRAF2-NIK-IKK complex (TRAF2: TNF receptor-associated factor 2; NIK: NF-κB-inducing kinase; IKK: IκB kinase complex).
Activation of the MAPK pathway by TRAILR1 or TRAILR2 involves a distinct complex of composed of TRAF2-MAP3K1-M2K4. MAP3K1 is MAP kinase kinase kinase 1. MAP2K4 is MAP kinase kinase 4. The activation of cell survival is the default pathway for the TNFR1 and TNFR2 proteins. When ligand (TNFα) binds, TNFR1 recruits several proteins to form a complex that ultimately activates NF-κB. This complex is composed of TRAF2, RIPK1 (receptor interacting Ser/Thr kinase 1), and the inhibitors of apoptosis proteins commonly referred to as cIAP1 (BIRC2) and cIAP2 (BIRC3). As described in the Table above, both cIAP1 and cIAP2 possess E3 ubiquitin ligase activity. Within the TNFR1 complex the kinase, RIPK1, is ubiquitylated by cIAP1 and cIAP2, a modification that is required for the subsequent downstream activation of NF-κB.
Table of Death Receptor Pathway Proteins
This Table is not intended to represent a complete list of all the known death receptors, their ligands, and downstream mediators
| Protein | Gene | Functions / Comments |
| TNF receptor superfamily member 1A | TNFRSF1A | also known as TNF receptor 1 (TNFR1) |
| TNF receptor superfamily member 1B | TNFRSF1B | also known as TNF receptor 2 (TNFR2) |
| TNF receptor superfamily member 25 | TNFRSF25 | also known as death receptor 3 (DR3) and thrombospondin-related apical membrane protein (TRAMP) |
| TNF receptor superfamily member 10a | TNFRSF10A | also known as death receptor 4 (DR4) and TNF-related apoptosis-inducing ligand receptor 1 (TRAILR1); expressed in most human tissues, including spleen, thymus, liver, peripheral blood leukocytes, activated T cells, small intestine, as well as several tumor cell lines |
| TNF receptor superfamily member 10b | TNFRSF10B | also known as death receptor 5 (DR5) and TNF-related apoptosis-inducing ligand receptor 2 (TRAILR2); ubiquitously expressed with highest levels seen in spleen, activated lymphocytes, and peripheral blood leukocytes |
| TNF receptor superfamily member 21 | TNFRSF21 | also known as death receptor 6 (DR6) |
| FAS cell surface death receptor | FAS | also known as TNF receptor superfamily member 6 (TNFRSF6), CD95 or APO-1 (apoptosis antigen-1); the cytoplasmic death domain (DD) interacts with the adapter protein FADD through the DD in FADD; FAS gene is ubiquitously expressed with highest levels in liver, heart, kidney, pancreas, brain, thymus, lymphoid tissues, and activated mature T lymphocytes; soluble FAS protein is generated from alternatively spliced mRNAs |
| FAS ligand | FASLG | also known as TNF superfamily member 6 (TNFSF6) and CD951 (CD95) ligand; expressed almost exclusively in activated T cells; forms a homotrimeric membrane complex that activates three FAS receptor proteins on the surface of target cells |
| TNFα | TNF | binds to TNFR1; activation normally induces cell survival, cytokine generation, and inflammation; apoptotic activity of TNFR1 activation occurs when the normal survival pathways are inhibited |
| TNF-related apoptosis-inducing ligand | TRAIL | binds to and activates the receptors encoded by the TNFRSF10A (TRAILR1 or DR4) and TNFRSF10B (TRAILR2 or DR5) genes |
| TNFRSF1A associated via death domain | TRADD | contains a death domain; interacts with TNFRSF1A |
| FAS-associated via death domain | FADD | contains a C-terminal death domain (DD); interacts with FASL as well as other death receptors such as TNFRSF10A and TNFRSF25; also contains a death effector domain (DED) that induces interaction with caspase 8 |
| apoptosis antagonizing transcription factor | AATF | also known as death effector domain (DED) |
| caspase 8 and FADD-like apoptosis regulator | CFLAR | also known as c-FLIP (cellular homolog of the viral FLICE-inhibitory protein, v-FLIP); structurally related to caspase 8 but lacks the catalytic Cys residue; multiple forms generated by alternative mRNA splicing; all the isoforms possess death effector domains (DED); the full-length form (cFLIPL) activates apoptosis, whereas, the short form (cFLIPS) inhibits or reduces TNFRSF-activated apoptosis |
Perforin and Granzyme-Mediated Apoptosis
Cytotoxic lymphocytes play a central role in both the innate and adaptive immune systems in that they are tasked with recognizing virus- and bacteria-infected or transformed cells and inducing their elimination via activated apoptosis. Cytotoxic lymphocytes are of two types, the natural killer cells (NK cells) and cytotoxic T lymphocytes (CTL). These cells exert their apoptosis inducing effects, in part, by synthesizing and secreting potent toxins that include the pore-forming protein, perforin and the serine proteases called granzymes. Granzyme proteins are so-called since they were initially characterized as components of the secretory granules of CTL. Perforin is encoded by the PRF1 gene. Humans express five granzyme genes identified as GZMA, GZMB, GZMH, GZMK, and GZMM.
Activation of CTL and NK cells results in the activation of perforin and granzyme synthesis and packaging in secretory vesicles and their migration to the immune synapse. The release of vesicle contents is triggered by target cell interaction which induces a signal transduction cascade resulting in increased cytosolic Ca2+. The mobilization of perforin and granzyme vesicles in CTL and NK cells involves several critical proteins encoded by the STX11, STXBP2, and UNC13D genes. The STX11 gene encodes the syntaxin 11 protein, a member of the syntaxin family of proteins required for exocytosis. The UNC13D gene encodes the UNC-13 homolog D protein which is also known as MUNC13-4 (mammalian UNC-13 homolog family member 4). The UNC family of genes (currently numbering 111), of the round worm C. elegans, are so-called because mutations in these genes result in “uncoordinated” movement (abnormal locomotion phenotype). The STXBP2 gene encodes syntaxin binding protein 2 which is also also known as MUNC18-2 (there are six MUNC-18 family member genes). Following vesicle fusion with the plasma membrane, the released perforin interacts with the target cell plasma membrane and forms a pore via which granzyme entry into the target cell is facilitated. The perforin pores in the membrane are internalized by the target cell, together with the granzyme molecules, via clathrin-dependent and dynamin-dependent endocytosis.
The predominant and most potent apoptosis inducing granzyme released from CTL and NK cells is granzyme B. Granzyme A also induces apoptosis but at a slower rate than that induced by granzyme B. Granzyme B cleaves procaspases (particularly procaspase 3) to their active forms setting off the apoptotic cascade. In addition, granzyme B cleaves the BID protein to release tBID, just as is the case for the action of caspase 8 on the BID protein. Following cleavage the tBID peptide migrates to the mitochondria where it inhibits the activity of BCL-2 and BCL-XL, thereby allowing cytochrome c release through the BAK and BAX complexes in the mitochondrial outer membrane (MOM). Granzyme M is also involved in triggering apoptosis by processing human FAS-associated death domain protein (FADD), causing FADD dimerization and increasing procaspase 8 processing to active caspase 8.
The clinical significances to the perforin/granzyme pathway of cell killing are that defects in numerous genes required for the processes are associated with a number of autosomal recessive immune-mediated diseases. These diseases are collectively referred to as the perforinopathies and can result from defects in the perforin gene (PRF1) as well as in the STX11, STXBP2, and UNC13D genes. The perforinopathies are classified into three groups. The acute perforinopathies are also known as the early-onset classic familial hemophagocytic lymphohistiocytosis, FHL. The subacute group includes atypical FHL or another pathology, which usually occurs after 5 years of age. The chronic group is characterized by heterozygosity and can predispose to disease in adults. In the acute cases, FHL invariably occurs before 2 years of age and is often a lethal disorder. The forms of FHL (termed type 2 FHL, FHL2) that result from mutations in the PRF1 gene account for approximately 50% of all FHL cases. Acute forms of perforinopathy are also the result of mutations in the UNC13, STX11, and STXBP2 genes. Mutations in the UN13D gene result in FHL3, mutations in the STX11 gene result in FHL4, and mutations in the STXBP2 gene result in FHL5.
Necroptosis
Necroptosis is a form of programmed cell death that encompasses aspects of apoptosis and necrosis. The process of necroptosis is mediated by death receptors and is activated when the caspase-8-dependent pathway of apoptosis is blocked.
The immune response activating ligands Fas, TNFα, and lipopolysaccharide (LPS) trigger the activation of receptor interacting kinase 3 (RIPK3) that then phosphorylates the protein, mixed lineage kinase domain like pseudokinase (MLKL). Once phosphorylated MLKL proteins oligomerize and then translocate to the plasma membrane from the cytosol through interaction with phosphatidylinositol phosphates. Once in the plasma membrane the oligomerized MLKL forms a pore in the membrane. The formation of the pore results in an inflammatory response involving secretion of chemokines and cytokines. The oligomerized MLKL also acts as a platform for the recruitment of Na+ and Ca2+ channels.
Due to tissue specific sites of expression of RIPK3 and MLKL, not all cells are capable of carrying out the necroptosis pathway of programmed cell death. In addition, when caspase-8 can be activated it blocks the process of necroptosis by cleaving receptor interacting kinase 1 (RIPK1) which, like RIPK3, stimulates necroptosis.
Ferroptosis
Ferroptosis is the term used to describe an iron-dependent form of regulated cell death (RCD) that is distinct from the classical pathways of caspase-mediated apoptosis, autophagy, and necrosis. Ferroptosis is characterized by iron-dependent accumulation of lipid peroxides. There are two major pathways via which ferroptosis can occur, the extrinsic and the intrinsic pathways.
The extrinsic pathway is the transporter dependent pathway of ferroptosis that involves inhibition of the heterodimeric cystine and glutamate antiporters that transport cystine in the opposite direction to that of glutamate. This transporter system is a member of the acidic amino acid transporter family and is commonly designated as the Xc– transporter system. The Xc– transporter is composed of a heavy chain (4F2hc) and a light chain (xCT). The 4F2hc protein is encoded by the SLC3A1 gene. The xCT protein is encoded by the SLC7A11 gene.
The intrinsic pathway of ferroptosis is the more well characterized and is also referred to as the enzyme regulated
pathway. Inhibition of glutathione peroxidase 4 (GPx4), starvation of cysteine, and the induction of the peroxidation of arachidonic acid all trigger the intrinsic pathway of ferroptosis. Inhibition of glutathione peroxidase activity results in a decrease in antioxidant capacity and an accumulation of lipid reactive oxygen species (ROS), ultimately leading to oxidative cell death.
The extrinsic and intrinsic pathways are closely linked through numerous metabolic pathways as well as through several subcellular organelles including the mitochondria, the endoplasmic reticulum (ER), the Golgi apparatus, the nucleus, the lysosomes, and the peroxisomes.
Ferroptosis is associated with metabolic processes involving iron, lipids, and thiols that lead to an iron-dependent
generation of lipid peroxidation which, in turn, results in cell death. The generation of lipid peroxides, and the process of ferroptosis can be prevented by two major antioxidant systems. One involves glutathione peroxidase 4 (GPx4) and the other involves ferroptosis suppressor protein 1 (FSP1). FSP1 is encoded by the AIFM2 (apoptosis inducing factor mitochondria associated 2) gene.
Glutathione (GSH) is an obligatory cofactor for GPx4. The synthesis of GSH requires the three amino acids cysteine, glutamate, and glycine. The role of the Xc– transporter system in the uptake of cystine, which is subsequently reduced to two moles of cysteine, and the synthesis of GSH explains how inhibition of this transporter system is involved in the triggering of ferroptosis. Inhibition of the SLC7A11 gene that encodes the light chain protein of the Xc– transporter also induced ferroptosis.
GPx4 catalyzes the reduction of lipid peroxides. FSP1 catalyzes the regeneration of reduced coenzyme Q10 (CoQ10) in the cytosol. The reduced form of CoQ10 is ubiquinol and the oxidized form is ubiquinone. Ubiquinol can function as a lipid peroxyl radical trap, thereby reducing membrane lipid oxidation.
The changes in protein expression, metabolic profile, and cell morphology that occur in response to ferroptosis allows for discrimination of this pathway of cell death from other forms of cell death. Cells undergoing ferroptosis adopt a characteristic rounded shape before cell death but without the cytoplasmic and organelle swelling and plasma membrane rupture that is typical of necrosis. Ferroptotic cell nuclei also do not exhibit altered structural integrity, chromatin accumulation against the nuclear membrane (referred to as chromatin margination), or apoptotic bodies as is typically observed when cells undergo apoptosis. In addition, ferroptotic cells do not exhibit double-membrane enclosed vesicles typical of the process of autophagy.
Ferroptosis is accompanied by metabolic changes that are regulated by specific genes. As the name of this cell death pathway implies, iron is an essential component of the process and it driving intracellular lipid peroxidation. Changes in GPx4 activity is also an indicator for ferroptosis. Regulation of genes related to iron metabolism, such as transferrin, iron response element-binding protein 2 (IREB2), nitrogen fixation 1 (NFS1), and nuclear receptor coactivator 4 (NCOA4) can prevent ferroptosis.
The role of lipid peroxidation in ferroptosis was demonstrated in several ways. First, inhibition of the lipoxygenase encoded by the ALOX12 (arachidonate 12-lipoxygenase; commonly just 12-LOX or 12(S)-LOX) gene could inhibit cell death mediated by glutamate toxicity. The ALOX12 encoded enzyme is an iron-containing lipid dioxygenase. Secondly, GPx4 can prevent ferroptosis via reduction in phospholipid hydroperoxides which effectively represses lipoxygenase-mediated lipid peroxidation.
In addition to ALOX12, other members of the lipoxygenase family of enzymes can promote ferroptosis in cell type-dependent processes including the enzymes encoded by the ALOX5, ALOX15, ALOX15B, and ALOXE3 genes. The ALOXE3 encoded enzyme synthesizes the epoxy alcohol, 8R-hydroxy-11R,12R-epoxyeicosa-5Z,9E,14Z-trienoic acid, from 12R-hydroperoxyeicosatetraenoic acid (12R-HpETE) which is derived from arachidonic acid.
In addition, the acyl-CoA synthetases encoded by the ACLS3 and ACSL4 (acyl-CoA synthetase long chain family member 3 and member 4, respectively) genes are critical contributors to ferroptosis. ACSL3 is involved in the production of monounsaturated fatty acids (MUFA) and in the metabolism of lipids through its role in promoting lipid droplet formation and maturation. MUFA reduce the susceptibility of plasma membrane lipids to peroxidation as a result of reductions in the levels of polyunsaturated fatty acids (PUFA) in the membrane. Thus, the activity of ACSL3 is protective of cells from ferroptosis.
ACSL4, on the other hand, is involved in the production of PUFA. The structure of PUFA is prone to oxidation because of the weak C-H bond at the bis-allylic methylene positions (–CH=H–CH2–CH=CH–). Allylic hydrogens are those on the carbons adjacent to carbon-carbon double bonds. Membrane PUFA are the primary targets for oxidation by reactive oxygen species (ROS). Accumulation of lipid peroxides in the membrane leads to membrane pore formation which disrupts the barrier function resulting in changes in membrane permeabilization. Thus, the level of ACSL4 activity promotes the process of ferroptosis.
Ascorbic acid (vitamin C) is a known antioxidant that contributes to reductions in lipid peroxidation. Recent work (2026) has demonstrated that vitamin C directly interacts with, and inhibits, the activity of ACSL4. The inhibition of ACSL4 by vitamin C represents an additional mechanism by which vitamin C can mitigate the negative effects of lipid peroxidation and thus reduce the potential for ferroptosis.
An imbalance between iron import, storage, and export, represents the critical role of iron in the process of ferroptosis. Studies have found that increased expression of the transferrin receptor leads to enhanced iron import which ultimately triggers ferroptosis. Enhanced degradation of the intracellular iron storage protein, ferritin, has also been found to be associated with induction of ferroptosis. Many of the enzymes involved in the generation of ROS are iron-containing (either heme or iron-sulfur centers). The oxidation of H2O2 by Fe2+, which is called the Fenton reaction, generates the potent hydroxyl free radical that contributes to non-enzymatic lipid peroxidation.
The orphan GPCR, GPR56, has recently been shown to bind the steroid, 17α-hydroxypregnenolone, resulting in suppression of ferroptosis in the liver. GPR56 is a member of the adhesion GPCR subfamily of the class B GPCR superfamily which is the secretin-like receptor class of GPCR. As such, GPR56 is encoded by the ADGRG1 (adhesion G protein-coupled receptor G1) gene.
The steroid hormone biosynthesis enzyme (P450c17) encoded by the CYP17A1 gene is a bifunctional enzyme that converts pregnenolone to 17α-hydroxypregnenolone (often designated 17-OH pregnenolone) and then to dehydroepiandrosterone (DHEA).
When 17-OH pregnenolone binds to GPR56 it stimulates the endocytosis of the membrane-localized fatty acid transporter identified as FAT/CD36. This effect of GPR56 results in a reduction in membrane-localized phospholipids containing polyunsaturated fatty acids (PUFA). As indicated, PUFA peroxidation is a major contributor to the activation of ferroptosis.
The 18-carbon fatty acid member of the octadecatrienoic acid, α-eleostearic acid (9Z,11E,13E-octadecatrienoic acid) has recently (2026) been found to play a role in the elimination of senescent cells via the induction of ferroptosis. α-Eleostearic acid (α-ESA) is enriched in the oils extracted from the nuts of the kalo nut tree (also known as the tung tree.
The ability of α-ESA to induce death in senescent cells results from an induction of ferroptosis, likely as a result of interaction with proteins in the ferroptotic pathway. The senolytic effects of α-ESA reduce overall tissue senescence resulting in healthspan extension, at least when assayed in experimental mice.
Cuproptosis
The roles of copper as an essential micronutrient in numerous biological processes is covered in detail in the Iron and Copper Homeostasis page. As discussed in that page, intracellular copper concentration is tightly regulated given the fact that even moderate increases can cause cytotoxicity and even lead to cell death. As detailed in the previous section covering Ferroptosis, excessive levels of trace elements, such as iron, can cause cell death via specific pathways.
Copper excess is associated with the induction of a novel type of cell death that has been termed cuproptosis. Cuproptosis represents a form of regulated cell death that is characterized by the aggregation of lipoylated mitochondrial enzymes and a loss of iron-sulfur (Fe–S) proteins. The distinction of the cell death pathway identified as cuproptosis has been demonstrated through the use of specific pharmacological inhibition of other known cell death pathways including ferroptosis (inhibited with ferrostatin-1) and apoptosis (inhibited with Z-VAD-FMK). The inhibition of these other cell death pathways failed to inhibit copper mediated cell death initiated with the use of a copper-specific ionophore. In contrast, the use of a copper chelator was shown to rescue cells from excess Cu2+-induced cell death.
The process of cuproptosis is linked to an alteration in the activity of several mitochondrial enzymes marking the mitochondrion as a major target of copper-induced cell death. The effect of excess copper on the mitochondria causes oxidative damage to the mitochondrial membrane and impaired function of enzymes in the TCA cycle. If electron transport is inhibited at complexes I and II the level of cuproptosis is significantly reduced.
The mechanism by which copper induces cell death involves a specific form of post-translational modification called lipoylation, a modification with lipoic acid. Only five proteins are known to be modified by lipoylation with three of the proteins being involved in the oxidation of pyruvate by the pyruvate dehydrogenase complex (PDHc) and the TCA cycle.
Two critical proteins, that are key regulators of copper toxicity, are involved in protein lipoylation. These proteins are mitochondrial ferredoxin 1 (FDX1) and lipoic acid synthetase (LIAS). Ferrodoxin 1 directly binds to lipoic acid synthetase, thereby regulating the activity of LIAS. The significance of these two proteins to the process of cuproptosis has been demonstrated by knocking out the genes in mice which results in inhibition of copper-induced cell death.
Lipoic acid synthetase, along with lipoyl (octanoyl) transferase 2 (encoded by the LIPT2 gene), and lipoyltransferase 1 (LIPT1) synthesize lipoic acid from the medium-chain fatty acid, octanoic acid. The synthesis of lipoic acid is directly coupled to the lipoylation of proteins. The five lipoylated proteins are the GCSH encoded H-protein of the glycine cleavage complex (GCC), the E2 subunit proteins of dehydrogenase complexes that includes the DLAT encoded protein of the pyruvate dehydrogenase complex (PDHc), the DLST encoded protein of the 2-oxoglutarate (α-ketoglutarate) dehydrogenase complex (OGDH), and the DBT encoded protein of the branched-chain keto acid dehydrogenase complex (BCKD). In addition to the E2 subunit of the PDHc, the PDHX encoded protein of this complex is also lipoylated.
Excess copper also disrupts the production of iron–sulfur (Fe–S) containing enzymes via a mechanism that involves inhibition of the mitochondrial assembly proteins. Most Fe–S proteins are required cofactors for enzymes involved in complexes of the electron transport chain.
Lipoylated proteins of the PDHc and the BCKD complex bind excess copper which results in their aggregation and loss of activity. The reduced pyruvate oxidation and TCA cycle activity causes a loss of energy production inducing mitochondrial stress and the activation of the process of cuproptosis.
In experimental conditions of gene knockout, several additional contributors to the sensitivity to cuproptosis have been identified. These genes include ATP7A (ATPase, Cu2+-transporting, alpha polypeptide), CDKN2A (cyclin dependent kinase inhibitor 2A), GLS (glutaminase), and MTF1 (metal regulatory transcription factor 1). The ATP7A encoded protein is a copper transporter involved in transport of dietary copper from intestinal enterocytes to the blood and cellular uptake of copper from the from the blood. Loss of function of ATP7A is the cause of Menkes disease. The CDKN2A encoded protein is a member of the INK4 (inhibitors of kinase 4) family of CDK inhibitors and as such is also identified as p16INK4A.
In contrast, enhanced activity of the transporter encoded by the SLC31A1 gene has been shown to be associated with intracellular copper accumulation and an increased likelihood of the induction of cuproptosis. The SLC31A1 encoded transporter is commonly referred to as copper transporter 1 (CTR1) and it functions in the uptake of dietary copper.
Inhibition of cuproptosis may represent a putative therapeutic target for the copper overload disease identified as Wilson disease. The excess copper deposition in the liver and brain in Wilson disease patients is the cause of cellular damage and the severe hepatic and neurological symptoms of the disease. Cuproptosis may be contributory to several neurodegenerative diseases, including Alzheimer disease (AD), Huntington disease (HD), and amyotrophic lateral sclerosis (ALS).
Oxeiptosis
Oxeiptosis refers to a reactive oxygen species (ROS)-sensitive, caspase independent, and non-inflammatory cell-death pathway. Reactive oxygen species include super oxide anion (O2•–), hydrogen peroxide (H2O2), hydroxyl radical (•OH), ozone (O3), singlet oxygen (1O2), and peroxy radicals (RO• and RO2•). Oxeiptosis is a pathway that is involved in protection against inflammation induced by ROS or ROS-generating agents. Oxeiptosis can induce apoptosis, autophagy, pyroptosis, necroptosis, and ferroptosis.
The process of oxeiptosis involves the activation of a signaling pathway consisting of KEAP1 (Kelch-like ECH associated protein 1), PGAM family member 5, mitochondrial serine/threonine protein phosphatase (PGAM5), and apoptosis inducing factor mitochondria associated 1 (AIFM1). This pathway is referred to as the KEAP1/PGAM5/AIFM1 pathway.
Several proteins contain Kelch (German for chalice) domains that are protein-protein interaction domains. This domain allows KEAP1 to interact with ubiquitin ligases of the Cullin family, specifically Cul3. Under normal physiological conditions KEAP1 ubiquitylates the transcription factor identified as NRF2 (nuclear factor erythroid 2-related factor 2) preventing it from entering the nucleus. NRF2 is encoded by the NFE2L2 gene. When Cys residues in KEAP1 become oxidized by increasing levels of ROS, NRF2 is released and migrates to the nucleus where it activates the expression of genes encoding proteins with cytoprotective functions. These NRF2-responsive genes contain an enhancer element termed To date there are over 250
Although the PGAM5 gene is a member of the phosphoglycerate mutase (PGAM) family of genes, the PGAM5 protein is a Ser/Thr phosphatase that is involved in mitochondrial health. The PGAM5 encoded protein also functions as a member of the histidine phosphatase family of enzymes. Histidine phosphatases are enzymes that have a His residue in the catalytic core that becomes phosphorylated during the course of the reaction.
Like NRF2, PGAM5 interacts with KEAP1 and when KEAP1 Cys residues are oxidized, PGAM5 is released and migrates into the mitochondria. Within the mitochondria, PGAM5 removes phosphate from its target substrates which can promote mitochondrial fusion and biogenesis thereby contributing to cell survival. Severe mitochondrial stress will lead to PGAM5 promoting mitochondrial fission and apoptosis leading to programmed cell death. One mechanism of PGAM5 triggering of apoptosis is via dephosphorylation of the AIFM1 protein which triggers its release from the mitochondria.
AIFM1 is a mitochondrial inner membrane-localized FAD-dependent NADH oxidase that functions to maintain the integrity of complex I of the electron transport chain (ETC). Within the mitochondria AIFM1 is also involved in the release of the pro-apoptotic proteins, cytochrome c and caspase-9. When AIFM1 is released from the mitochondria it migrates to the nucleus where it functions to triggers chromatin condensation and DNA fragmentation.