

Last Modified: April 10, 2026
Introduction
Very nearly all human proteins undergo some form of modification during and/or following their synthesis. Protein modifications can involve the relatively simple process of cleavage or, as in the case of the large majority of proteins, the modification can be one or more of a wide variety of co-and posttranslational modifications. Many co-translational modifications include carbohydrate addition and these types of reactions are detailed in the Glycoproteins: Synthesis and Clinical Consequences page.
Protein Activation via Proteolytic Cleavage
Most proteins undergo proteolytic cleavage following translation. The simplest form of this is the removal of the initiation methionine. Many proteins are synthesized as inactive precursors that are activated under proper physiological conditions by limited proteolysis. Pancreatic enzymes and enzymes involved in clotting are examples of the latter. Inactive precursor proteins that are activated by removal of polypeptides are termed proproteins. If a precursor protein is synthesized via association with the endoplasmic reticulum (ER), as described above, it is targeted to that location by the N-terminal signal sequence (signal peptide or leader peptide) which is proteolytically removed after the association with the ER. These latter proteins are referred to as preproteins. A protein that begins with a leader peptide and also must undergo further proteolysis to be functional is termed a preproprotein.
A complex example of post-translational processing of a preproprotein is the cleavage of prepro-opiomelanocortin (POMC) synthesized in the pituitary (see the Peptide Hormones page for discussion of POMC). This preproprotein undergoes complex cleavages, the pathway of which differs depending upon the cellular location of POMC synthesis.
Another example of a preproprotein is insulin. Since insulin is secreted from the pancreas it has a signal sequence (leader peptide) making it a preprotein. Following cleavage of the 24 amino acid signal peptide the protein folds into proinsulin. Proinsulin is further cleaved yielding active insulin (thus it is synthesized as a preproprotein) which is composed of two peptide chains linked together through disulfide bonds.
Still other proteins, that are enzymes, are synthesized as inactive precursors called zymogens. Zymogens are activated by proteolytic cleavage such as is the situation for several proteins of the blood clotting cascade.
Protein Methylation
Post-translational methylation of proteins occurs on nitrogen and oxygen atoms in various amino acids, primarily lysine and arginine. The activated methyl donor for these reactions is S-adenosylmethionine (SAM). The most common methylations are on the ε-amine of the R-group of lysine residues and the guanidino moiety of the R-group of arginine. Methylation of lysine residues in histones in the nucleosome is an important regulator of chromatin structure and consequently of transcriptional activity.
For more complete information on the functions of histone protein methylation and demethylation go to the Regulation of Gene Expression page.
Additional nitrogen methylations are found on the imidazole ring of histidine and the R-group amides of glutamate and aspartate. Methylation of the oxygen of the R-group carboxylates of glutamate and aspartate also takes place and forms methyl esters. Proteins can also be methylated on the thiol R-group of cysteine.
As indicated below, many proteins are modified at their C-terminus by prenylation near a cysteine residue in the consensus CAAX. Following the prenylation reaction the protein is cleaved at the peptide bond of the cysteine and the carboxylate residue of the cysteine is methylated by a prenylated protein methyltransferase.
Protein Methylation “Writers”
Humans express six large families of methyltransferases that methylate a wide array of substrates that in addition to proteins includes DNA, RNA, lipids, and small molecules. The six methyltransferase families are identified as the homocysteine methyltransferase family, the lysine methyltransferase family, the radical S-adenosylmethionine domain containing family, the seven-beta-strand (7BS) methyltransferase motif containing family, the SET domain containing family, and the SPOUT methyltransferase domain containing family.
The SPOUT nomenclature is derived from the identification of sequence homology between the SpoU and the TrmD methyltransferases, where Trm refers to tRNA methyltransferase. SpoU was originally identified as TrmH. Several of the seven-beta-strand methyltransferase motif containing family enzymes and SPOUT methyltransferase domain containing family enzymes methylate nucleotides in tRNA, mRNA, and DNA.
The homocysteine methyltransferase family contains three genes. These three genes include MTR which encodes methionine synthase (official name is 5-methyltetrahydrofolate-homocysteine methyltransferase), and the two betaine-S-homocysteine methyltransferase encoding genes, BHMT and BHMT2.
The lysine methyltransferase family contains 34 genes.
The radical S-adenosylmethionine domain containing family contains nine genes. This family of enzymes include enzymes that are involved in the synthesis of lipoic acid (LIAS) and the synthesis of the hypermodified guanosine [termed wybutosine(yW)] found in a phenylalanine tRNA.
The seven-beta-strand (7BS) methyltransferase motif containing family is composed of four subfamilies, two of which are themselves composed of subfamilies. The 7BS methyltransferase motif family is the largest of the methyltransferase families consisting of a total of 125 genes in humans. This methyltransferase family is often referred to as the Class I family. The enzymes in this large family methylate lysine and arginine residues in proteins, nucleotides in DNA and RNA, and various small molecules such as glycine and histamine.
The SET domain containing family is composed of 35 genes and one subfamily (PR/SET domain family) that itself contains 19 genes. Many of the SET domain containing family enzymes are lysine methyltransferase (KMT) enzymes. The major substrates of the SET domain family methyltransferases are the histones and various ribosomal proteins.
The SPOUT methyltransferase domain containing family contains 8 genes. The SPOUT family enzymes function in the methylation of RNA. These enzymes carry out the methylation of the N1 atom of guanosine, the N3 atom of uridine, and and the 2′-O of ribose.
Lysine Methylation Writers
Lysine methylation was originally thought to be a permanent covalent mark, providing long-term signaling, including the histone-dependent mechanism for transcriptional memory. However, it has become clear that lysine methylation, similar to other covalent modifications, can be transient and dynamically regulated by an opposing demethylation activity. Methylation of lysine residues affects gene expression not only at the level of chromatin modification, but also by modifying the activity of numerous transcription factors. Lysine methylation can result in monomethyl lysine, dimethyl lysine, or trimethyl lysine.
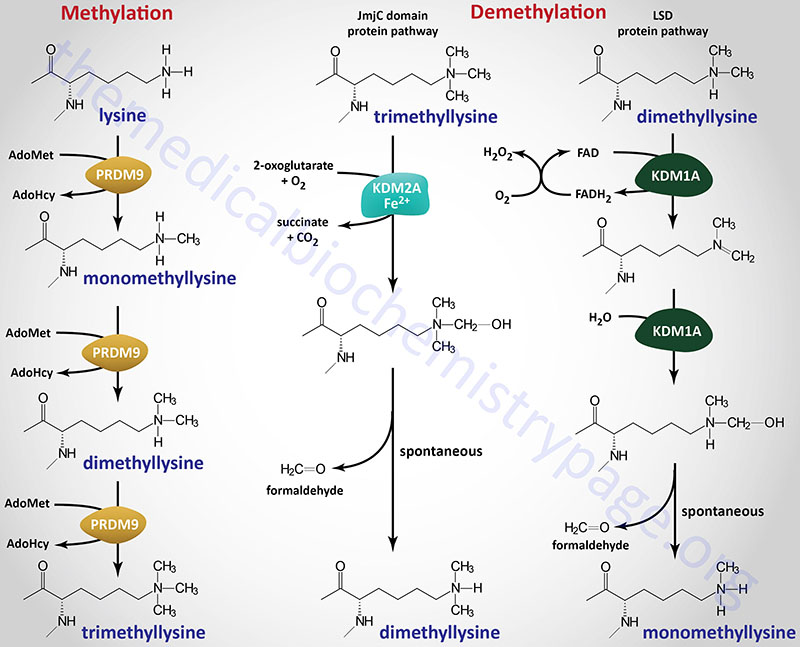
Within the context of protein methylation the lysine methyltransferases are organized into the lysine methyltransferase family (34 genes), the SET domain containing family (19 of the 35 genes in the family), and the 7BS protein lysine methyltransferase subfamily (16 genes). The 7BS protein lysine methyltransferase family is composed of 16 genes. These enzymes are a subfamily of the 7BS protein methyltransferase family which contains both lysine and arginine methyltransferase subfamilies. The 7BS protein methyltransferase family is itself a subfamily of the seven-beta-strand (7BS) methyltransferase motif containing family of genes.
Many of the lysine methyltransferase encoding genes use the KMT [lysine (K) MethylTransferase] nomenclature. Several of the histone lysine methyltransferases are also identified as HMTases (for Histone MethylTransferases). Not all of the human protein lysine methyltransferase encoding genes encode enzymes that methylate histones.
The enzymes that carry out histone lysine methylation are all members of the SET-domain-containing family of methyltransferases except for one enzyme, DOT1 (disruptor of telomeric silencing 1) like histone lysine methyltransferase which is encoded by the DOT1L gene.
Arginine Methylation Writers
Arginine methylation is found on approximately 0.5% of the arginine residues of the total protein complement of human tissues. Arginine methylation is known to occur on both nuclear and cytoplasmic proteins and is predominantly found in glycine- and arginine-rich (GAR) motifs. Histone arginine methylation, like histone lysine methylation, is known to regulate chromatin structure and consequently transcriptional activity.
Humans express 11 protein arginine methyltransferase (PRMT) expressing genes. These 11 genes belong to the 7BS protein arginine methyltransferase subfamily. The 7BS protein arginine methyltransferase enzymes are a subfamily of the 7BS protein methyltransferase family which contains both arginine and lysine methyltransferase subfamilies. The 7BS protein methyltransferase family is itself a subfamily of the seven-beta-strand (7BS) methyltransferase motif containing family of genes. The arginine methyltransferases are classified into three groups defined as Types I, II, and III. These classifications are determined by the type of methylation a particular PRMT catalyzes.
Arginine methyltransferases catalyze a methyl transfer reaction similar to that of lysine methyltransferases (KMT) where the methyl donor is S-adenosylmethionine (AdoMet) and the products are S-adenosylhomocysteine (AdoHcy) and methylated arginine. Three different types of methylated arginine residues have been identified in mammalian cells, monomethylarginine (MMA), asymmetric dimethylarginine (ADMA: methyl groups attached to the same guanidino nitrogen), and symmetric dimethylarginine (SDMA; symmetrical addition of methyl groups on each of the guanidino nitrogen atoms).
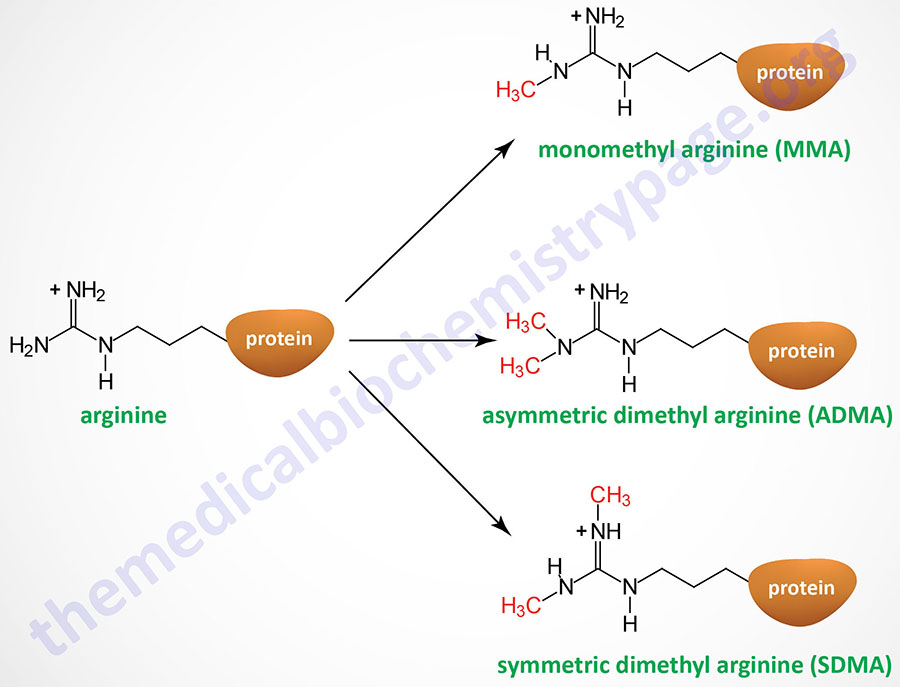
All three classes of PRMT can generate MMA. Type I PRMT are also responsible for generating ADMA from MMA. Type II PRMT also responsible for generating SDMA from MMA. Type III PRMT, which consists of a single enzyme encoded by the PRMT7 gene, only catalyzes the formation of MMA.
Histidine Methylation Writers
Histidine methyltransferases are identified by the acronym: PHMT (protein histidine methyltransferase). Similar to all methyltransferases the PHMT utilize S-adenosylmethionine (AdoMet) as the methyl donor. Methylation of histidine occurs on either of the two nitrogen atoms in the imidazole ring and can result in the generation of N1-methylhistidine (1MeH) and N3-methylhistidine (3MeH).
Within the large superfamily of methyltransferases, three have been shown to be PHMT enzymes encoded by the SETD3, METTL9, and METTL18 genes. The SETD3 gene encodes the enzyme identified as SET domain containing 3, actin N3(tau)-histidine methyltransferase. The METTL9 gene encodes the enzyme identified as methyltransferase 9, His-X-His N1(pi)-histidine. The METTL18 gene encodes the enzyme identified as methyltransferase 18, RPL3 N3(tau)-histidine.
The SETD3 enzyme belongs to the SET domain family of methyltransferases. Both the METTL9 and METTL18 enzymes belong the the 7BS methyltransferase motif containing family.
SETD3 methylates the N3 (3MeH) of His73 in actin which is likely to be the sole substrate for this PHMT. The consequences of SETD3 activity are regulation in actin polymerization. METTL18 generates 3MeH in the large ribosomal protein, RPL3, on His243. The consequences of METTL18 activity are regulation of ribosomal assembly and, therefore, the dynamics of protein synthesis. The METTL9 PHMT methylates the N1 (1MeH) numerous substrates that possess the H-x-H (HxH) motif where x refers to any small amino acid. One important target for METTL9 is the protein S100A9.
Protein Methylation “Readers”
Proteins that recognize the state of protein modification, such as lysine methylation, are often referred to as “readers”. The largest class of methylation readers are the proteins that contain a domain termed the plant homeodomain (PHD) finger. The PHD finger is a zinc-finger (specifically Cys4-His-Cys3) domain originally identified in the plant homeodomain-containing proteins HAT3.1 (Arabidopsis thaliana) and ZmHox1A (Zea mays: maize). This domain is distinct from another protein domain with the same acronym, the proly hydroxylase domain (PHD). Humans express at least 90 genes that encode proteins containing the PHD finger domain with there likely being more than 120 proteins in this family.
The PHD finger proteins are often found in complexes with histone methyltransferases (writers) or histone demethylases (erasers). The PHD finger proteins generally recognize unmodified or methylated lysines with the majority of the proteins in the family binding to histone H3 tails either methylated at K4 (H3K4), or unmodified in that position. Some PHD proteins are readers of trimethylated histone H3, H3K9 (H3K9me3) and H3K3 (H3K36me3).
Some members of the PHD finger protein family are themselves histone modifying enzymes such as the writers lysine acetyltransferase 6A (encoded by the KAT6A gene) and lysine methyltransferase 2A (encoded by the KMT2A gene), and the eraser lysine demethylase 2A (encoded by the KDM2A gene).
Clinical significance is associated with the PHD finger proteins given that several genes have been found to be mutated in breast cancers and leukemias.
In addition to the PHD family of methylation readers there are at least seven additional families of methylation reader proteins. These seven family are the chromo domain family, the tudor domain family, the PWWP domain family, the MBT domain family, the BAH domain family, the SPIN protein family, and the CW domain family. The chromodomain, tudor, PWWP, and MBT family proteins constitute what is referred to as the Royal Family of methylation readers due to the fact that these proteins bind both methylated DNA and methylated lysine in histone proteins.
The chromo domain (chromatin organization modifier) is a structural domain consisting of 40-50 amino acid residues that is responsible for the recognition and interaction with methylated residues, particularly methylated lysine residues in histone proteins.
The Tudor domain was originally identified in the Tudor protein which is a Drosophila protein involved in early embryonic development. Mutations in the gene encoding Tudor are lethal and the gene was therefore, given the name for the Tudor King Henry VIII and the several miscarriages experienced by his wives. Tudor domain proteins bind to methylated lysine and arginine residues.
The PWWP domain is so named because of the conserved Pro-Trp-Trp-Pro sequence motif that resides in a domain that encompasses 90-130 amino acids. The PWWP domain was first described as a structural motif in the protein encoded by the gene NSD2 (nuclear receptor binding SET domain protein 2), mutations in which are responsible for Wolf–Hirschhorn syndrome.
The MBT (malignant brain tumor) domain was originally identified in the protein encoded by the lethal(3) malignant brain tumor [l(3)mbt] gene in Drosophila. This protein contains three tandem repeats of a motif consisting of 99–103 amino acids which was termed the “mbt-repeat”.
The BAH (Bromo-Adjacent Homology) domain is a domain in several proteins that serves as a protein-protein interaction domain. Some BAH domain containing proteins interact with histone deacetylases and thus, contribute to transcriptional silencing.
The SPIN family proteins are so named for spindlin referring to a protein that was originally shown to interact the mitotic spindle during gametogenesis.
The CW (Cys-Trp) domain is a zinc binding domain, composed of approximately 50-60 amino acid residues with four conserved cysteine (C) and two to four conserved tryptophan (W) residues.
Protein Methylation “Erasers”
Numerous enzymes catalyze lysine demethylation reactions with one of the largest families of methylation erasers being the Jumonji C (JmjC) domain containing demethylases. All of the JmjC methylation erasers are members of a large family of at least 80 enzymes that are 2-oxoglutarate and Fe2+-dependent dioxygenases (2OG-oxygenases).
All enzymes that demethylate methylated lysines, particularly in histone proteins, are identified as KDM family enzymes where KDM stands for lysine (K) DeMethylase. Humans express 25 genes in the KDM family. There are currently eight KDM subfamilies of enzymes divided based upon factors such as substrate preference, presence of certain domains, and cofactor requirements.
Currently only a single arginine demethylase has been identified. This enzyme is identified as the Jumonji domain containing 6, arginine demethylase and lysine hydroxylase which is encoded by the JMJD6 gene. The JMJD6 encoded protein is related to the JmjC family of hydroxylases which, like the JmjC family demethylases, are members of the large family of 2OG-oxygenases.
Protein Acetylation
Post-translational acetylation of proteins occurs on the ε-amine of lysine residues the same as for the methylation of lysine residues in proteins. In addition, a large number of proteins (more than 80% of human proteins) are acetylated on the N-terminal amino acid.
The enzymes that catalyze protein acetylation of lysine residues are classified as lysine (K) acetyltransferases and denoted by the nomenclature KAT. Humans express 17 genes encoding KAT enzymes. The activated acetyl donor for the KAT enzymes is acetyl-CoA. The role of acetyl-CoA in the acetylation of proteins places this post-translational processing event at the crossroads of metabolic regulation and the control of gene expression.
Physiological and pathophysiological conditions that result in increases or decreases in the production and utilization of acetyl-CoA will, therefore, have profound effects on the ability of KAT enzymes to carry out their functions.
Lysine Acetylation
Acetylation of lysine residues in histones in the nucleosome is an important regulator of chromatin structure and consequently of transcriptional activity. Like the reversibility of lysine methylation, protein lysine acetylation is also reversible. The enzymes that carry out removal of the acetyl group are broadly classified into two primary groups. One group is identified as the histone deacetylases (HDAC), which are Zn2+-dependent enzymes and the other group is identified as the sirtuins (SIRT) which are NAD+-dependent protein deacetylases. More than 1,750 proteins in human tissues have been shown to be modified by acetylation. Greater detail on histone acetylation-deacetylation can be found in the Regulation of Gene Expression page. The discussion here will focus on metabolic regulation via reversible acetylation.
Protein lysine acetylation is observed on proteins in most all compartments of the cell. Recent evidence has demonstrated that numerous enzymes, that control a vast array of metabolic processes, have their activity modulated by reversible lysine acetylation. Within the liver, nearly 1,000 different proteins (not including nuclear proteins) have been shown to be acetylated with many of the proteins functional in the processes of metabolic regulation. Of these nearly 1,000 proteins, more than 150 are found in the mitochondria of hepatocytes. An astounding outcome of the work on metabolic regulation, via protein acetylation, is that very nearly all of the enzymes involved in glycolysis, glycogen metabolism, gluconeogenesis, the TCA cycle, fatty acid oxidation, the urea cycle, and nitrogen metabolism, and have been shown to be acetylated. In addition, several enzymes involved in oxidative phosphorylation and amino acid metabolism have also been found to be acetylated.
The acetylation of metabolic enzymes results in alterations in their activities by several different mechanisms. Acetylation can lead to subsequent ubiquitylation and proteasomal degradation of the modified protein. Acetylation can also result in destruction of the modified protein via the lysosomes. Protein degradation is not the only mechanism whereby lysine acetylation can be used to regulate an enzymes level of activity. Numerous enzymes, including metabolic enzymes, that are acetylated have altered catalytic activity. Acetylation can lead to neutralization of an active site lysine or the acetylation can lead to blockade of the action of an allosteric activator. Numerous other lysine acetylation-mediated effects on enzyme activity have been documented including the blocking of substrate binding, blocking of metabolite binding, and modifying the subcellular localization of an enzyme.
Table of Several Metabolic Enzymes Regulated by Reversible Acetylation
| Enzyme Name | Gene | Acetylase | Deacetylase | Comments |
| Acetyl-CoA acetyltransferase 1 | ACAT1 | unknown | SIRT3 | mitochondrial enzyme involved in ketone body utilization; major activity is the cleavage of acetoacetyl-CoA into two acetyl-CoA units; acetylation down-regulates the activity of the enzyme; K260 and K265 deacetylated by SIRT3 but K187 is not |
| Acyl-CoA dehydrogenase, long chain | ACADL | unknown | SIRT3 | mitochondrial fatty acid β-oxidation enzyme; acetylation down-regulates the activity of the enzyme |
| Acyl-CoA synthetase 1 | ACSL1 | unknown | SIRT3 | major liver and adipose tissue enzyme involved in the activation of fatty acids for β-oxidation; enzyme contains at least 15 sites of acetylation that are acetylated differentially dependent upon physiological status; acetylation of K285 is known to down-regulate the activity of the enzyme |
| Aldehyde dehydrogenase 2 | ALDH2 | unknown | SIRT3 | the mitochondrial aldehyde dehydrogenase; multiple sites of acetylation; acetylation increase the activity of the enzyme; K370 is deacetylated by SIRT3 but K453 is not |
| acyl-CoA synthetase short chain family member 1 | ACSS1 | unknown | SIRT3 | mitochondrial enzyme; also identified as AceCS2; catalyzes conversion of acetate to acetyl-CoA; important in energy homeostasis during periods of fasting; acetylation results in down-regulation of enzyme activity |
| acyl-CoA synthetase short chain family member 2 | ACSS2 | KAT3A (CBP) | SIRT1 | cytoplasmic enzyme; also identified as AceCS1; catalyzes conversion of acetate to acetyl-CoA; acetate stimulates interactions between ACSS2, CBP [derived from CREB (cAMP-response element binding protein)-binding protein], and the hypoxia induced factor, HIF-2 (see the Hypoxia and Metabolism page for more details on the hypoxia induced pathway); CPB protein is encoded by the CREBBP gene which is also identified by the standard KAT nomenclature as KAT3A; acetylation of ACSS2 results in down-regulation of enzyme activity by interference with the active site; ACSS2 generates lactoyl-CoA from lactate which is utilized for protein lysine lactylation |
| argininosuccinate lyase | ASL | unknown | unknown | urea cycle enzyme; acetylation results in down-regulation of enzyme activity by interference with the active site |
| carbamoylphosphate synthetase I | CPS1 | unknown | SIRT5 | urea cycle enzyme; acetylation results in down-regulation of enzyme activity |
| carnitine palmitoyltransferase 2, CPT2 | CPS1 | unknown | unknown | mitochondrial enzyme involved in transport of activated fatty acids into the mitochondria for β-oxidation; consequences of acetylation of four sites (K104, K453, K537, and K544) yet to be determined |
| glyceraldehyde-3-phosphate dehydrogenase | GAPDH | KAT2B | HDAC5 | glycolytic enzyme; KAT2B was originally identified as PCAF (p300/CBP-associated factor); lysine residues K117, K227, K251 and K254 are acetylated; acetylation of K227 causes an interaction of GAPDH and one of the seven in abstentia (Sia) homolog (SIAH) ubiquitin ligases resulting in cytoplasmic to nuclear translocation; the Sia gene was originally identified in Drosophila as being required for the specification of R7 cell fate in the eye; humans express three SIAH genes identified as SIAH1, SIAH2, and SIAH3; acetylation of K254 results in increased enzyme activity in response to increased glucose concentration |
| glutamate dehydrogenase | GLUD1 | unknown | SIRT3 | major enzyme of overall nitrogen homeostasis and regulator of energy status; |
| glutaminase | GLS2 | unknown | unknown | enzyme involved in overall nitrogen homeostasis; acetylation of K329 results in down-regulation of enzyme activity |
| 3-hydroxy-3-methylglutaryl CoA synthase 2 | HMGCS2 | unknown | SIRT3 | mitochondrial enzyme involved in synthesis of the ketone bodies; acetylation results in down-regulation of enzyme activity; K310 is deacetylated by SIRT3 but K354 is not |
| isocitrate dehydrogenase 2 | IDH2 | unknown | SIRT3 | mitochondrial enzyme involved in the production of NADPH in response to oxidative stress; acetylation results in down-regulation of enzyme activity |
| malate dehydrogenase 2 | MDH2 | unknown | unknown | mitochondrial enzyme of the TCA cycle; lysines K185, K301, K307, and K314 are acetylated; acetylation results in up-regulation of enzyme activity; acetylation of MDH2 increases under conditions of increased fatty acid intake |
| ornithine transcarbamylase | OTC2 | unknown | SIRT3 | mitochondrial enzyme involved in urea cycle; lysine K88 in the active site is a primary target for acetylation; acetylation of K88 inhibits enzyme activity by decreasing affinity for substrate, carbamoyl phosphate; mutation of K88 to asparagine (K88N mutation) found in some patients suffering from OTC deficiency |
| phosphoenolpyruvate caboxykinase 1 | PCK1 | EP300 | SIRT2 | cytoplasmic form of the enzyme (also known as PEPCK-c) involved in gluconeogenesis; the EP300 gene encodes the p300 protein (adenovirus E1A binding protein p300) that is a close relative of the CBP acetyltransferase; EP300 is also identified by the KAT nomenclature as KAT3B; CPB protein is encoded by the CREBBP gene which is also identified by the standard KAT nomenclature as KAT3A; acetylation of PEPCK-c results in down-regulation of enzyme activity via interaction with the UBR5 ubiquitin ligase (ubiquitin ligase E3 component N-recognin 5) |
| phosphoglycerate mutase 1 | PGAM1 | unknown | SIRT1 | cytoplasmic enzyme involved in glycolysis; at least nine lysines shown to be acetylated in PGAM1; the major sites of acetylation are K251, K253, and K254; acetylation results in up-regulation of enzyme activity |
| pyruvate kinase, muscle isoform | PKM | KAT2B | unknown | cytoplasmic enzyme involved in glycolysis; the PKM gene produces two PKM isoforms (PKM1 and PKM2) as a result of alternative mRNA splicing; expression of the gene is induced in proliferating cells and all human cancers; expression of PKM2 and synthesis of the PKM2 isoform of the enzyme results in reduced oxidation of glucose to pyruvate resulting in the accumulation of glycolytic intermediates which promotes the production of macromolecules from glucose carbons; acetylation of K305 is stimulated in the presence of high glucose; acetylation results in down-regulation of enzyme activity as a result of the lysosomal degradation pathway referred to as chaperone-mediated autophagy, CMA |
| succinate dehydrogenase complex subunit A | SDHA | unknown | SIRT3 | mitochondrial enzyme that is one of four subunits of the SDH complex; involved in the TCA cycle and in oxidative phosphorylation; acetylation results in down-regulation of enzyme activity |
| superoxide dismutase 2 | SOD2 | unknown | SIRT3 | mitochondrial matrix enzyme involved in removal of super oxide anions; catalyzes reduction of super oxide anion to hydrogen peroxide; acetylation results in down-regulation of enzyme activity |
| sphingosine kinase 1 | SPHK1 | p300/CBP | unknown | cytoplasmic enzyme involved in synthesis of the bioactive lipid sphingosine-1-phosphate, S1P; acetylation results in stabilization of the protein leading to up-regulation of enzyme activity |
N-Terminal Acetylation
The acetylation of proteins on the N-terminal amino acid occurs in greater than 80% of all human proteins. The modification is identified as Nt-acetylation. In most proteins where the initiator methionine remains at the N-terminus, this amino acid is acetylated. When the initiator methionine is removed, as is the case for all secreted, transmembrane, and glycoproteins due to removal of the leader peptide in the lumen of the ER, the protein can still be Nt-acetylated. The most commonly occurring amino acids at the N-terminus that are acetylated are alanine (A), serine (S), cysteine (C), threonine (T), and valine (V).
The enzymes that incorporate an acetyl group onto the N-terminal amino acid of human proteins are referred to as N-acetyltransferases (NAT). These enzymes represent a distinct family of acetyltransferases that distinguishes them from the lysine acetyltransferases (KAT). Like the KAT enzymes, the NAT enzymes utilize acetyl-CoA as the acetyl donor for the acetyltransferase reaction.
There are seven NAT complexes in human cells identified as NatA–NatF and NatH. The NatA, NatB, NatC, NatD, and NatE complexes are associated with the ribosomal machinery where they function in the process of co-translational N-terminal acetylation. The NatF complex is associated with the Golgi and the NatH complex is associated with actin filaments and both of these complexes function in the process of post-translational N-terminal acetylation.
Functional NAT enzymes are heterotrimeric complexes where the α-subunit of the complex is the catalytic protein. The catalytic α-subunits are encoded by a family of 13 genes identified as N-alpha-acetyltransferases (NAA). Eight of the NAA gene, NAA10, NAA11, NAA20, NAA30, NAA40, NAA50, NAA60, and NAA80 encode catalytic subunits. Five of the NAA genes, NAA15, NAA16, NAA25, NAA35, and NAA38 encode auxiliary subunits.
The NatA complex can be generated through the association of different NAA proteins that includes NAA10, NAA11, NAA15, NAA16, and NAA50 where two are catalytic subunits (NAA10 and NAA50) and two are auxiliary subunits (NAA15 and NAA16). The NatA complex predominantly acetylates small N-terminal amino acids that are exposed as a result of the removal of the initiator methionine. An addition protein found associated with the NatA complex is encoded by the HYPK (huntingtin interacting protein K) gene. The HYPK protein serves as a chaperone for the Huntingtin (Htt) protein allowing it to ne Nt-acetylated. Evidence indicates that HYPK is essential for the normal activity of the NatA complex, not just for the acetylation of Htt. The activity of the NatA complex accounts for almost 40% of all Nt-acetylated proteins.
The NatB complex contains the catalytic NAA20 encoded protein and the auxiliary NAA25 encoded protein. The activity of the NatB complex accounts for just over 20% of all Nt-acetylated proteins.
The NatC complex is composed of the catalytic (NAA30 encoded) protein and the two auxiliary (NAA35 or NAA38 encoded) proteins, where the NAA35 protein is the ribosomal anchor for the complex.
The NatD, NatE, and NatF complexes each contain a single NAA protein, NAA40, NAA50, and NAA60, respectively. The NatH complex consists of the NAA80 protein.
The NatB, BatC, NatE, and NatF complexes acetylate the N-terminal Met residues of proteins. NatB shows preference for proteins that contain acidic or amidic amino acids after the N-terminal Met residue. The NatC, NtE, and NatF complexes acetylate N-terminal Met residues when it is followed by hydrophobic or amphipathic amino acids.
The attachment of an acetyl group to the N-terminus of a protein creates a hydrophobic domain that plays a role in the folding, localization, stability, and various interactions of the modified protein. N-terminal acetylation is required for the regulation of a wide array of cellular processes that includes metabolism, differentiation, proliferation, stress responses, and migration.
In many cases, the presence of the Nt-acetylation creates a specific degradation signal referred to as a degron, specifically the Ac/N-degron. This signal is most often N-terminally acetylated Met, Ser, Val, Ala, Thr, or Cys. The presence of the degron signal then targets the protein for ubiquitylation via the ubiquitin-dependent N-end rule pathway. The ubiquitylated proteins are then degraded in the proteasome. Components of the N-end-rule pathway are referred to as N-recognins. It is the N-recognins that are the ubiquitin ligases (UBR: for ubiquitin ligase E3 component N-recognin) that ubiquitylate the Nt-acetylated protein.
The primary human E3 ubiquitin ligase that recognizes Ac/N-degron is encoded by the MARCHF6 (membrane associated ring-CH-type finger 6) gene. In addition to the MARCHF6 gene, humans express seven UBR family ubiquitin E3 ligases (UBR1–UBR5, UBR7, and FBXO11).
When proteins are N-terminally acetylated by the NatC complex they are protected from degradation via the Ac/N-degron pathway. Conversely, if these proteins are not correctly acetylated by the NatC complex they are targeted for degradation by ubiquitin ligases of the N-recognin family. Experiments performed in the fruit fly, Drosophila melanogaster, have demonstrated that the NatC-mediated N-terminal acetylation pathway is associated with longevity.
When the N-terminal amino acid that is acetylated is a cysteine it can be oxidized by nitric oxide (NO) followed by arginine attachment via the action of arginyltransferase 1 encoded by the ATE1 gene.
An example of a metabolic enzyme that is targeted for ubiquitylation via the N-recognin pathway is the cytosolic phosphoenolpyruvate carboxykinase (PEPCK-c) as indicated in the Table above. In the case of PEPCK-c the ubiquitin ligase is UBR5.
Protein Lysine Acylation
Post-translational acylation of proteins occurs on the ε-amine of lysine residues the same as for the acetylation and methylation of lysine residues in proteins. A large number of different protein lysine acylation events have been identified with many being catalyzed by enzymes and several resulting from non-enzymatic attachment. Many protein lysine acylations have been identified on histone proteins as well as numerous other non-histone proteins.
The various protein lysine acylations include butyrylation (Kbu), crotonylation (Kcr), β-hydroxybutyrylation (Kbhb), lactylation (Kla), malonylation (Kmal), propionylation (Kpr), succinylation (Ksucc), 2-hydroxyisobutyrylation (Khib), and glutarylation (Kglu). Lysine lactylation is also termed lactoylation.
Protein lysine acylation involves acyl-CoA molecules as the activated substrates. Many of these protein lysine acylations are covered in detail in the Histone Modifications, Chromatin Structure, Transcriptional Regulation section of the Regulation of Gene Expression page.
Although the majority of protein lysine acylation occurs through the action of enzymes, some of the acyl-CoAs, such as succinyl-CoA and glutaryl-CoA, undergo spontaneous intramolecular catalysis to form reactive intermediates that non-enzymatically modify lysine residues. Additional acyl-CoA molecules that have been shown to be involved in non-enzymatic protein lysine acylation are 3-hydroxy-3-methylglutaryl(HMG)-CoA (HMGylation), 3-methylglutaconyl-CoA (MGcylation), and 3-methylglutaryl-CoA (MGylation).
Protein Lysine Butyrylation (Kbu)
Colonic bacteria generate short-chain fatty acids (SCFA) through fermentation of soluble fiber. These SCFA include acetate, propionate, and butyrate which are absorbed by colonocytes. Metabolically the gut bacteria-derived SCFA can be used for oxidation or diverted into the ketogenesis pathway. In addition to hepatocytes, gut epithelial cells are the only other cell to express the HMGCS2 gene allowing them to contribute to ketone synthesis. However, gut-derived SCFA also exert other important cell signaling effects. Butyrate has been shown to inhibit the activity of histone deacetylases (HDAC).
Gut butyrate is transported into intestinal epithelial cells via several transporters including SLC16A1 (commonly identified as monocarboxylate transporter 1, MCT1), SLC16A3 (commonly identified as monocarboxylate transporter 4, MCT4), SLC5A8 (also known as sodium-coupled monocarboxylate transporter 1, SMCT1), and ABCG2 (also known as breast cancer resistance protein, BCRP). Most of the gut-derived butyrate is metabolized by intestinal epithelial cells.
Butyrate that enters the portal circulation is taken up by hepatocytes and metabolized such that the circulating levels are generally quite low. Within intestinal epithelial cells and hepatocytes butyrate is converted to butyryl-CoA through the actions of acyl-CoA synthetase 2, encoded by the ACSS2 gene. However, there is conflicting data regarding validity of ACSS2 as functioning butyryl-CoA generating enzyme.
Like all protein lysine acylations, butyryl-CoA represents the substrate for lysine butyrylation (Kbu) of histone proteins as well as other non-histone proteins. Butyryl-CoA is generated via mitochondrial β-oxidation of fatty acids and also via mitochondrial fatty acid synthesis.
Mitochondrial butyryl-CoA is converted to butyryl-carnitine by carnitine O-acetyltransferase (encoded by the CRAT gene). The transport of butyryl-carnitine from the mitochondria to the cytosol is carried out carnitine acylcarnitine translocase, CACT. The CACT transporter is a member of the SLC family of transporters and as such is encoded by the SLC25A20 gene. The carnitine acylcarnitine translocase is located in the inner mitochondrial membrane where it facilitates acylcarnitine transport across the outer and inner mitochondrial membranes in exchange for free carnitine. Succinate is transported out of the mitochondria via the action of SLC25A10.
Accumulation of butyryl-CoA is characteristic of short-chain acyl-CoA dehydrogenase deficiency (SCADD). Indeed, measurement for elevated plasma butyryl-carnitine is a diagnostic feature in patients with SCADD.
Histone and non-histone protein lysine butyrylation is carried out via the actions of several acetyltransferases including p300, CBP, GCN5, PCAF, and MOF. Removal of lysine butyrylation is most likely the result of the sirtuins, SIRT1, SIRT2, and SIRT3.
Protein Lysine β-Hydroxybutyrylation (Khbh)
Like butyrate, the ketone, β-hydroxybutyrate (BHB), has also been shown to inhibit the activity of HDAC. The effects of β-hydroxybutyrate-mediated HDAC inhibition are enhanced expression of genes that reduce the level of oxidative stress.
In addition to altering the patterns of gene expression through modification of HDAC activity, β-hydroxybutyrate (BHB) can alter gene expression patterns by serving as a direct modifier of lysine residues in histones and many other non-histone proteins resulting in lysine β-hydroxybutyrylation. The effects histone β-hydroxybutyrylation on gene expression represents a novel form of epigenetic control. The level of histone β-hydroxybutyrylation is similar to the level of the more well studied epigenetic modification, histone acetylation.
In order for BHB to be utilized for lysine β-hydroxybutyrylation it must first be activated by CoA attachment. The most likely candidate enzyme for this reaction is acyl-CoA synthetase short chain 2, encoded by the ACSS2 gene. The β-hydroxybutyrylation reaction is catalyzed by the acyltransferase identified as histone acetyltransferase p300, encoded by the EP300 gene. This enzyme is also responsible for acetylation, propionylation, and crotonylation of numerous proteins. Although EP300 does indeed carry out lysine β-hydroxybutyrylation there are likely to be additional acetyltransferase involved in this important post-translational modification. Removal of BHB from sites of lysine β-hydroxybutyrylation is most likely catalyzed by histone deacetylases with HDAC1 and HDAC2 being the most likely enzymes.
An important consequence of histone β-hydroxybutyrylation is altered gene expression profiles in the liver. Experiments have shown that increases β-hydroxybutyrylation in hepatocytes occur in response to prolonged fasting. These effects of BHB are found to be associated with starvation-responsive genes that effectively couples ketogenic metabolism with the control of gene expression.
Histone lysine β-hydroxybutyrylation has been shown to be associated with changes in expression of numerous genes such as the gene for the transcriptional co-activator, PGC-1β (gene symbol: PPARGC1B) which is itself involved in the regulation of expression of numerous genes involved in energy homeostasis, the insulin receptor substrate 2 (IRS2) gene whose encoded protein is involved in insulin signaling, and the carnitine palmitoyltransferase 1A (CPT1) gene whose encoded protein regulates the ability of the mitochondria to oxidize long-chain fatty acids.
Within the liver, in addition to histones, more than 250 proteins have been identified as targets for lysine β-hydroxybutyrylation. These proteins are involved in fatty acid and amino acid metabolic pathways, one-carbon metabolism, and pathways of cellular detoxification. Genes that are expressed in the liver in response to starvation have been identified as associated with β-hydroxybutyrylation of lysine 9 in histone H3 (H3K9bhb).
Protein Lysine Lactylation (Kla)
Glycolysis serves as the metabolic pathway for the generation of lactate and its production is a balance between glycolysis and mitochondrial metabolism. Conditions such as hypoxia and bacterial infection induce the production of lactate via glycolysis. The role of intracellular lactate in histone, and other protein, modification was demonstrated by inhibition of the pyruvate dehydrogenase complex (PDHc) and inhibition of lactate dehydrogenase (LDH) which results in reduced levels of protein lactylation (also referred to as lactoylation). Histones, and many other proteins, undergo lysine lactylation by both enzymatic and non-enzymatic mechanisms.
Because studies have identified that protein lactylation can occur non-enzymatically, and that the product of that process consists of the D-isomer of lactate, a nomenclature has been adopted to distinguish these two types of post-translational modification. The products of enzymatic lactylation consist of the L-isomer and so it is more correct to designate the modified Lys residues as KL-la. The non-enzymatically lactylated Lys residues are designated as KD-la
The clinical significance of protein lactylation is demonstrated by the fact that one of the hallmarks of the metabolic changes associated with cancer is the increased metabolism of glucose to lactate. Indeed, in many cancers the expression of the LDHA gene is significantly upregulated. The increased levels of LDH contribute to increased lactate production and consequently an increase in protein lactylation.
Recent studies have demonstrated that production of lactoyl-CoA (specifically the stereoisomeric L-lactoyl-CoA form) from lactate is catalyzed by an enzyme of the acyl-CoA synthetase family, specifically the enzyme encoded by the ACSS2 (acyl-CoA synthetase short-chain family member 2) gene. Another recently identified lactoyl-CoA synthetase activity is a complex of the GTP-specific form of succinyl-CoA synthetase (GTPSCS; encoded by the SUCLG2 gene) of the TCA cycle and the histone acetyltransferase, p300. This complex lactylates histone H3 at lysine 18 (H3K18la).
When cells in culture are irradiated to induce double-strand DNA breaks the level of protein lysine lactylation significantly increases. Recently it has been shown that an increase in the lactylation of lysine (specifically K388) in the nibrin protein (encoded by the NBN gene) correlates to poor cancer survival. The nibrin protein (also identified as NBS1) is involved in the process of double-strand break repair and increased lactylation of the protein enhances DNA repair. This function of nibrin results in resistance of cancer cells to the effects of DNA damage-inducing chemotherapeutics and thus, correlates to the observed poor survival rates.
With respect to the nibrin protein, the KAT5 (lysine acetyltransferase 5) encoded protein (also known as TIP60: Tat-Interactive Protein 60kDa) has been shown to be the lysine lactyltransferase and the histone deacetylase encoded by the HDAC3 gene, has been found to be the de-lactylase.
Another member of the lysine acetyltransferase (KAT) family that has been shown to function as a lactyltransferase is the KAT2A encoded enzyme which was originally identified as GCN5. The MYST family lysine acetyltransferase, KAT5 (TIP60: Tat-Interactive Protein, 60 kDa, where Tat is a gene in the HIV-1 genome) is also known to be a lactylgtransferase. The activities of ACSS2 and KAT2A function in concert to lactylate histone H3 resulting in altered expression of several genes whose encoded proteins allow tumor cells to escape immune system detection.
Enzymatic histone lysine lactylation has also been shown to be carried out by the histone acetyltransferase (HAT), p300/CBP. Histone lysine lactylation was initially identified in histone H3. In a model of bacterial challenge with macrophages in culture it has been shown that over 1200 genes can be identified associated with lactylation of histone H3. In addition to histone H3, histone H4 and H2B have also been found to be lactylated.
Two of the class II aminoacyl-tRNA synthetases, encoded by the AARS1 and AARS2 genes, have been shown to function not only in the process of amino acid activation for translation, but also in the process of protein lysine lactylation. The AARS1 gene encodes the cytoplasmic alanyl-tRNA synthetase 1, whereas the AARS2 gene encodes the mitochondria-localized alanyl-tRNA synthetase 2. The ability of these two enzymes to function as lactylation writers stems, in part, from the fact that alanine and lactate are structurally very similar.
Several members of the histone deacetylase family, including HDAC1, HDAC2, and HDAC3 are known to function as lactylation erasers. Enzymes of the SIRT family of NAD+-dependent deacetylases (SIRT1, SIRT2, and SIRT3) have also been shown to function as lactylation erasers.
The non-enzymatic lactylation of Lys residues involves S-D-lactylglutathione (D-lactyl-GSH; also abbreviated LGSH) resulting in a post-translational modification referred to as lysine D-lactylation (KDla). This process is initiated by the glycolytic intermediates, glyceraldehyde-3-phosphate (G3P) and dihydroxyacetone phosphate (DHAP). Both G3P and DHAP spontaneously eliminate their phosphate generating methylglyoxal (MGO). The enzymes glyoxalase 1 (Glo1) and glyoxalase 2 (Glo2) detoxify MGO by converting it to D-lactate with the use of glutathione (GSH) forming the intermediate, S-D-lactylglutathione (D-lactyl-GSH). D-lactyl-GSH non-enzymatically modifies Lys residues forming D-lactyllysine residues. The primary targets for Lys D-lactylation are glycolytic enzymes, the consequences of which are inhibition of their activity.
Metabolic enzymes have also been shown to be modified by lysine lactylation such as aldolase A (fructose-1,6-bisphoshate aldolase; encoded by the ALDOA gene), fatty acid synthase (FAS), carnitine palmitoyltransferase 2 (CPT2), acyl-CoA synthetase long-chain family member 1 (ACSL1), HADHA and HADHB. HADHA and HADHB encode the α-subunits and β-subunits of mitochondrial trifunctional protein (MTP).
Lactylation of aldolase A results in reduced levels of activity, particularly in cancer cells. Lactylation of PKM2 isoform of pyruvate kinase in immune cells results in increased activity of the enzyme. Fatty acid synthase has been shown to be lactylated on three different Lys residue, the consequences of which are suppression of de novo fatty acid synthesis.
Protein Lysine Crotonylation (Kcr)
The post-translational modification of proteins by the attachment of crotonic acid (but-2-enoic acid) to lysine residues was first identified in 2011 in the context of histone proteins, and subsequently shown to be a modification in numerous other proteins. This modification is referred to as lysine crotonylation. Crotonic acid (crotonate) is a short-chain unsaturated fatty acid that is found in the diet, predominantly from plants, and is also an intermediate, as crotonyl-CoA, in the metabolism of the amino acids tryptophan and lysine, and the metabolism of certain fatty acids. It has been suggested that dietary crotonate is converted to crotonyl-CoA via the action of acyl-CoA synthetase 2, encoded by the ACSS2 gene. However, there is conflicting data regarding validity of ACSS2 as functioning crotonyl-CoA generating enzyme so it may be that the ACSS1 or ACSS3 encoded enzymes may be responsible for crotonyl-CoA synthesis. The transporter responsible for transport of dietary crotonyl-CoA into the nucleus and mitochondria, and metabolism derived crotonyl-CoA out of the mitochondria is currently unknown.
In the context of tryptophan and lysine catabolism, as well as fatty acid metabolism, the crotonyl-CoA that is generated in the mitochondria is converted to β-hydroxybutyryl-CoA by enoyl-CoA hydratase encoded by the ECHS1 (enoyl-CoA hydratase, short chain 1; also identified as SCEH) gene. The ECHS1 encoded enzyme is often referred to as crotonase. Within the nucleus crotonyl-CoA is metabolized to β-hydroxybutyryl-CoA by the CDYL (chromodomain Y like) encoded enzyme.
Lysine crotonylation is catalyzed by crotonyltransferases and its removal is catalyzed by decrotonylases. Histone
acetyltransferases (HAT) have been shown to have histone crotonyltransferase (HCT) activity. There are three major families of HAT enzymes p300/CBP, GNAT, and MYST. The first HAT complex identified as being able to carry out histone crotonylation was p300/CBP. In in vitro experiments p300/CBP-mediated histone crotonylation was shown to enhance transcription to a greater level than acetylation by p300/CBP. Subsequent to the identification of p300/CBP as being able to carry out histone crotonylation, members of the MYST family, specifically the acetyltransferase encoded by the KAT8 gene (also known as MOF: Males absent Of the First), were also found to catalyze histone crotonylation.
Histone deacetylases (HDAC) have been identified as possessing histone decrotonylase (HDCR) activity. The first HDAC shown to possess HDCR activity was histone deacetylase 3 (HDAC3). Subsequently the sirtuins, SIRT1 and SIRT2, were shown to be able to decrotonylate histones.
Recognition of crotonylated proteins is associated with proteins possessing a double PHD finger (DPF) domain as well as members of the YEATS domain family, both of which are known to interact with acetylated proteins. Members of the YEATS domain protein family have a much higher affinity for crotonylated proteins compared with acetylated proteins. The PHD (plant homeodomain) finger domain is a type of zinc finger (Cys4-His-Cys3) originally found in plant homeodomain containing proteins. The YEATS domain was originally identified as a domain found in five yeast proteins (Yaf9, ENL, AF9, Taf14, and Sas5), hence the derivation of the acronym.
Protein Lysine Propionylation (Kpr)
Propionyl-CoA is the substrate for protein lysine propionylation. Propionyl-CoA is generated predominantly within the mitochondria from the oxidation of the amino acids methionine, threonine, isoleucine, and valine, and from the oxidation of fatty acids with an odd number of carbon atoms. Propionyl-CoA is also generated in the peroxisomes from the oxidation of branched-chain fatty acids such as phytanic acid. The major source of propionyl-CoA used in propionylation of histone proteins is the oxidation of isoleucine.
Mitochondrial propionyl-CoA is transferred out of the mitochondria by carnitine acylcarnitine translocase, CACT. The CACT transporter is a member of the SLC family of transporters and as such is encoded by the SLC25A20 gene. The carnitine acylcarnitine translocase is located in the inner mitochondrial membrane where it facilitates acylcarnitine transport across the outer and inner mitochondrial membranes in exchange for free carnitine. The propionyl-CoA is then transported into the nucleus where it serves as the substrate for histone propionylation.
The processes of protein propionylation and de-propionylation are catalyzed by many of the same enzymes that carry out histone acetylation and deacetylation. Histone (as well as other protein), propionylation has been demonstrated to occur through the enzymatic actions of enzymes of three of the HAT families, GCN5/PCAF, p300/CBP, and MYST. Specifically GCN5 (KAT2A), PCAF, p300, CBP, and MOF have been shown to propionylate histones. Histone de-propionylation has been shown to be carried out by the sirtuin family member enzymes, SIRT1 and SIRT2.
Protein Lysine Succinylation (Ksucc)
Like all protein lysine acylations, succinyl-CoA represents the substrate for lysine succinylation (Ksucc) of histone proteins as well as other non-histone proteins. Succinyl-CoA can be derived from several sources and pathways with the most prevalent being from the TCA cycle. Succinyl-CoA is also produced from propionyl-CoA which is an intermediate in the catabolism of the amino acids isoleucine, valine, methionine, and threonine, and from the catabolism of fatty acids with an odd number of carbon atoms, and from the peroxisomal oxidation of dicarboxylic acids. The predominant site of protein succinylation is within the mitochondria and then the nucleus. However, there is ample evidence of cytoplasmic protein succinylation.
The transport of 2-oxoglutarate (α-ketoglutarate) from the mitochondria to the cytosol is carried out by SCL25A11. Succinyl-carnitine is transported out of the mitochondria by carnitine acylcarnitine translocase, CACT. The CACT transporter is a member of the SLC family of transporters and as such is encoded by the SLC25A20 gene. The carnitine acylcarnitine translocase is located in the inner mitochondrial membrane where it facilitates acylcarnitine transport across the outer and inner mitochondrial membranes in exchange for free carnitine. Succinate is transported out of the mitochondria via the action of SLC25A10.
Cytosol 2-oxoglutarate is transported into the nucleus where nuclear-localized 2-oxoglutarate dehydrogenase complex (OGDHc; also known as α-ketoglutarate dehydrogenase) oxidizes it to succinyl-CoA. Cytoplasmic succinyl-carnitine and succinate are converted to succinyl-CoA, most likely via the action of one or more members of the acyl-CoA synthetase family of enzymes. The succinyl-CoA is then transported into the nucleus.
Peroxisomal succinyl-CoA is hydrolyzed to succinate via the action of peroxisomal succinyl-CoA thioesterase which is encoded by the ACOT4 gene. The succinate is then transported to the cytosol where is can be converted to succinyl-CoA again and transported into the nucleus.
Succinyl-CoA is a sufficiently energetic compound that non-enzymatic succinylation can occur. Despite this, enzymatic succinylation has been described. The “writer” for enzymatic lysine succinylation has been shown to be the GCN5/PCAF family member GCN5 (KAT2A). The nuclear OGDHc interacts with GCN5 allowing the succinyl-CoA that is generated to be directly accessible by the acetyltransferase.
De-succinylation of mitochondrial and nuclear succinylated proteins has been shown to be catalyzed by two members of the sirtuin family, SIRT5 and SIRT7. SIRT5 activity is the major mitochondrial de-succinylase but also functions within the nucleus. SIRT7 functions as a histone desuccinylase in the processes of DNA damage repair. SIRT7 is recruited to sites of double-strand break (DSB) by polyADP-ribose polymerase 1 (PARP1) where it de-succinylates lysine 122 of histone H3 (H3K122). The de-succinylation of H3 promotes chromatin condensation and efficient DSB repair.
Protein Lysine Malonylation (Kmal)
Malonyl-CoA is the product of acetyl-CoA carboxylation via the action of the acetyl-CoA carboxylases, ACC1 and ACC2. Malonyl-CoA a major substrate for fatty acid synthase (FAS) in the de novo synthesis of fatty acids. ACC2 is closely associated with the outer mitochondrial membrane localized enzyme, carnitine palmitoyltransferase 1 (CPT1). The generation of malonyl-CoA by ACC2 allows for rapid inhibition of the activity of CPT1, thereby limiting the mitochondrial oxidation of newly synthesized fatty acids. Given its function in activation of fatty acid synthesis and inhibition of fatty acid oxidation, malonyl-CoA is critical regulator of overall fatty acid homeostasis. Within the mitochondria, malonyl-CoA is generated by the enzyme encoded by the ACSF3 (acyl-CoA synthetase family member 3) gene in the process of mitochondrial fatty acid synthesis.
Lysine malonylation occurs predominantly non-enzymatically. Numerous proteins have been identified as being malonylated including many metabolic enzymes. Several enzymes of glycolysis, including glucose-6-phosphate isomerase (encoded by the GPI gene), phosphoglycerate kinase (encoded by the PGK1 gene), aldolase A (encoded by the ALDOA gene), and enolase (encoded by the ENO1 gene) are modified by lysine malonylation. The TCA cycle enzyme, malate dehydrogenase (encoded by the MDH2 gene) has also been shown to undergo lysine malonylation.
Removal of lysine malonylation has been shown to occur through the action of the sirtuin, SIRT5. Although lysine malonylation of many proteins, including all four of the nucleosomal histones (H2A, H2B, H3, and H4), the functional significance of these modifications has not yet been fully characterized.
Protein Lysine Lipoylation
Protein lysine lipoylation is an important post-translational modification involving lipoic acid. Lipoic acid synthetase, along with lipoyl (octanoyl) transferase 2 (encoded by the LIPT2 gene), and lipoyltransferase 1 (LIPT1) synthesize lipoic acid from the medium-chain fatty acid, octanoic acid. The synthesis of lipoic acid is directly coupled to the lipoylation of proteins.
The five lipoylated proteins are the GCSH encoded H-protein of the glycine cleavage complex (GCC), the E2 subunit proteins of dehydrogenase complexes that includes the DLAT encoded protein of the pyruvate dehydrogenase complex (PDHc), the DLST encoded protein of the 2-oxoglutarate (α-ketoglutarate) dehydrogenase complex (OGDH), and the DBT encoded protein of the branched-chain keto acid dehydrogenase complex (BCKD). In addition to the E2 subunit of the PDHc, the PDHX encoded protein of this complex is also lipoylated.
The lipoylation of proteins occurs in the context of mitochondrial lipoic acid synthesis. The initial substrate for lipoic acid synthesis is the octanoic acid intermediate of mitochondrial fatty acid synthesis that is attached to the ACP sulfhydryl of the mitochondrial fatty acid synthesis protein encoded by the NDUFAB1 (NDUFAB1 (NADH:ubiquinone oxidoreductase subunit AB1) gene as described in the Mitochondrial Fatty Acid Synthesis section of the Synthesis of Fatty Acids page.
The ACP of NDUFAB1, as well as several other lipid biosynthetic enzymes, is 4′-phosphopantetheine, an intermediate in the synthesis of CoASH. The 4′-phosphopantetheine from CoA is covalently attached to a specific Ser residue via a phosphodiester linkage. This reaction is catalyzed by L-aminoadipate-semialdehyde dehydrogenase-phosphopantetheinyltransferase which is encoded by the AASDHPPT gene. During the course of the attachment, 3′-phosphoadenosine 5′-phosphate (PAP) is released.
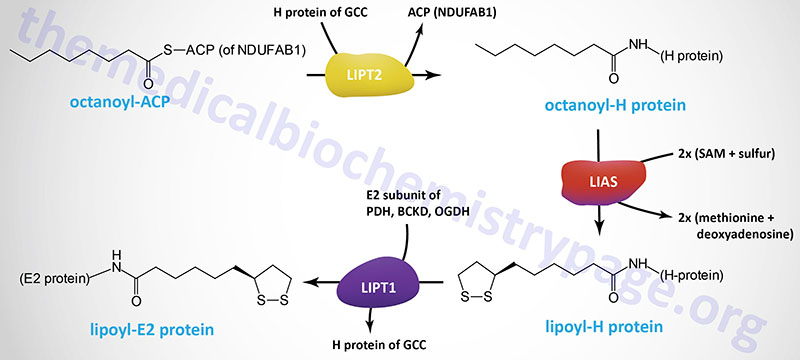
In addition to the important role of protein lysine lipoylation in overall energy homeostasis, this post-translational modification is involved in the mechanism by which copper induces cell death in the process referred to as cuproptosis. Two critical proteins, that are key regulators of copper toxicity, are involved in protein lipoylation. These proteins are mitochondrial ferredoxin 1 (FDX1) and lipoic acid synthetase (LIAS). Ferrodoxin 1 directly binds to lipoic acid synthetase, thereby regulating the activity of LIAS. The significance of these two proteins to the process of cuproptosis has been demonstrated by knocking out the genes in mice which results in inhibition of copper-induced cell death.
Protein Cysteine Modification
Protein Cysteine Lactylation
Protein lysine lactylation represents the bulk the post-translational modification of proteins via the enzymatic attachment of lactate to Lys residues. However, studies have shown that there are proteins that undergo non-enzymatic attachment of lactate to cysteine residues, a process referred to as protein S-lactylation. The process of S-lactylation, like that of non-enzymatic Lys D-lactylation, is stimulated by the glycolytic intermediate, G3P.
Currently the only identified protein to undergo S-lactylation is KEAP1 (Kelch-like ECH-associated protein 1). Several proteins contain Kelch (German for chalice) domains that are protein-protein interaction domains. This domain allows KEAP1 to interact with ubiquitin ligases of the Cullin family. Under normal physiological conditions KEAP1 ubiquitylates the transcription factor identified as NRF2 (nuclear factor erythroid 2-related factor 2) preventing it from entering the nucleus. NRF2 is encoded by the NFE2L2 gene. When KEAP1 is S-lactylated it dissociates from NRF2 which then migrates to the nucleus and activates the expression of genes encoding proteins with cytoprotective functions.
Protein Cysteine Persulfidation
Protein persulfidation (formation of RSSH which represents the oxidized thiol group in cysteine) is a post-translational modification of the thiol of the R-group of cysteine residues in various proteins via the reactivity of hydrogen sulfide, H2S. The process of protein persulfidation was originally termed sulfhydration but since there is no involvement of water the name is no longer be used. The term protein S-sulfhydration is also used to describe the post-translational modification process of persulfidation. Another commonly used term for persulfidation is hydropersulfidation.
The production of H2S in human cells is primarily catalyzed by the cysteine biosynthesis enzyme, cystathionine γ-lyase (also called cystathionase). Another enzyme in the cysteine biosynthesis pathway, cystathionine β-synthase, is also involved in the production of H2S. The synthesis of H2S is also generated by one of the cysteine catabolic enzymes, mercaptopyruvate sulfurtransferase (encoded by the MPST gene). The MPST encoded enzyme is also known as cysteine aminotransferase/mercaptopyruvate sulfurtransferase.
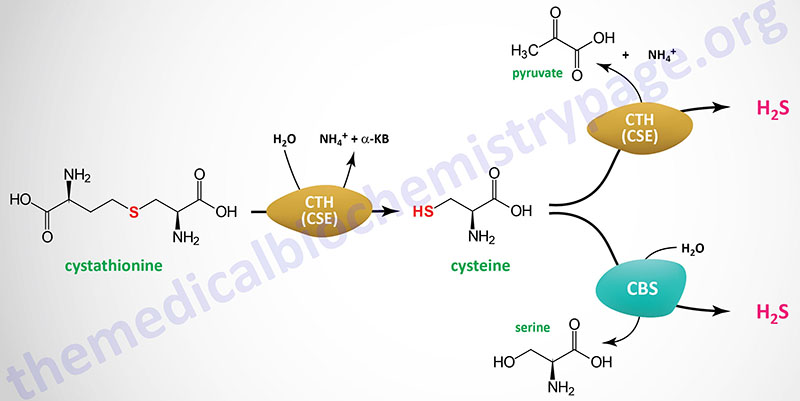
A major function of protein persulfidation is to serve as an intermediate, activated step in the metabolic pathways that require the transfer of sulfur. These pathways include, but are not limited to, the synthesis of lipoic acid and the synthesis of iron-sulfur centers in proteins.
One of the first proteins to be found to exhibit an altered function as a result of persulfidation was the cytosolic version of glyceraldehyde-3-phosphate dehydrogenase (GAPDH; encoded by the GPD1 gene). The GPD1 encoded enzyme is often designated cGPDH. When persulfidated the enzymatic activity of cGAPDH increases several fold. When the CTH gene is knocked out in mice there results an approximately 35% reduction in the level of cGAPDH activity.
Another protein whose activity has been shown to be increased by persulfidation is the ubiquitin E3 ligase, parkin which is encoded by the PARK2 gene. Parkin is so-called due to it being associated with Parkinson disease. The function of parkin is in the process of mitochondrial autophagy, a process that is known to be defective in Parkinson disease. On the other hand, one of the first proteins demonstrated to exhibit reduced activity as a result of persulfidation is another protein involved in autophagy identified as autophagy related protein, WD repeat domain phosphoinositide interacting 1 (encoded by the WIPI1 gene).
Protein Cysteine Sulfenylation
The post-translational modification of the sulfhydryl group of cysteine residues (Cys-SH) that involves oxidation to sulfenic acid (Cys-SOH) is referred to as cysteine sulfenylation. This modification is catalyzed by reactive oxygen species (ROS), particularly hydrogen peroxide, H2O2. Given that the sulfenylation of Cys residues is catalyzed by ROS, this post-translational modification serves to function as a redox-sensitive switch that controls protein function.
Numerous proteins (over 700) involved in signal transduction, in enzymatic processes, in antioxidant processes, and in the processes of hemostasis (blood coagulation) have been shown to be modified by cysteine sulfenylation. The Src tyrosine kinase undergoes sulfenylation on at least two Cys residues, the consequences of which lead to activation of the kinase activity of the enzyme. Additional signaling proteins whose activities are altered by sulfenylation include the phosphoinositide phosphatase, PTEN, the protein tyrosine phosphatase, PTP1B, the Ser/Thr kinase AKT2, and the EGF receptor (EGFR).
The sulfenylation of the mitochondrial inner membrane transporter identified as UCP1 (encoded by the SLC25A7 gene), in response to cold exposure, leads to enhanced thermogenesis consequent to sympathetic stimulation.
Metabolic enzymes shown to undergo cysteine sulfenylation include several enzyme of glycolysis such as PKM2 isoform of pyruvate kinase, glyceraldehyde-3-phosphate dehydrogenases (GAPDH), phosphoglycerate kinase (PGK), phosphoglycerate mutase (PGAM1), enolase, and triosephosphate isomerase (TPI).
Protein Cysteine Succination
Unlike the post-translational enzyme catalyzed addition of succinate, from succinyl-CoA, to lysine residues in proteins, cysteine succination is a non-enzymatic post-translational modification. Protein cysteine, and glutathione (GSH), succination involves fumarate which is derived primarily via the TCA cycle as well as via the urea cycle. The spontaneous addition of fumarate occurs via a reaction referred to as a “Michael addition”. During the addition, the double bond of fumarate is reduced forming succinate which is why the process is termed succination. The resulting residue in proteins and GSH is termed 2-(succino)cysteine (2SC).
Critical proteins that have been found to undergo succination, thus altering their activities, have been identified in numerous types of cancer, particularly in fumarate hydratase (fumarase) deficient cancers. Several of these proteins include nuclear factor erythroid 2-related factor 2 (NRF2), phosphatase and tensin homolog (PTEN), and SWI/SNF-related, matrix-associated, actin-dependent regulator of chromatin subfamily C member 1 (SMARCC1). NRF2 is a transcription factor, PTEN is a phosphatidylinositol-3-phosphate phosphatase, and SMARCC1 is a core component of what is identified as the SWI/SNF chromatin remodeling complex. SWI/SNF refers to switching of mating type/sucrose non-fermenting, a chromatin remodeling complex originally identified in yeast.
Metabolic enzymes have also been found to be subject to succination under conditions of excess cytosolic fumarate. Both glyceraldehyde-3-phosphate dehydrogenase (GAPDH) of glycolysis and glucose-6-phosphate dehydrogenase (G6PDH) of the pentose phosphate pathway (PPP) are subject to succination. The effect of this post-translational modification is increased glycolytic flux and increase PPP activity. Both of these effects are beneficial to rapidly proliferating cells, such as cancer cells.
Recent (2024) evidence has discovered that the urea cycle enzyme, argininosuccinate synthetase, encoded by the ASS1 gene, plays a role in the tumor suppressor p53-mediated responses to DNA damage. The loss of expression of the ASS1 gene has been correlated to increased DNA damage in colorectal carcinoma cells as well as in fibroblasts in patients with ASS1 deficiency (known as citrullinemia type 1, CTLN1). When DNA damage is sensed and p53 is activated to migrate to the nucleus, there is a concomitant increase in both argininosuccinate synthetase and argininosuccinate lyase localization to the nucleus. Within the nucleus these two enzymes increase the level of fumarate which in turn enhances the succination of the SMARCC1 protein. The consequences of the succination of SMARCC1 is a decrease in the level of expression of p53 target genes and the cessation of cell cycle progression.
The importance of argininosuccinate synthetase to the suppression of cancer progression is evidenced by the fact that ASS1 promoter hypermethylation, and thus transcriptional silencing, has been shown to occur in numerous types of cancer.
Protein Cysteine CoAlation
Cysteine modification in proteins by thiolation with coenzyme A (CoA) has become recognized as a critical modification in the regulation of cellular responses to oxidative stress and metabolic stress induced by nutrient deprivation. The process of the formation of a disulfide bond between the –SH groups of cysteine residues in proteins and CoA is referred to as CoAlation. Similar to other post-translational modifications, the process of CoAlation is reversible.
Although numerous proteins have been identified that are modified by CoAlation, the bulk of modified proteins are metabolic enzymes. With respect to metabolic enzymes, changes in the level of CoAlation is associated with oxidative stress, high fat diets, and nutrient derivation such as occurs during starvation.
Metabolic enzymes that undergo cysteine CoAlation include those in glycolysis, the TCA cycle, fatty acid oxidation, ketone body metabolism, and amino acid metabolism. Metabolic enzymes shown to possess higher levels of CoAlation in response to oxidative stress include, but is not limited to, glyceraldehyde-3-phosphate dehydrogenase, malate dehydrogenase, isocitrate dehydrogenase, HMG-CoA lyase, malonyl-CoA decarboxylase, β-hydroxybutyrate dehydrogenase, propionyl-CoA carboxylase, and glutamate dehydrogenase. Many of these same enzymes also undergo changes in CoAlation levels in response to nutrient deprivation.
Protein Cysteine S-Nitrosylation
Protein S-nitrosylation is a post-translational modification that serves as a mechanism to transfer nitric oxide (NO)-mediated signals. Nitric oxide is produced by one of three nitric oxide synthases (NOS) , endothelial NOS (eNOS; also known as NOS3), neuronal NOS (nNOS; also known as NOS1) and inducible NOS (iNOS; also known as NOS2). The major mechanism of NO-induced signaling is via binding to, and activating, the soluble form of guanylyl cyclase leading to the generation of the second messenger, cyclic GMP (cGMP). Like cAMP, cGMP exerts numerous effects directly but it also activates a kinase called cGMP-dependent protein kinase PKG.
The other mechanism of signal transduction elicited by NO is through S-nitrosylating target proteins. Indeed, protein S-nitrosylation represents the major mechanism by which NO exerts numerous critical effects across a wide array of tissues. Protein S-nitrosylation exerts effects on protein conformational, protein-protein interactions, and additional post-translational modifications that include phosphorylation, acetylation, and ubiquitylation. The S-nitrosylation of proteins has been shown to be involved in a diverse array of processes that includes , transcription regulation, DNA damage repair, cellular growth and differentiation, and apoptosis.
Protein S-nitrosylation involves the attachment of the nitrosyl group to the reactive thiol group of a cysteine in a target protein generating an S-nitrosothiol (SNO) in the protein. In addition to the role of the NOS enzymes in generating NO, S-nitrosylation of proteins involves enzymes called SNO synthases and tansnitrosylases. SNO synthases and transnitrosylases can both be classified as protein S-nitrosylases. In mammals there are likely to be several different S-nitrosylases but two proteins that are known to participate in the generation of S-nitrosothiol intermediates are hemoglobin and cytochrome c. Various transnitrosylases have also been identified that includes glutathione (GSH), the Ca2+– and Zn2+-binding proteins S100A8 and S100A9, and the glycolytic enzyme, glyceraldehyde-3-phosphate dehydrogenase (GAPDH).
The SNO synthases convert NO into SNO and then the transnitrosylases transfer NO to the target cysteine generating the S-nitrosylated protein (SNO-protein). Proteins that undergo S-nitrosylation directly interact with NOS. The cysteine that is S-nitrosylated is found to reside in a consensus motif I/L-X-C-X2-D/E, where X refers to any amino acid.
Glutathione is S-nitrosylated, generating S-nitrosoglutathione (GSNO), in the mitochondria when the nitrosyl group is transferred from the heme iron of cytochrome c to glutathione. GSNO is the most abundant S-nitrosothiol and the major endogenous NO donor (S-nitrosylase) for proteins throughout the cell. Following the formation of GSNO in the mitochondria it translocates to various subcellular locations and participates in the transnitrosylation of numerous interacting proteins such as AKT/PKB, NF-κB, the EGF receptor (EGFR), and the IGF-1 receptor (IGF-1R). The S-nitrosylation of AKT/PKB, EGFR, and IGF-1R, results in the inhibition of their phosphorylation-dependent activation.
The proteins identified as S100A8 and S100A9 form a heterodimer that is identified as calprotectin. Because these two proteins function as a heterodimer the designation is most often S100A8/A9. These proteins are members of the S100 family of proteins that contain two EF-hand domains. S100A8/A9 plays a major role in the regulation of inflammatory processes, elicited by neutrophils, by forming a complex with arachidonic acid that is released in response to tissue injury. This complex interacts with NADPH oxidase (NOX) which is then activated to produce reactive oxygen species. As an S-nitrosylase, S100A8/A9 has been found to transnitrosylate over 100 distinct proteins which includes hemoglobin and GAPDH. When S100A8/A9 is S-nitrosylated it function it is converted to an anti-inflammatory complex that inhibits mast cell activation and leukocyte interaction with the endothelium of the vasculature.
Following S-nitrosylation on Cys150 by the S100A8/A9 S-nitrosylase, GAPDH translocates to both the nucleus and the mitochondria, locations where it functions in the regulation of transcription and the regulation of apoptosis. The S-nitrosylation of GAPDH enables it to interact with the E3 ubiquitin ligase SIAH1. SIAH1 possesses a nuclear localization signal which allows GAPDH to be co-translocated into the nucleus. In the nucleus GAPDH transnitrosylates proteins that are involved in transcriptional regulation and also in DNA repair such as SIRT1 and DNA-activating protein kinase (DNA-PK). Under stress conditions S-nitrosylated GAPDH translocates to mitochondria where it is involved in the transnitrosylation mitochondrial proteins that are involved in the regulation of mitochondrial membrane permeability such as voltage-dependent anion channel 1 (VDAC1).
Proteins undergo removal of the S-nitrosylation via the action of denitrosylases. Denitrosylases are of two main types. There are low molecular weight (LMW) thiol cofactor-dependent denitrosylases and thioredoxin-related denitrosylases. The thioredoxin-related denitrosylases are sometimes referred to as as high molecular weight (HMW) denitrosylases. Low molecular weight thiols, primarily glutathione (GSH) and coenzyme A (CoASH), are S-nitrosylated generating S-nitrosoglutathione (GSNO) and S-nitroso-coenzyme A (SNO-CoA), respectively, both of which are designated as LMW-SNOs. The LMW denitrosylases act on LMW SNOs to regenerate the Cys-SH group in the protein.
LMW denitrosylases include GSNO reductase. The primary GSNO reductase in humans is encoded by the ADH5 [alcohol dehydrogenase 5 (class III), chi polypeptide] gene. The archetypal human SNO-CoA reductase is encoded by the AKR1A1 (aldo-keto reductase family 1 member A1) gene.
The HMW denitrosylases are thioredoxin (encoded by the TXN gene) and thioredoxin-related protein of 14kDa, TRP14 [encoded by the TXNDC17 (thioredoxin domain containing 17) gene]. Thioredoxin and TRP14 catalyze the denitrosylation of SNO-proteins via the generation of nitroxyl (HNO) while simultaneously being oxidized. The reduction of oxidized thioredoxin involves the NADPH-dependent thioredoxin reductases.
Protein Cysteine Glutathionylation
The antioxidant tripeptide, glutathione (GSH), which harbors a cysteine sulfhydryl (-SH) group, has been shown to form disulfide bonds with the -SH group of cysteine residues in numerous proteins. The formation of GSH-modified cysteine residues in proteins is referred to as protein S-glutathionylation (often abbreviated GS-ylation or SSG).
Proteomic analysis of various cell types has found that protein S-glutathionylation occurs on numerous proteins. The S-glutathionylation of enzymes of glycolysis, the TCA cycle, and oxidative phosphorylation result in regulation of metabolic processes. In addition to metabolic regulation, the process of protein S-glutathionylation plays a role in the regulation of protein synthesis, calcium homeostasis, signal transduction by kinases and phosphatases, and G-proteins, as well as inflammation, and apoptosis.
Metabolic enzymes shown to be S-glutathionylated under various conditions, such as reoxygenation following ischemia, include glyceraldehyde 3-phosphate dehydrogenase (GAPDH), lactate dehydrogenase (LDH), phosphoglycerate kinase (PGK1), phosphoglycerate mutase (PGAM1), and the α-subunit of propionyl-CoA carboxylase (encoded by the PCCA gene).
Enzymes of energy homeostasis that increase in S-glutathionylation under similar conditions of reoxygenation include the succinate dehydrogenase (SDH) subunit encoded by the SDHC gene, mitochondrial malate dehydrogenase (encoded by the MDH2 gene), the NDUFS1 encoded subunit of complex I, and the D subunit of ATP synthase (complex V) which is encoded by the ATP5PD (also known as ATP5H) gene.
Protein Serine Phosphopantetheinylation
Protein phosphopantetheinylation refers to the post-translational modification of proteins by the addition of the 4′-phosphopantetheine group (4′-PP) derived from coenzyme A (CoA). Humans express only six proteins that have been identified as being phosphopantetheinylated, three cytosolic enzymes and three mitochondrial enzymes.
The 4′-phosphopantetheine serves as the acyl carrier portion (ACP) of several lipid biosynthetic enzymes that includes cytosolic fatty acid synthase and the mitochondrial NDUFAB1 encoded protein (NADH:ubiquinone oxidoreductase subunit AB1) which serves as the ACP in mitochondrial fatty acid synthesis. The role of the NDUFAB1 encoded enzyme in lipoic acid synthesis was discussed in the Protein Lysine Lipoylation section above.
The 4′-phosphopantetheine group also functions in the context of the folate cycle via the cytosolic ALDH1L1 (aldehyde dehydrogenase 1 family member L1) encoded enzyme and the functionally related mitochondrial ALDH1L2.
An additional enzyme modified by 4′-phosphopantetheine attachment is the cytosolic AASDH (aminoadipate-semialdehyde dehydrogenase; 2-aminoadipic-6-semialdehyde dehydrogenase) encoded enzyme. The AASDH encoded enzyme is also sometimes identified as ACSF4 (acyl-CoA synthetase family member 4) and as β-alanine activating enzyme.
The mitochondria-localized DHRS2 encoded enzyme is another phosphopantetheinylated enzyme. The DHRS2 encoded enzyme is a member of the short-chain dehydrogenase/reductase (SDR) family of oxidoreductases.
The attachment of the 4′-phosphopantetheine, from CoA, to the proteins indicated, occurs on a conserved Ser residue via a phosphodiester linkage. This reaction is catalyzed by L-aminoadipate-semialdehyde dehydrogenase-phosphopantetheinyltransferase which is encoded by the AASDHPPT gene. During the course of the attachment, 3′-phosphoadenosine 5′-phosphate (PAP) is released.
Protein N-Terminal Arginylation
The addition of the amino acid arginine the N-terminus of proteins, primarily to N-terminal glutamate or aspartate residues, is a process that is termed protein arginylation. Subsequent to the identification of N-terminal arginylation, it was demonstrated that arginylation of the side-chain of internal glutamate residues can also occur. Protein arginylation occurs post-translationally and is a tRNA-dependent reaction catalyzed by the enzyme identified as arginyltransferase 1.
Arginyltransferase 1 is encoded by the ATE1 gene. The ATE1 gene is located on chromosome 10q26.13 and is composed of 19 exons that generate five alternatively spliced mRNAs, each of which encode a distinct protein isoform.
The dependence upon tRNA for protein arginylation reflects the fact that the substrate for the process is the arginine aminoacyl-tRNA, Arg-tRNA (tRNAArg). The synthesis of Arg-tRNA is catalyzed by an Arg-tRNA synthetase just as would be for the activation of Arg for protein synthesis.
Protein arginylation at the N-terminus is associated with, but not exclusive to, the process of protein degradation via the ubiquitin-dependent N-end rule pathway. In numerous proteins the process of arginylation is associated with important biological functions. Indeed, the knock-out of the mouse ATE1 gene is associated with impaired cardiovascular development resulting in embryonic lethality.
Over 100 proteins have been found to be substrates for arginylation and includes secreted proteins, membrane-associated proteins, and nuclear-localized proteins. Intracellular arginylated proteins function at the level of both structural and metabolic regulation. Extracellular regulatory proteins and hormones that have been found to be arginylated include angiotensin II, beta-melanocyte stimulating hormone (β-MSH), and insulin.
Protein Ubiquitylation (Ubiquitination)
The attachment of the peptide ubiquitin to proteins, a process referred to as ubiquitylation or ubiquitination, is most often a means to tag the protein for degradation in the proteasome. Exceptions to this are the histones where the ubiquitylation alters their functions in organizing chromatin structure.
Details of the mechanisms of protein ubiquitylation are presented in the Protein, Organelle, and Cell Turnover page.
Protein O-GlcNAcylation
Multiple nuclear and cytoplasmic proteins, including transcription factors, histones, cytoskeletal proteins, oncogenes, and kinases, are post-translationally modified on serine and threonine residues with β-N-acetylglucosamine (GlcNAc). This modification is referred to as O-GlcNAcylation. The O-GlcNAcylation of proteins is reversible just as is the case for protein hydroxyl phosphorylation.
The details of the synthesis of UDP-GlcNAc, the activated form of GlcNAc, and the incorporation into, and removal from, proteins is covered in detail in the Glycoproteins: Synthesis and Clinical Consequences page. The role of histone O-GlcNAcylation is detailed in the Regulation of Gene Expression page.
Protein Hydroxyl Phosphorylation
Post-translational phosphorylation is one of the most common reversible protein modifications that occurs in animal cells. Most of the protein phosphorylation events occurs on the hydroxyl oxygen of the R-group of the amino acids serine, threonine, and tyrosine. As discussed in the next section, the amino acid histidine is also an important site for phosphorylation. The vast majority of phosphorylations occur as a mechanism to regulate the biological activity of an enzyme or protein and as such are transient. In other words a phosphate (or more than one in many cases) is added by a specific kinase and later removed by a specific phosphatase.
Physiologically relevant examples are the phosphorylations that occur in glycogen synthase and glycogen phosphorylase in hepatocytes in response to glucagon release from the pancreas. Phosphorylation of glycogen synthase inhibits its activity, whereas, the activity of glycogen phosphorylase is increased. These two events lead to increased hepatic glucose delivery to the blood.
The enzymes that phosphorylate proteins are termed kinases and those that remove phosphates are termed phosphatases. For a more detailed discussion of kinases and phosphatases go to the Signal Transduction Pathways: Phosphatases page. Protein kinases catalyze reactions of the following type:
ATP + protein → phosphoprotein + ADP
As indicated, in human cells the amino acids Ser, Thr, and Tyr are the amino acids that are predominantly subject to phosphorylation-dephosphorylation. The largest group of kinases are those that phosphorylate either serine or threonine residues and as such are termed protein serine/threonine kinases. The ratio of phosphorylation of the three different amino acids is approximately 1000/100/1 for serine/threonine/tyrosine.
Although the level of tyrosine phosphorylation is minor, the importance of phosphorylation of this amino acid is profound. As an example, the activity of numerous growth factor receptors is controlled by tyrosine phosphorylation.
Protein Histidine Phosphorylation
Phosphorylation of the oxygen in the hydroxyl of the amino acids Ser, Thr, and Tyr, as well as the hydroxylysines in collagens is a well documented and commonly utilized post-translational modification designed to regulate the activity and functions of thousands of different proteins. Phosphorylation of the imidazole nitrogens of His has long been known to be a critical modification in prokaryotic proteins, however, its role in the regulation of eukaryotic proteins is less well documented. Nonetheless, there are several well characterized examples of His phosphorylation (identified as pHis) in eukaryotic proteins, such as the phosphorylation of His299 in the α-subunit of succinyl-CoA synthetase encoded by the SUCLG1 gene and the phosphorylation of His11 in phosphoglycerate mutase encoded by the PGAM1 gene. Indeed, the His phosphorylation of succinyl-CoA synthetase was the first reported example of pHis in a eukaryotic protein and was found in bovine liver mitochondria.
Phosphohistidine is unique as a phosphoamino acid given that either of the imidazole nitrogen atoms (N1 or N3) can be phosphorylated forming two different isomeric pHis isoforms: 1-phosphohistidine (1-pHis) and 3-phosphohistidine (3-pHis). In addition, the chemistry of the pHis bonds are high energy phosphoramidate (P–N) bonds, whereas the pSer, pThr, and pTyr bonds are the more stable phosphoester bonds (P–O).
Two mammalian enzymes have been shown to possess histidine kinase (protein histidine kinase, PHK) activity. These enzymes are members of the nucleoside diphosphate kinase family and are encoded by the NME1 and NME2 genes. Three pHis phosphatases have also been identified and they are encoded by the PHPT1, LHPP, and PGAM5 genes. Each of these enzymes are described in the Table below.
In addition to SUCLG1 and PGAM1, numerous substrate proteins have been found to be modified by pHis with several of the more well characterized outlined in the following Table. Studies indicate that there may be nearly 800 human proteins that are modified by 1-pHis or 3-pHis modifications.
Table of Eukaryotic Proteins Involved in or Modified by Histidine Phosphorylation
| Protein Name | Gene | Functions / Comments |
| ATP-citrate lyase | ACLY | functions as a homotetramer; functional in the cytosol where it hydrolyzes acetyl-CoA from citrate generating acetyl-CoA in the cytosol for the synthesis of fatty acids and cholesterol; functional in the nucleus where it generates the acetyl-CoA necessary for histone acetylation; nuclear ACLY is involved in DNA-damage-induced histone acetylation; phosphorylated on His760 |
| G-protein subunit beta 1 | GNB1 | component of heterotrimeric G-proteins; one of five β-subunit encoding genes; phosphorylated on His266 |
| histone cluster 1 H4 family member a | HIST1H4A | component of the nucleosome; gene contained in the large HIST1 cluster located on chromosome 6p22.2; phosphorylated on His18 and His75 |
| potassium calcium-activated channel subfamily N member 4 | KCNN4 | also identified as KCa3.1; functions as a calcium-activated potassium channel predominantly in erythrocytes and T lymphocytes; phosphorylated on His358 |
| phospholysine phosphohistidine inorganic pyrophosphate phosphatase | LHPP | major function is to regulate the activity of the NME1/NME2 phosphorylated proteins; considered a tumor suppressor |
| nicotinamide phosphoribosyltransferase 2 | NAMPT | functions in the recycling of nicotinamide to NAD+; phosphorylation of His247 forming the 1-pHis isoform results in dramatic increases in catalytic activity |
| NME/NM23 nucleoside diphosphate kinase 1 | NME1 | functions as a hexameric protein composed of A and B isoforms; the A isoform is encoded by the NME1 gene and the B isoform is encoded by the NME2 gene; both the NME1 and NME2 genes located on chromosome 17q21.33; the NME1 and NME2 encoded proteins form the only known mammalian histidine kinases; these proteins autophosphorylate forming the 1-pHis isoform exclusively which activates the nucleoside diphosphate kinase activity of the complex; in addition to histidine kinase and nucleoside diphosphate kinase activity the NME1/NME2 complexes also function as a Ser/Thr protein kinase, as geranyl and farnesyl pyrophosphate kinases, and also possesses 3′-5′ exonuclease activity |
| NME/NM23 nucleoside diphosphate kinase 2 | NME2 | functions as a hexameric protein composed of A and B isoforms; the A isoform is encoded by the NME1 gene and the B isoform is encoded by the NME2 gene; both the NME1 and NME2 genes located on chromosome 17q21.33; the NME1 and NME2 encoded proteins form the only known mammalian histidine kinases; these proteins autophosphorylate forming the 1-pHis isoform exclusively which activates the nucleoside diphosphate kinase activity of the complex; in addition to histidine kinase and nucleoside diphosphate kinase activity the NME1/NME2 complexes also function as a Ser/Thr protein kinase, as geranyl and farnesyl pyrophosphate kinases, and also possesses 3′-5′ exonuclease activity |
| NME/NM23 nucleoside diphosphate kinase 4 | NME4 | mitochondrially localized enzyme; also identified as nucleoside diphosphate kinase D; functions as a hexomeric enzyme |
| NME/NM23 nucleoside diphosphate kinase 7 | NME7 | localized to the centrosome; phosphorylated on His206 |
| phosphoglycerate mutase 1 | PGAM1 | functions in the pathway of glycolysis; His11 is phosphorylated by the PKM2 form of pyruvate kinase utilizing phosphoenolpyruvate (PEP) as the phosphate donor; the formation of pHis11 in PGAM1 occurs at a higher rate in highly proliferative cells and in all types of cancers which alters the metabolism of glucose in these cells; this occurs in the context of what is referred to as the Warburg effect |
| phosphoglycerate mutase 5 | PGAM5 | PGAM gene family member; functions to dephosphorylate the autophosphorylated pHis form of the NME1/NME2 complex; is itself phosphorylated on His105; also functions as a phosphatase for pSer and pThr residues; |
| phosphohistidine phosphatase 1 | PHPT1 | phosphohistidine phosphatase that functions to dephosphorylate the pHis residues in the GNB1, ACLY, and KCNN4 encoded proteins |
| phospholipase D1 | PLD1 | one of the two (including PLD2) major phospholipase D family member enzymes; is phosphorylated on His94; preferentially hydrolyzes phosphatidylcholines (PC) to phosphatidic acid and choline |
| succinyl-CoA synthetase α-subunit | SUCLG1 | functions in the TCA cycle; forms a heterodimeric complex with the β-subunit proteins encoded by the SUCLA2 and SUCGL2 genes; phosphorylated on His299 |
| transient receptor potential cation channel subfamily V member 5 | TRPV5 | major calcium uptake transporter expressed in the apical membranes of epithelial cells in the distal convoluted tubule of the nephron; also expressed in the apical membranes of small intestinal enterocytes where it is involved in calcium absorption |
Protein Hydroxylation
Hydroxylation of proteins represents and enzymatic modification resulting in the incorporation of a single oxygen atom to create an alcohol or a hydroxyl group. The majority of enzymes that carry out the hydroxylation of proteins are members of the large family of 2-oxoglutarate (α-ketoglutarate) and Fe2+-dependent dioxygenases (2-OGDD).
The most well studied protein hydroxylation events are those involved in the modification and maturation of collagen proteins by proly and lysyl hydroxylases. Another family of hydroxylating enzymes, the prolyl hydroxylase domain (PHD) containing enzymes, are involved in the regulation of cellular responses to hypoxia and stress.
The amino acids that are known to be hydroxylated include proline, lysine, asparagine, arginine, aspartate, and histidine. The protein identified as factor inhibiting hypoxia-induced factor 1 (FIH1), which is encoded by the HIF1AN (hypoxia inducible factor 1 alpha subunit inhibitor) gene, was the first to be identified as undergoing histidine hydroxylation.
Two well characterized 2-OGDD that catalyze ribosomal protein histidine hydroxylation are encoded by the RIOX1 and RIOX2 genes. RIOX1 was originally identified as NO66 for nucleolar protein 66. RIOX2 was originally identified as MINA53 for myc-induced nuclear antigen 53. Both of these enzymes belong to the JmjC domain-containing subfamily of 2-OGDD. However, unlike most other JmjC domain-containing 2OGDD, the RIOX1 and RIOX2 encoded enzymes do not possess any other obvious functional domains. Both RIOX1 and RIOX2 hydroxylate the C-3 carbon of His residues in ribosomal proteins. RIOX1 hydroxylates His-216 of the large ribosomal subunit protein, RPL8. RIOX2 hydroxylates His-39 of the large ribosomal protein, RPL27A.
Protein Fatty Acid Acylation
Protein fatty acylation, like other forms of protein modification with lipid such as prenylation, serve to modify both localization and function of the modified protein. Protein fatty acylation occurs via thioester (S-acylation of cysteine), amide (N-acylation such as N-terminal glycine and ε-amine of internal lysines), and ester (O-linkage of serine and threonine) linkages.
Despite the fact that the initiator methionine is very often hydrolyzed following protein synthesis (catalyzed by methionine aminopeptidases), acylation of the N-terminus still occurs. N-terminal acetylation is catalyzed by a family of N-terminal acetyltransferases (NAT), as discussed above, using acetyl-CoA as the acetyl donor for these reactions.
Protein fatty acylation at the N-terminus most often involves attachment of the 14-carbon saturated fatty acid, myristic acid, to an N-terminal glycine residue, referred to as N-myristoylation. Another common fatty acylation of proteins utilizes the 16-carbon fatty acid palmitic acid (C16:0) which is attached to the sulfhydryl group of internal and N-terminal cysteine residues and is, therefore, referred to as S-palmitoylation.
Fatty N-acylation of lysine residues most often involves the 15-carbon monomethyl branched-chain fatty acid, 13-methyltetradecanoic acid (isoC15:0) but acylation with the polyunsaturated fatty acid (PUFA), eicosapentaenoic acid (EPA) has been identified.
Although O-acylation of serine is much less abundant than the acylation of glycine and lysine, a broad range of fatty acylations from C14 to C18 have been identified including acylation with EPA.
Although other long, medium, and short chain fatty acids are found attached to either the N-terminal amino acid (such as N-terminal propionylation) or to internal amino acids, N-myristoylation and S-palmitoylation represent the bulk of protein fatty acylations.
One physiologically relevant example of internal protein acylation is the hormone ghrelin. Ghrelin is a stomach-derived hormone that is acylated, a modification required for its biological activity, with octanoic acid on a specific serine residue. The ghrelin acylation is catalyzed by an enzyme that is a member of the multi-pass transmembrane acyltransferase family termed MBOAT for membrane-bound O-acyltransferase. The ghrelin acyltransferase is encoded by the MBOAT4 gene which was originally identified as ghrelin O-acyltransferase, GOAT.
Protein N-Myristoylation
N-terminal myristoylation is catalyzed by N-terminal myristoyltransferases (NMT). Humans express two NMT genes identified as NMT1 and NMT2. Incorporation of myristic acid onto an N-terminal glycine residue occurs predominantly as a co-translational event, although post-translational N-myristoylation has been shown to occur in apoptotic cells. Within the human proteome, it has been shown that approximately 0.5% of all proteins are N-myristoylated.
Protein S-Palmitoylation (S-Acylation)
Similar to protein N-myristoylation, protein S-palmitoylation is an important post-translational modification involved in the regulation of membrane attachment, intracellular trafficking, and membrane subdomain localization. The bulk of S-palmitoylation occurs on the sulfhydryl of internal cysteine residues, however, important examples of N-terminal palmitoylated proteins are known.
In addition to the attachment of palmitic acid (C16:0), S-palmitoylation is a term used to describe the S-acylation of proteins with stearic acid (C18:0), oleic acid (C18:1), arachidonic acid (C20:4), and eicosapentaenoic acid (C20:5; EPA). More than 700 proteins have been identified as undergoing S-acylation. Indeed, current analysis estimates that 10% of the human proteome is subject to S-acylation.
S-acylation of proteins is catalyzed by a family of protein acyltransferases (PAT) that are members of the Asp-His-His-Cys-containing protein acyltransferase family, identified as DHHC-PAT. Due to the DHHC motif forming a zinc-finger domain the proteins encoded by these genes, the enzymes are termed zinc finger DHHC type containing (ZDHHC) with a number designating the specific gene. Currently 23 human ZDHHC genes have been identified and characterized, ZDHHC1–ZDHHC9, ZDHHC11–ZDHHC24 (there is no ZDHHC10 gene).
Cellular uptake of several free fatty acids (FFA; non-esterified fatty acids, NEFA) involves the activity of a plasma membrane-localized transporter identified as FAT (fatty acid transporter). FAT is also known as CD36 and so the transporter is often designated FAT/CD36. FAT/CD36 undergoes S-palmitoylation by the ZDHCC5 encoded palmitoyltransferase which promotes the localization of the transporter to the plasma membrane.
N-terminal palmitoylation is known to occur on the α-subunit of Gs-type G-proteins as well as on the sonic hedgehog (SHH) protein. N-terminal palmitoylation of SHH is catalyzed by a specific enzyme encoded by the HHAT (hedgehog acyltransferase) gene. The HHAT protein belongs to the MBOAT family of multipass transmembrane acyltransferases. In addition to N-terminal S-palmitoylation, SHH is modified by the attachment of cholesterol to the C-terminus, a modification required to limit the spread of the protein across the anteroposterior axis of the developing neural tube and the developing limb bud.
The human homolog of the Drosophila melanogaster segment polarity gene porcupine, encoded by the PORCN gene, is also a member of the MBOAT family of acyltransferases. The PORCN encoded enzyme S-palmitoylates the Wnt family proteins, a modification that is required for correct distribution of the gradients of these important development regulating growth factors.
Several proteins involved in lipid droplet homeostasis, and thus the process of lipid metabolism, have been identified as undergoing S-acylation. The rate-limiting enzyme of triglyceride metabolism, ATGL, is modified by S-acylation. Additional proteins involved in lipid droplet homeostasis such as the ELMOD2 (ELMO domain containing 2; where ELMO refers to engulfment and motility domain) encoded protein, and the small monomeric G-proteins, Rab10 and Rab18, which are required for proper lipid droplet targeting, are also S-acylated. All of these lipid droplet homeostasis proteins are S-palmitoylated.
Removal of palmitic acid from proteins containing the S-acylation modification, such as FAT/CD36, is catalyzed by an enzyme referred to as acyl-protein thioesterase 1 (APT1). The human APT1 enzyme is encoded by the LYPLA1 gene. When fatty acids interact with FAT/CD36 this activates the kinase activity of the associated kinase, LYN (LCK/YES-related novel tyrosine kinase) which then phosphorylates FAT/CD36. The phosphorylation of FAT/CD36 triggers endocytosis, thereby internalizing the associated fatty acid.
Clinical significance is associated with the activity of the APT1 enzyme given that studies have shown changes in the level of activity in pancreatic β-cells of individuals with type 2 diabetes. Loss of APT1 activity in experimental animals is associated with hypersecretion of insulin resulting from the elevated S-palmitoylation of a protein (secretory carrier membrane protein 1, SCAMP1) associated with insulin secretory vesicles.
Protein Prenylation
Prenylation refers to the addition of the 15 carbon farnesyl group or the 20 carbon geranylgeranyl group to acceptor proteins, both of which are isoprenoid compounds derived from the cholesterol biosynthetic pathway. The isoprenoid groups are attached to cysteine residues at the carboxy terminus of proteins in a thioether linkage (C–S–C). A common consensus sequence at the C-terminus of prenylated proteins has been identified and is composed of CAAX, where C is cysteine, A is any aliphatic amino acid (except alanine) and X is the C-terminal amino acid. More than 120 human proteins have been identified that are modified by the addition of a prenyl group. These proteins include the γ-subunit of numerous heterotrimeric G-proteins, members of the Ras superfamily of small GTPases, the nuclear lamins, and several protein kinases and protein phosphatases.
In the course of the prenylation reaction, the prenyl group (either farnesyl or geranylgeranyl) is added to the cysteine in the CAAX motif at the C-terminus of target proteins and the AAX tripeptide is subsequently removed. These prenylation reactions are carried out, in humans, by one of several CAAX isoprenylation enzymes. The major isoprenylation enzymes are farnesyltransferase and geranylgeranyltransferase type I.
Farnesyltransferase and geranylgeranyltransferase function as heterodimers composed of a common α-subunit and a distinct β-subunit. The common α-subunit is encoded by the FNTA gene (farnesyltransferase, CAAX box, alpha). The β-subunit of farnesyltransferase is encoded by the FNTB gene. The β-subunit of geranylgeranyl transferase type I is encoded by the PGGT1B gene (protein geranylgeranyltransferase type I subunit beta).
Following protein isoprenylation the AAX tripeptide is removed by CAAX proteases. The major CAAX protease in humans is encoded by the RCE1 gene (Ras converting CAAX endopeptidase 1).
The last step in protein isoprenylation involves the methylation of the carboxylate group of the prenylated cysteine in a reaction utilizing S-adenosylmethionine as the methyl donor. Humans express three enzymes that carry out the isoprenylcysteine methyltransferase reaction with the most abundant being encoded by the isoprenylcysteine carboxyl methyltransferase (ICMT) gene. The protein encoded by the ICMT gene is also known as protein-S-isoprenylcysteine O-methyltransferase
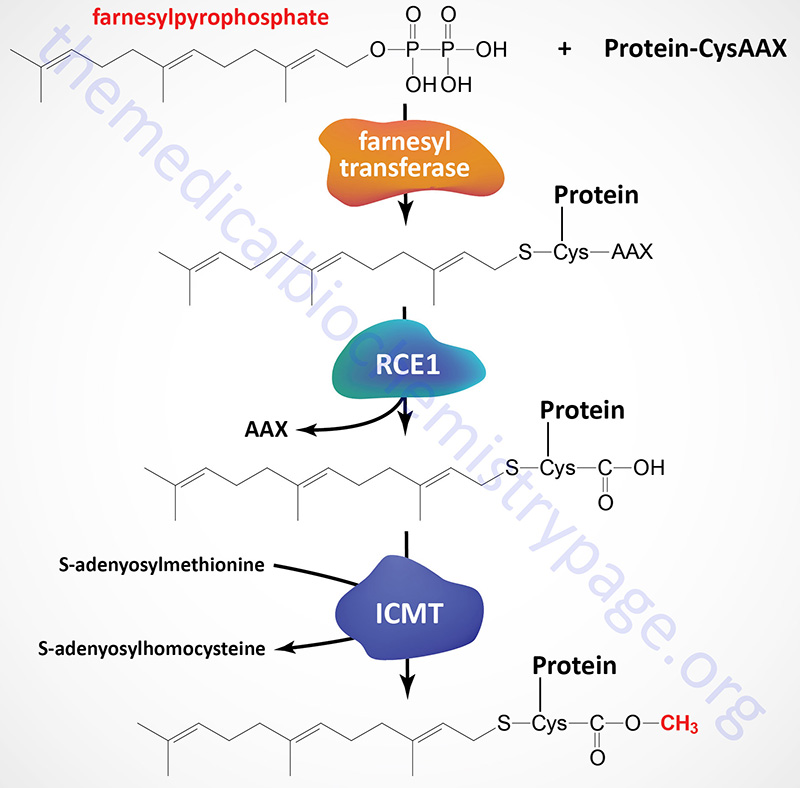
In addition to numerous prenylated proteins that contain the CAAX consensus, prenylation is known to occur on proteins of the RAB family of RAS-related G-proteins. There are 65 proteins in this family that are prenylated at either a CC or CXC element in their C-termini. The RAB family of proteins are involved in signaling pathways that control intracellular membrane trafficking.
Some of the most important proteins whose functions depend upon prenylation are those that modulate immune responses. These include proteins involved in leukocyte motility, activation, and proliferation and endothelial cell immune functions. It is these immunomodulatory roles of many prenylated proteins that are the basis for a portion of the anti-inflammatory actions of the statin class of cholesterol synthesis-inhibiting drugs due to a reduction in the synthesis of farnesylpyrophosphate and geranylpyrophosphate and thus reduced extent of inflammatory events. Other important examples of prenylated proteins include the oncogenic GTP-binding and hydrolyzing protein RAS and the γ-subunit of the visual protein transducin, both of which are farnesylated. In addition, as indicated above, numerous heterotrimeric G-proteins have their γ-subunits modified by geranylgeranylation.
Protein ADP-Ribosylation
Protein ADP-ribosylation is a form of posttranslational modification that was originally identified in the eary 1960s as occurring on eukaryotic elongation factor 2 (eEF-2) as a consequence of infection by Corynebacterium diphtheriae. Subsequent to this initial characterization of an ADP-ribosylating reaction catalyzed by a bacterial toxin, numerous proteins have been identified that undergo ADP-ribosylation as a natural mechanism of regulating their function.
The process of protein ADP-ribosylation is a reversible posttranslational modification that results in the covalent attachment of either a single ADP-ribose unit (monoADP-ribose: MAR; referred to as MARylation) or polymers of ADP-ribose units (polyADP-ribose: PAR; referred to as PARylation) on the side chain of asparagine, aspartic acid, glutamic acid, arginine, lysine, or cysteine resides within target proteins. Some target proteins harboring a MAR are substrates for polyADP-ribose polymerases which then generates PAR modified proteins.
Protein ADP-ribosylation is catalyzed by a diverse group of ADP ribosyl transferase (ADPRT; also designated ART) enzymes that use ADP-ribose units derived from NAD+ to catalyze the ADP-ribosylation reaction. In addition to the bacterial toxins there are three different ADP-ribosyltransferase families in yeast and animals. These three families are the arginine-specific ecto-enzymes (ARTC), the sirtuins (SIRT), and the polyADP-ribose polymerases (PARP).
These various ADP-ribosylating enzymes are referred to as ADP-ribose “writers”.
In the context of ADP-ribosylation these enzymes release nicotinamide, NAM. The NAM is salvaged for the resynthesis of NAD+ as described in detail in the Vitamin B3: Metabolism and Functions page.
PolyADP-Ribose Polymerases: PARP
The polyADP-ribose polymerase (PARP) enzymes represent a group of 17 enzymes in humans that posttranslationally transfer the negatively charged ADP-ribose group from donor NAD+ molecules onto their target proteins. The PARP enzymes can be characterized based upon structural domains and functions as well as by whether they catalyze monoADP ribosylation or polyADP ribosylation. Using the terminology of histone modifications and mRNA modifications the PARP enzymes are termed “writers” of ADP-ribose. Recognition of monoADP-ribose (MAR) or polyADP-ribose (PAR) in proteins is the role of “reader” proteins. The removal of MAR or PAR residues is the function of “eraser” enzymes.
The 17 PARP family enzymes are related to the the ADP-ribosyltransferase produced by the Corynebacterium diphtheriae bacterium and so these enzymes are also referred to as ADP-ribosyltransferase diphtheria toxin-like enzymes which use the ARTD acronym.
The process of protein ADP-ribosylation is most commonly referred to as PARylation. The PARylation of target proteins in mammals is necessary to control a wide array of cellular processes, such as DNA repair, modulation of chromatin structure, transcriptional regulation, RNA interference, mitochondrial function, modulation of inflammation, and regulation of the cell cycle. The process of PARylation has also been shown to have a critical role during the cellular responses to stress, responses that require rapid adaptation. The responses mediated by PARylation include the inhibition of protein-protein or protein-DNA interactions, the formation of scaffolds that alter protein localization and promote protein interactions, and the regulation of other protein modifications, such as ubiquitylation.
Although there are 17 genes in the human PARP family, the functions and mechanisms of PARylation have been best characterized for the enzymes encoded by the PARP1 and PARP2 genes as well as the TNKS and TNKS2 (the tankyrases) genes. The TNKS gene is also known as PARP5A and the TNKS2 gene is also known as PARP5B.
The DNA-dependent PARP are encoded by the PARP1, PARP2, and PARP3 genes. The PARP1, PARP2, and PARP3 genes have also been referred to as ARTD1, ARTD2, and ARTD3 respectively. These three PARP enzymes contain a DNA-binding domain at the N-terminus. Upon binding to the ends of DNA, the activation sites in their enzymatic domains are fully exposed, which activates the PARylation reaction. Although PARP3 does not have a distinctive DNA binding domain it is classified in this subfamily of PARP enzymes since it is activated in the presence of DNA in vitro.
The tankyrases are so-called due to the characterization of the original enzyme as TERF1-interacting ankyrin-related ADP-ribose polymerase (where TERF1 stands for telomere repeat-binding factor 1). The tankyrases are encoded by the TNKS (PARP5A) and TNKS2 (PARP5B) genes. The tankyrases contain multiple ankyrin repeat clusters that function as protein-protein interaction domains that facilitate their interaction with appropriate substrates.
The Cys-Cys-Cys-His zinc finger (CCCH) domain-containing and Trp-Trp-Glu (WWE) domain-containing enzymes are encoded by the PARP7, PARP12, and ZC3HAV1 genes. The ZC3HAV1 gene encoded mRNA undergoes alternative splicing resulting in two distinct protein isoforms that were originally identified as PARP13.1 and PARP13.2. The CCCH domain is an RNA-binding domain and the WWE domain is a protein-protein interaction domain. Through the interaction with RNA the CCCH domain-containing PARP enzymes regulate the stability and translation of specific target mRNAs and modulate the miRNA silencing pathway. The activity of the ZC3HAV1 encoded enzymes have been shown to be important in modulating cellular responses to stress and to restrict the activity of oncogenic viruses. The ZC3HAV1 enzyme also represses the activity of the prosurvival cytokine receptor, tumor necrosis factor (TNF)-related apoptosis-inducing ligand receptor 4 (TRAILR4). This latter activity suggests that ZC3HAV1 activity may exert protective effects against malignant transformation and cancer development.
The PAR-binding macrodomain-containing (macroPARP) enzymes are encoded by the PARP9, PARP14, and PARP15 genes. The macroPARP enzymes contain multiple macro domains at the N-terminus as well as an ADP-ribosyltransferase domain at the C-terminus. The macro domain functions as an ADP-ribose binding module that allows the macroPARP domain to bind to ADP-ribosylated substrates. Through the C-terminal ADP-ribosyltransferase domain the macroPARP then add additional ADP-ribose residues to target proteins. The precise activity of the PARP15 encoded enzymes remains largely uncharacterized, whereas the PARP9 and PARP14 encoded enzymes regulate gene transcription most critically during the processes of macrophage activation.
In addition to classification based upon functional domains, the PARP family enzymes can also be categorized according to their catalytic activities. This classification divides the family into monoADP ribosyltransferases and polyADP ribosyltransferases. However, the full activity of all of the proteins encoded by the 17 PARP genes has yet to be realized.
The monoADP ribosyltransferases are commonly identified as MART and include the enzymes encoded by the PARP3, PARP4, PARP6, PARP10, PARP14, PARP15, and PARP16 genes. As the identification implies the MART enzymes catalyze the addition of a single ADP-ribose unit on target proteins, a process that is referred to as MARylation.
The polyADP ribosylating enzymes are encoded by the PARP1, PARP2, TNKS, and TNKS2 genes. These enzymes catalyze the polymerization of ADP-ribose units through α(1→2) O-glycosidic bonds in linear or branched chains in a process referred to as PARylation.
Table of Human PolyADP-Ribose Polymerase (PARP) Enzymes
| Common Name | Gene | Transferase Name | Localization | Comments |
| PARP1 | PARP1 | ARTD1 | nucleus | is the major polyADP-ribosylating (PAR) enzyme in humans; localized to the nucleus; recruited to double-strand and single-strand breaks in DNA in response to DNA damage; functions as a major mediator of DNA damage repair; activation of PARP1 at sites of DNA damage induces long polyADP-ribose chains on PARP1 itself and other DNA damage response proteins; recruitment of PAR-binding proteins to the site of damage such as ATM (ataxia telangiectasia mutated kinase) and XRCC1 (X-ray repair cross-complementing protein 1) |
| PARP2 | PARP2 | ARTD2 | nucleus | polyADP-ribosylating (PAR) enzyme; localized to the nucleus; functions in the base excision repair (BER) process of DNA damage repair; |
| PARP3 | PARP3 | ARTD3 | nucleus | monoADP-ribosylating (MAR) enzyme; localized to the nucleus; functions in the repair of single-strand DNA breaks; also involved in base-excision repair (BER) of damaged DNA |
| PARP4 | PARP4 | ARTD4 | cytosol | monoADP-ribosylating (MAR) enzyme; localized to the cytosol |
| PARP5A | TNKS | ARTD5 | nucleus and cytosol | polyADP-ribosylating (PAR) enzyme; TNKS refers to tankyrase which is derived from telomeric repeat binding factor 1 (TERF1)-interacting ankyrin-related ADP-ribose polymerase; localized to the cytosol; involved in the regulation of Wnt signaling, vesicular trafficking, and in telomere maintenance; Wnt signal regulation occurs through PARsylation of AXIN1 and AXIN2 which are involved in the β-catenin destruction complex; telomere length regulation occurs through PARsylation of TERF1 |
| PARP5B | TNKS2 | ARTD6 | nucleus and cytosol | polyADP-ribosylating (PAR) enzyme; TNKS2 refers to tankyrase 2 which is derived from telomeric repeat binding factor 1 (TERF1)-interacting ankyrin-related ADP-ribose polymerase 2; localized to the cytosol; activities that are highly similar to those of the TNKS encoded enzyme |
| PARP6 | PARP6 | ARTD17 | monoADP-ribosylating (MAR) enzyme; localized to the cytosol | |
| PARP7 | TIPARP | ARTD14 | monoADP-ribosylating (MAR) enzyme; localized to the cytosol and nucleus; TIPARP refers to TCDD-inducible polyADP ribosyltransferase (where TCDD is the chemical compound: 2,3,7,8-tetrachlorodibenzodioxin) | |
| PARP8 | PARP8 | ARTD16 | monoADP-ribosylating (MAR) enzyme; localized to the cytosol | |
| PAPR9 | PARP9 | ARTD9 | likely to be a monoADP-ribosylating (MAR) enzyme; localized to the cytosol and the nucleus; binds to polyADP ribose units; functions in association with the E3 ubiquitin ligase, DTX3L; involved in DNA damage repair; also involved in interferon-mediated antiviral defense | |
| PARP10 | PARP10 | ARTD10 | nucleus and cytosol | monoADP-ribosylating (MAR) enzyme; localized to the cytosol and the nucleus |
| PARP11 | PARP11 | ARTD11 | monoADP-ribosylating (MAR) enzyme; localized to the cytosol; involved in nuclear envelope stability and nuclear remodeling during spermatogenesis | |
| PARP12 | PARP12 | ARTD12 | cytosol | monoADP-ribosylating (MAR) enzyme; localized to the cytosol |
| PARP13 | ZC3HAV1 | ARTD13 | cytosol | inactive as an ADP-ribosylating enzyme; alternative splicing generates a full-length version (identified as PARP13.1) and a truncated version (identified as PARP13.2); functions as an antiviral protein by recruitment of the cellular RNA degradation machinery; the activity of the PARP13.1 isoform is a more potent as an antiviral than is the PARP13.2 isoform; viral targets include retroviridae (HIV-1), filoviridae (Ebola virus and Marburg virus), togaviridae (sindbis virus and Ross river virus) |
| PARP14 | PARP14 | ARTD8 | cytosol | monoADP-ribosylating (MAR) enzyme; localized to the cytosol and the nucleus; negatively regulates pro-inflammatory cytokines via MARylation of the transcription factor, STAT1 (signal transducer and activator of transcription 1) which reduces the ability of STAT1 to be phosphorylated; also MARylates STAT6 leading to enhanced STAT6-dependent transcription |
| PARP15 | PARP15 | ARTD7 | cytosol | monoADP-ribosylating (MAR) enzyme; localized to the cytosol; likely to be a transcriptional repressor |
| PARP16 | PARP16 | ARTD15 | monoADP-ribosylating (MAR) enzyme; localized to the cytosol; may be involved in the unfolded protein response (UPR) |
The 17 human ADP-ribosylating enzymes described in the preceding Table are organized into several groupings referred to as clades (five clades for human enzymes) based primarily on structure characteristics. Clade 1 consists of PARP1, PARP2, and PARP3. Clade 3 consists of PARP7, PARP9, PARP10, PARP11, PARP12, PARP13, PARP14, and PARP15. Clade 4 consists of PARP5A and PARP5B. Clade 5 consists of PARP4. Clade 6 consists of PARP6, PAPR8, PARP16. Clade 2 consists of plant PARP enzymes.
Sirtuins: SIRT
The sirtuin (SIRT) family of proteins are homologues of the yeast protein silent information regulator 2 (Sir2). Members of the mammalian SIRT family function as NAD+-dependent protein deacetylases or ADP-ribosyltransferases. There are seven mammalian sirtuins encoded by the SIRT1–SIRT7 genes. SIRT1 catalyzes lysine deacetylation of target proteins coupled to the hydrolysis of NAD+. Major targets of SIRT1 (as well as SIRT3) are the histones, deacetylation of which results in altered chromatin structure and changes in gene expression. The sirtuins also deacetylate transcription factors and transcription co-regulators leading to changes in gene expression.
The sirtuins encoded by the SIRT1, SIRT2, SIRT3, and SIRT7 genes function as NAD+-dependent protein deacetylases. The SIRT4 encoded enzyme is a mitochondria localized ADP-ribosyl transferase. The SIRT5 encoded enzyme, in addition to possessing deacetylase activity, functions as a demalonylase and a desuccinylase. The SIRT6 encoded enzyme, in addition to possessing deacetylase activity, functions as a demyristoylase, a depalmitoylase, and an ADP-ribosyl transferase. The deacetylase activity of the sirtuins is not only directed to histones but also to many other acetylated proteins.
The human SIRT1 protein is localized to the nucleus and cytosol. The SIRT2 protein is localized to the cytoplasm. SIRT3, SIRT4, and SIRT5 are localized to the mitochondria, although SIRT3 has been shown to be in the nucleus and the cytoplasm as well. SIRT6 and SIRT7 are only found in the nucleus with SIRT7 in the nucleolus.
A major function of the sirtuins is in the cell survival pathway. Indeed, in studies on the longevity effects of calorie restriction it was found that a major contributor to the positive effects was the activation of the SIRT1 gene. The sirtuins, specifically SIRT1 and SIRT7, inhibit apoptosis via their ability to deacetylate the tumor suppressor protein, p53. Deacetylation of p53 represses its transcriptional activity which decreases its ability to activate apoptotic gene expression pathways.
Sirtuins are also involved in pathways that inhibit inflammation and regulate overall cellular metabolic rates. SIRT1 and SIRT3 activation leads to deacetylation of the kinase identified as LKB1 (also called STK11 and PJS kinase). Deacetylation of LKB1 results in its activation leading to phosphorylation and activation of the master metabolic regulatory kinase, AMPK.
Another major target of sirtuins that results in metabolic regulation is PGC-1α. Activation of PGC-1α by deacetylation results in the activation of gluconeogenic genes and inhibition of glycolytic genes. PGC-1α also activates mitochondrial oxidative phosphorylation in skeletal muscle. Adipose tissue metabolic processes are also regulated by sirtuin function. SIRT1 in conjunction with the transcriptional corepressor complex NCOR1 represses the transcriptional activation of PPARγ resulting in reduced adipogenesis.
Arginine-Specific Ecto-Enzymes (ARTC)
The ARTC family enzymes are related to the ADP-ribosyltransferase produced by the Vibrio cholerae bacterium and so these enzymes are also referred to as ADP-ribosyltransferase cholera toxin-like which uses the same ARTC acronym. This family of ADP-ribose writers are found associated with the plasma membrane or secreted (ARTC5) which accounts for the “ecto” in their nomenclature.
The ADP-ribosylation of arginine (Arg: R) occurs on the guanidino group of arginine through the action of the specific subfamily of ADPRT termed the arginine-specific ecto-enzymes (ARTC). Humans possess four ARTC genes identified as ART1 (also called ARTC1), ART3 (also called ARTC3), ART4, and ART5 (also called ARTC5). The human ART3 gene encodes an enzymatically inactive protein. The human ART4 gene encodes a protein with a monoADP-ribosylation motif but the encoded protein has not been shown to possess enzymatic activity. The ART4 encoded protein represents the Dombrook blood group antigens.
Although there is predicted to be many proteins that undergo ADP-ribosylation on Arg residues, the two most well characterized human proteins modified by Arg-specific ADP-ribosylation are P2X7 and HNP-1. The P2X7 protein is a member of the P2X family of receptors. The P2X receptors are ATP-gated non-selective ion channels (ligand-gated ion channels). Each of the P2X receptors are homo- or heterotrimeric channels that primarily transport Na+, K+, or Ca2+ ions in response to the binding of extracellular ATP. There are seven subtypes of P2X receptors in humans identified as P2X1–P2X7. Arg-specific ADP-ribosylation of P2X7 activates the ion gating of the receptor promoting ion flux through the receptor channel.
The HNP-1 protein is a member of the human neutrophil peptide (HNP) family. Epithelial cells lining human airways and cells recruited to airways participate in innate immune responses, in part, by releasing HNP. ADP-ribosylation of HNP-1 modifies its activity and the modification is only seen in inflammatory conditions or disease such as in the case of patients with asthma, idiopathic pulmonary fibrosis, or a history of smoking. The Arg-specific ADP-ribosylation of HNP-1 is not seen in the airways of normal healthy individuals.
Arginine ADP-ribosylation can be fully reversed by specific ADP-ribosylhydrolases. Humans express a single ADP-ribosylhydrolase encoded by the ADPRH (ADP-ribosylarginine hydrolase) gene and two ADP-ribosylhydrolase-like genes (ADPRHL1 and ADPRHL2).ADP-Ribose “Readers”
In order for the incorporated mono- or polyADP ribose units to be functional they must be detectable by other proteins. These ADP-ribose-binding proteins are referred to as “readers”. ADP-ribose reader proteins contain domains referred to as ADP-ribose-binding domains, ARBD. There are several well characterized types of ARBD including PAR-binding motifs (PBM), PAR-binding zinc finger (PBZ) modules, and WWE domains. The WWE domain is named after the three conserved amino acids in the domain, Tryp (W) and Glu (E). Other ARBD that are not as well characterized include RNA recognition modules (RRM), BRCA1 C-terminal (BRCT) domains, arginine (R)- and glycine (G)-rich motifs (RGG) which are also referred to as glycine–arginine-rich (GAR) domains, and the phosphopeptide-binding Forkhead-associated (FHA) domain.
The PBM, which are short (∼20 amino acid) sequences, bind to an ADP-ribose monomer or the terminal
ADP-ribose moiety in a polyADP ribose (PAR) chain. The PBZ are also short (∼30 amino acids) domains bind the ADP-ribose–ADP-ribose junctions of PAR chains as well as ADP-ribose monomers. The WWE domain binds exclusively
to ADP-ribose oligomers or polymers.
ADP-Ribose “Feeders”
Given that the polyADP ribose polymerases (PARP) are NAD+-dependent enzymes, they require a source of NAD+ in all of the cellular compartments in which they function. The enzymes of NAD+ synthesis and salvage are described in detail in the Vitamin B3: Metabolism and Functions page. The nicotinamide nucleotide adenylyltransferase (NMNAT) enzymes serve as major “feeder” enzymes through their synthesis of NAD+ from nicotinamide mononucleotide (NMN) and ATP. NMNAT-1 is localized to the nucleus. Within the nucleus NMNAT-1 is recruited to chromatin by PARP1 which allows for NMAT1 to directly “feed” NAD+ to PARP1. NMNAT-2 is localized to the cytoplasm and Golgi, and NMNAT-3 is localized to the mitochondria.
ADP-Ribose “Erasers”
The ADP-ribosylation of proteins is not a permanent post-translational modification, indeed PAR polymers turn over rapidly in the cell. There are a number of ADP-ribose “eraser” enzymes encoded in the human genome. The erasers include ADP-ribosyl hydrolase 3 (ARH3), PAR glycohydrolase (PARG), O-acyl-ADP-ribose deacylase 1 (OARD1; also called TARG1), mono-ADP ribosylhydrolase 1 (MACROD1) and MACROD2, and members of the Nudix hydrolase (NUDT) family, specifically NUDT9 and NUDT16. The NUDT family contains 24 genes in the human genome. The term nudix is derived from nucleoside diphosphate-linked moiety X.
The PARG and ARH3 encoded enzymes catalyze the degradation of PAR chains via both endoglycosidic and exoglycosidic activities. The activity of these two enzymes cleaves ribose–ribose bonds but leaves a terminal ADP-ribose moiety attached to the acceptor amino acid residue of the substrate. The OARD1, MACROD1, and MACROD2 encoded enzymes can hydrolyze the ester bond between the ribose and acceptor aspartate and glutamate residues in the target protein which results in complete removal of the ADP-ribose moiety. The NUDT family of hydrolases can hydrolyze PAR chains by targeting the phosphodiester bond in the ADP-ribose unit directly attached to the acceptor amino acid in the target protein. The action of the NUDT enzymes, thus, results in the formation of a phosphoribose moiety attached to the acceptor amino acid.
The ADP-ribose “erasers” have different subcellular localizations and thus, distinct functions within the cell. The MACROD1 and ARH3 proteins localize to the mitochondria. The MACROD2 and OARD1 proteins are predominantly found within the nucleus.
Protein Sulfation
Sulfate modification of proteins occurs at tyrosine residues. As many as 1% of all tyrosine residues present in the eukaryotic proteome are modified by sulfate addition making this the most common tyrosine modification. Tyrosine sulfation is accomplished via the activity of tyrosylprotein sulfotransferases (TPST) which are membrane-associated enzymes of the trans-Golgi network. There are two known TPSTs identified as TPST-1 and TPST-2. The universal sulfate donor for these TPST enzymes is 3′-phosphoadenosyl-5′-phosphosulphate (PAPS). Addition of sulfate occurs almost exclusively on secreted and trans-membrane spanning proteins. Since sulfate is added permanently it is necessary for the biological activity and not used as a regulatory modification like that of tyrosine phosphorylation.
At least 34 human proteins have been identified that are tyrosine sulfated although the total number that are predicted is much higher. In all vertebrates a total of 310 tyrosine sulfated proteins have been identified. It is predicted that the mouse proteome is likely to contain over 2000 tyrosine sulfated proteins. The addition of sulfate to tyrosine is believed to play a role in the modulation of protein-protein interactions of secreted and membrane-bound proteins. The process of tyrosine sulfation has been shown to be critical for the processes of blood coagulation, various immune functions, intracellular trafficking, and ligand recognition by several G-protein-coupled receptors (GPCRs). Some well-known tyrosine sulfated proteins are the coagulation protein factor VIII, and the gut peptides gastrin and cholecystokinin (CCK).
Vitamin C-Dependent Protein Modifications
Modifications of proteins that depend upon vitamin C as a cofactor include proline and lysine hydroxylations and carboxy terminal amidation of neuroendocrine peptides. The hydroxylating enzymes are identified as prolyl hydroxylases and lysyl hydroxylases. The most important hydroxylated proteins are the collagens. Within collagens specific proline residues are hydroxylated by prolyl 3-hydroxylase and prolyl 4-hydroxylase and specific lysine residues are hydroxylated by lysyl hydroxylases.
Humans express three distinct prolyl 3-hydroxylase genes (P3H1, P3H2, and P3H3). Human prolyl 4-hydroxylases are functional as heterotetrameric enzymes composed of two α-subunits (the catalytic subunits) and two β-subunits. Humans express four distinct prolyl 4-hydroxylase α-subunit genes (P4HA1, P4HA2, P4HA3, and P4HTM) and one β-subunit gene (P4HB). The protein encoded by the P4HTM gene is a transmembrane protein localized to the endoplasmic reticulum. The P4HTM protein functions in the modulation of cellular responses to hypoxia by altering the activity of hypoxia inducible factor 1 (HIF1) as a result of stabilization of one of the specific subunits of HIF termed HIF-1α. The proly 3- and prolyl 4-hydroxylases all belong to the large family of 2-oxoglutarate and Fe2+-dependent dioxygenases (2-OGDD or 2OG-oxygenases) whose members are most notable for their roles in histone demethylation and the regulation of cellular responses to hypoxia initiated by HIF1.
Humans express three distinct lysyl hydroxylase genes identified as PLOD1, PLOD2, and PLOD3. PLOD stands for procollagen-lysine, 2-oxoglutarate 5-dioxygenase. The PLOD1 gene is located on chromosome 1p36.22 and is composed of 21 exons that encode a precursor protein of 727 amino acids. The PLOD2 gene is located on chromosome 3q24 and is composed of 22 exons that generate two alternatively spliced mRNAs encoding two isoforms of the PLOD2 enzyme. The PLOD3 gene is located on chromosome 7q22 and is composed of 17 exons that encode a 736 amino acid precursor protein. Each of the lysyl hydroxylase enzymes, like the prolyl hydroxylases, are members of the large family of 2-oxoglutarate and Fe2+-dependent dioxygenases (2OG-oxygenases).
During the process of C-terminal protein amidation the modified amino acid is glycine that is found within the context –XGXX–COOH where X can be any amino acid. The amidation of the C-terminus results in the neutralization of negative charges. The glycine amidation process is a two-step process carried out by the enzyme peptidylglycine α-amidating monooxygenase which is encoded by the PAM gene. The PAM encoded protein is expressed as a preproprotein which is proteolytically processed and possesses two distinct enzymatic activites required for protein amidation. The two activities of the PAM encoded protein are contained within the peptidylglycine α-hydroxylating monooxygenase (PHM) domain and the peptidyl-α-hydroxyglycine α-amidating lyase (PAL) domain. The PAM gene is located on chromosome 5q21.1 and is composed of 28 exons that generate six alternatively spliced mRNAs that are predicted to encode precursor proteins that may all be proteolytically processed to an active enzyme similar to the 973 amino acid precursor identified as isoform e. Several peptide hormones such as oxytocin and vasopressin have C-terminal amidation.
Vitamin K-Dependent Protein Modifications
Vitamin K is a cofactor in the carboxylation of glutamic acid residues catalyzed by the enzyme gamma-glutamyl carboxylase (γ-glutamyl carboxylase). The result of this type of reaction is the formation of a γ-carboxyglutamate (gamma-carboxyglutamate), referred to as a gla residue. The gene encoding γ-glutamyl carboxylase is identified as GGCX and is located on chromosome 2p11.2. The GGCX gene spans 13 kbp and consists of 15 exons encoding a 758 amino acid protein. The γ-glutamyl carboxylase protein is an integral membrane protein with three transmembrane spanning domains associated with microsomal membranes.
The overall reaction, resulting in the incorporation of a gla-residue, actually involves a series of three distinct reactions. The reaction catalyzed by γ-glutamyl carboxylase is the one that incorporates the gla-residue but two additional enzyme activities are required to convert vitamin K back to its active hydroquinone (quinol) form. The latter two reactions are catalyzed by vitamin K epoxide reductase (VKORC1). These latter two reactions involve a dithiol conversion to a disulfide. An additional enzyme called vitamin K quinone reductase (VKQR) can also carry out the conversion of the quinone form of vitamin K (as formed by the action of VKORC1 or as obtained from the diet) to the hydroquinone form. This latter reaction utilizes NADH as a co-factor.
The formation of gla residues within several proteins of the blood clotting cascade is critical for their normal function. The presence of gla residues allows the protein to chelate calcium ions and thereby render an altered conformation and biological activity to the protein. The coumarin-based anticoagulants, warfarin and dicumarol function by inhibiting the second and third enzymes of the overall carboxylation reaction.
Selenoproteins
Selenium is a trace element and is found as a component of several prokaryotic and eukaryotic enzymes that are involved in redox reactions. Two critical re-dox enzyme families that require selenocysteine residues are the glutathione peroxidase and thioredoxin reductase families.
Glutathione peroxidase is a critical enzyme involved in the protection of red blood cells from reactive oxygen species (ROS). This enzyme is a component of a re-dox system that also involves the enzyme glutathione reductase and NADPH as the terminal electron donor. This system is required for the continued reduction of oxidized glutathione (GSSG) and represents the single most significant system requiring continued glucose metabolism via the Pentose Phosphate Pathway in erythrocytes as the means for the production of the NADPH. Glutathione (GSH) becomes oxidized in the context of reducing various ROS and peroxides and to continue in this capacity the oxidized form needs to be continuously reduced.
Glutathione Peroxidases
Humans express eight different glutathione peroxidase genes identified as GPX1 through GPX8.
- The GPX1 gene is located on chromosome 3p21.3 and is composed of 2 exons that generate two alternatively spliced mRNAs. The GPX1 coding region contains a polyalanine tract in the N-terminal region of the protein. There are several alleles of this gene that have five, six, or seven alanine repeats. The allele with five alanine repeats has been shown to be highly correlated to increased risk for development of breast cancer. The enzyme encoded by the GPX1 gene (GPx1) is found in the cytosol of nearly all cell types in humans. GPx1 functions almost exclusively to reduce hydrogen peroxide (H2O2) to water.
- The GPX2 gene is located on chromosome 14q24.1 and is composed of 4 exons.
- The GPX3 gene is located on chromosome 5q33.1 and is composed of 5 exons. The protein encoded by the GPX3 gene, GPx3, is an extracellular enzyme found primarily in the plasma.
- The GPX4 gene is located on chromosome 19p13.3 and is composed of 8 exons. The GPX4 encoded enzyme, GPx4, is localized to the intestines and is an extracellular enzyme as well.
- The GPX5 gene is located on chromosome 6p22.1 and is composed of 7 exons. The resultant GPX5 mRNA does not contain the canonical selenocysteine codon (UGA) and thus, the resulting protein does not contain a selenocysteine residue. Expression of the GPX5 gene is regulated by androgens and the gene is expressed exclusively in the epididymis in the male reproductive tract where the expressed protein, GPx5, is involved in protecting spermatozoa membranes from the damaging effects of lipid peroxidation.
- The GPX6 gene is located on chromosome 6p22.1 and is composed of 5 exons. GPX6 expression is restricted to embryonic tissues and the adult olfactory system.
- The GPX7 gene is located on chromosome 1p32 and is composed of 3 exons.
- The GPX8 gene is located on chromosome 5q11.2 and is composed of 3 exons.
Thioredoxin Reductases
As the name of the enzyme implies, thioredoxin reductase is involved in the reduction of thioredoxin which itself is principally involved in the reduction of oxidized disulfide bonds in proteins. The reduction of these disulfide bonds results in oxidation of thioredoxin which then is reduced by thioredoxin reductase. The overall process, like the glutathione peroxidase system, requires NADPH as the terminal electron donor for the reduction process. A critically important reaction that is coupled to the thioredoxin system is the formation of deoxynucleotides.
Humans contain three thioredoxin reductase genes that encode three distinct enzymes identified as TrxR1, TrxR2, and TrxR3.
- The TrxR1 enzyme is encoded by the TXNRD1 gene located on chromosome 12q23.3 and is composed of 18 exons that generate seven alternatively spliced mRNAs encoding five different isoforms of TrxR1. The TrxR1 enzyme is functional in the cytosol and is primarily involved in the maintenance of the ribonucleotide reductase system.
- The TrxR2 enzyme is encoded by the TXNRD2 gene located on chromosome 22q11.21 and is composed of 19 exons that generate two alternatively spliced mRNAs resulting in two different isoforms of TrxR2. The TrxR2 enzyme is functional in the mitochondria where it is principally involved in the detoxification of reactive oxygen species (ROS) produced in this organelle.
- The TrxR3 enzyme is encoded by the TXNRD3 gene located on chromosome 3q21.3 and is composed of 16 exons that generate two alternatively spliced mRNAs resulting in two different isoforms of TrxR3. TrxR3 is a testes-specific isoform of the enzyme.
The enzymes of the deiodinase family are also important selenocysteine-containing enzymes. Clinically relevant enzymes in this family are the thyroid deiodinases that are critical for the maturation and catabolism of the thyroid hormones. Humans express three different thyroid deiodinase genes identified as DIO1, DIO2, and DIO3.
- The DIO1 gene is located on chromosome 1p33–p32 and is composed of 4 exons that generate four alternatively spliced mRNAs. The enzyme encoded by the DIO1 gene, thyroxine deiodinase type I (also called iodothyronine deiodinase type I) is involved in the peripheral tissue conversion of thyroxine (T4) to bioactive form of thyroid hormone, tri-iodothyronine (T3). In addition to its role in the generation of T3, thyroxine deiodinase I is involved in the catabolism of thyroid hormones.
- The DIO2 gene is located on chromosome 14q24.2–q24.3 and is composed of 6 exons that generate four alternatively spliced mRNAs. The enzyme encoded by the DIO2 gene, iodothyronine deiodinase type II, is also involved in the conversion of T4 to T3 but does so within the thyroid gland itself. The activity of iodothyronine deiodinase II has been associated with the thyrotoxicosis of Graves disease.
- The DIO3 gene is located on chromosome 14q32 and is an intronless gene (is a single exon gene) that encodes a protein of 304 amino acids. The enzyme encoded by the DIO3 gene is involved only in the inactivation (catabolism) of T3 and T4. Expression of the DIO3 gene is highest the female uterus during pregnancy and in fetal and neonatal tissue suggesting a role for this enzyme in the regulation of thyroid hormone levels and functions during early development.
Co-Translational Incorporation of Selenocysteine
Selenocysteine incorporation in eukaryotic proteins occurs co-translationally at UGA codons (normally stop codons) via the interactions of a number of specialized proteins and protein complexes. In addition, there are specific secondary structures in the 3′ untranslated regions of selenoprotein mRNAs, termed SECIS (selenocysteine insertion sequence) elements, that are required for selenocysteine insertion into the elongating protein. One of the complexes required for this important modification is comprised of a selenocysteinyl tRNA [(Sec)-tRNA(Ser)Sec] and its specific elongation factor identified as selenoprotein translation factor B (SelB). SelB is also commonly called eukaryotic elongation factor, selenocysteine-tRNA-specific (EEFsec or EFsec). The protein that is involved in the interaction of the SECIS element with the (Sec)-tRNA(Ser)Sec is referred to as SECIS binding protein 2, SECISBP2. Additional proteins involved in the selenocysteine biosynthesis pathway include two selenophosphate synthetases, SEPHS1 and SEPHS2, Sep (O-phosphoserine) tRNA:Sec (selenocysteine) tRNA synthase (encoded by the SEPSECS gene and originally identified as soluble
Table of Selenocysteine Containing Proteins
The following Table is not intended to represent a complete list of all known selenocysteine containing proteins, it is just a representative list.
| Protein Name(s) | Gene(s) | Functions / Comments |
| glutathione peroxidases 1, 2, 3, 4, and 6 | GPX1, GPX2, GPX3, GPX4, GPX6 | humans express eight glutathione peroxidases, five of which are selenocysteine containing enzymes; major class of anti-oxidant enzymes; involved in numerous reaction pathways involved in the reduction of hydrogen peroxide (H2O2) such as is critical in the erythrocyte |
| iodothyronine (thyroxine) deiodinases 1, 2, and 3 | DIO1, DIO2, DIO3 | catalyze the conversion of the thyroid hormone thyroxine (T4) to triiodothyronine (T3); also involved in the catabolism of T3 and T4 to inactive molecules |
| methionine sulfoxide reductase B1 | MSRB1 | functions in the protection of cells from oxidative stress; catalyzes the reduction of methionine-R-sulfoxides to methionine; highly expressed in the liver and kidneys |
| selenophosphate synthetase 2 | SEPHS2 | catalyzes the synthesis of selenophosphate from selenium (as the selenide ion: Se2–) and ATP; selenophosphate is the selenium donor in the co-translational incorporation of selenocysteine residues |
| selenoprotein F | SELENOF | exact function has not been determined; found in the endoplasmic reticulum (ER) associated with UDP-glucose:glycoprotein glucosyltransferase (UGTR; encoded by the UGGT1 gene); UGTR is key component of the glycoprotein ER quality control (ERQC) system |
| selenoprotein H | SELENOH | a nucleolar localized enzyme involved in regulating redox status of cells |
| selenoprotein I | SELENOI | CDP-alcohol phosphatidyltransferase class-I family; catalyzes the transfer of phosphoethanolamine from CDP-ethanolamine to diacylglycerol in the synthesis of phosphatidylethanolamine, PE; enzyme commonly called CDP-ethanolamine:1,2-diacylglycerol ethanolaminephosphotransferase and also called diacylglycerol ethanolaminephosphotransferase |
| thioredoxin reductases 1, 2, and 3 | TXNRD1, TXNRD2, TXNRD3 | these enzymes reduce oxidized thioredoxin such as that which is generated during the synthesis of deoxynucleotides via the action of ribonucleotide reductase |