
Last Updated: February 13, 2026
Introduction to Propionic Acidemia
Propionic acidemia is a member of the family of disorders termed organic acidemias. Propionic acidemia is an autosomal recessive disorder that results from mutations in either of the two genes encoding the subunits of the mitochondrial enzyme, propionyl-CoA carboxylase (PCC). The incidence of propionic acidemia is approximately 1:100,000 live births.
Propionic acidemia is a clinically heterogeneous disorder divided into a neonatal lethal form and a late-onset form that appears in older children or adults. The neonatal-onset form manifests shortly after birth in an otherwise healthy infant. The neonatal-onset form of the disease is the more common form of propionic acidemia.
Molecular Biology of Propionic Acidemia
The metabolism of four amino acids (valine, methionine, isoleucine, and threonine), as well as the oxidation of fatty acids with an odd number of carbon atoms, results in the generation of the organic acid, propionyl-CoA. The propionyl-CoA is converted, to succinyl-CoA via a series of three mitochondrially-localized enzymes. The succinyl-CoA can then enter the TCA cycle for further oxidation.
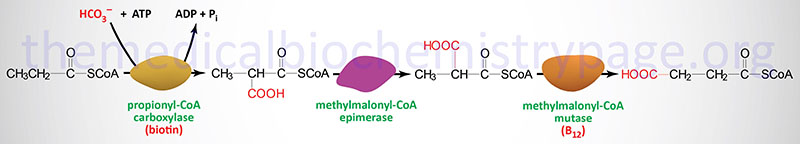
The enzymes required for this conversion are propionyl-CoA carboxylase, methylmalonyl-CoA epimerase, and methylmalonyl-CoA mutase, respectively. Propionyl-CoA carboxylase is called an ABC enzyme due to the requirements for ATP, Biotin, and CO2 for the reaction. The biotin-dependent carboxylating enzymes in mammals are multifunctional and contain three distinct enzymatic activities that may be contained in a single protein or in different subunits of the multisubunit enzymes. These three enzymatic activities are the biotin carboxylase (BC), the carboxyltransferase (CT), and the biotin carboxyl carrier protein (BCCP) activities.
The clinical significance of methylmalonyl-CoA mutase in this pathway is that it is one of only two enzymes that requires a vitamin B12-derived co-factor for activity. The other B12-requiring enzyme is methionine synthase (see the Amino Acid Biosynthesis page). This propionyl-CoA conversion pathway is remembered by the mnemonic as the VOMIT pathway, where V stands for valine, O for odd-chain fatty acids, M for methionine, I for isoleucine, and T for threonine.
Propionyl-CoA carboxylase functions as a heterododecameric enzyme (subunit composition: α6β6) and the two different subunits are encoded by the PCCA and PCCB genes, respectively. The α-subunit of PCC contains the three activities of a typical biotin-dependent carboxylase. The BC domain of the α-subunit resides in the N-terminal region and the BCCP domain resides in the C-terminal region. Propionyl-CoA carboxylase has a domain, called the biotin transfer (BT) domain, that is not found in the other biotin-dependent carboxylase family enzymes. The BT domain is found in the middle of the α-subunit and is required for interactions between the α- and β-subunits.
The PCCA gene is located on chromosome 13q32.3 and is composed of 34 exons that generate eleven alternatively spliced mRNAs, each of which encode a unique protein isoform.
The PCCB gene is located on chromosome 3q22.3 and is composed of 17 exons that generate two alternatively spliced mRNAs encoding precursor proteins of 539 amino acids (isoform 1) and 559 amino acids (isoform 2).
Numerous mutations have been identified in both the PCCA and PCCB genes associated with propionic acidemia. These mutations include missense and nonsense mutations as well as splice site mutations. Null mutations in the PCCA gene result in the most severe forms of propionic acidemia whereas splice site mutations result in milder forms. Two PCCA null mutations are common in propionic acidemia. These mutations result in conversion of an Arg codon at amino acid 288 to a stop codon (designated R288X) and a Ser codon at 537 being converted to a stop codon (designated S537X). The most common mutation identified in the PCCB gene is a missense mutation at codon 168 that changes an Asp to a Lys (designated (E168K). Several missense mutations in the PCCB gene interfere with the interactions of the α- and β-subunits. These mutations are A497V, R512C, and L519P. The nonsense mutation at codon 531 (W531X) also disturbs subunit interactions.
Metabolic Disturbances in Propionic Acidemia
As the name of the disorder implies, defects in the function of propionyl-CoA carboxylase (PCC) lead to accumulation of propionyl-CoA. Measurement of propionyl-CoA levels in humans is difficult so the assay that is used is to measure for the carnitine ester (propionyl-carnitine) in blood and urine. Additional metabolites that are elevated in propionic acidemia, and can be assayed in the urine, include methylcitrate and 3-hydroxypropionate. Methylcitrate is formed by conjugation of propionyl-CoA with oxaloacetic acid from the TCA cycle. Production of 3-hydroxypropionate is thought to result from β-oxidation of propionic acid.
The accumulation of propionyl-CoA, within the cell, leads to inhibition of mitochondrial metabolism thereby resulting in reduced production of ATP, GTP, and citrate. The normal product of the action of PCC is the TCA cycle intermediate, succinyl-CoA. Thus, PCC participates in anaplerotic replenishment of TCA intermediates. This anaplerotic function of PCC is particularly important in the brain due to the diversion of the TCA cycle intermediate, 2-oxoglutarate (α-ketoglutarate) to the synthesis of the neurotransmitter glutamate and subsequent conversion of glutamate to the neurotransmitter GABA.
In addition to the direct contribution of PCC to TCA cycle anaplerosis being disturbed in patients with propionic acidemia, there are also disturbances in additional functions of the TCA cycle. Additionally, propionic acidemia results in disturbances of other mitochondrial functions such as oxidative phosphorylation. Propionic acid, as well as the toxic metabolites that accumulate in propionic acidemia, have been shown to inhibit the pyruvate dehydrogenase complex (PDHc), the 2-oxoglutarate dehydrogenase complex (OGDH; also referred to as α-ketoglutarate dehydrogenase), and succinyl-CoA synthetase. Methylcitrate has been shown to inhibit the function of isocitrate dehydrogenase (encoded by the IDH3 gene) and citrate synthase.
The toxic metabolites in propionic acidemia can also interfere with the important transaminases, alanine transaminase (ALT) and aspartate transaminase (AST). Reduced ALT and AST function will lead to reduced levels of oxaloacetate, malate, and pyruvate which in turn will lead to reduced levels of other TCA cycle intermediates.
Propionyl-CoA also directly inhibits carbamoyl phosphate synthase 1 (CPS1) of the urea cycle. Propionyl-CoA has also been shown to inhibit the activity of N-acetylglutamate synthase (NAGS), resulting in a form of secondary NAGS deficiency. NAGS is the enzyme which generates the critical activator of CPS1, N-acetylglutamate. The result of these urea cycle dysfunctions by propionyl-CoA is hyperammonemia which is highly detrimental to brain function.
Due to the role of PCC in the oxidation of fatty acids with an odd number of carbon atoms these fatty acids accumulate in patients with propionic acidemia. Measurement, in blood, of the levels of pentadecanoic acid (C15:0), heptadecanoic acid
(C17:0), and 2-pentadecenoic acid (C15:1) are useful in the diagnosis of propionic acidemia. In addition, these fatty acids are found in the membranes of erythrocytes in propionic acidemia patients.
Propionic acid, the carnitine metabolite, propionyl-carnitine, and methylcitrate have been shown to interfere with the normal function of cardiac potassium channels, specifically the delayed rectifier channel identified as I(Kr). This channel is encoded by the KCNH2 gene and the encoded protein associates with a regulatory subunit encoded by the KCNE2 gene. The I(Kr) channel is normally responsible for the repolarization of cardiomyocytes, in particular ventricular cardiomyocytes. The effect of these metabolites on the potassium channel can explain the cardiac arrhythmias seen in propionic acidemia patients. Propionyl-CoA and propionyl-carnitine are also essential sources of energy for the heart during ischemia.
Clinical Features of Propionic Acidemia
The neonatal-onset form of propionic acidemia is characterized by poor feeding, vomiting, and fatigue in the first days of life in a previously healthy infant. The early onset symptoms of the neonatal form include vomiting, irritability, and temperature instability. If the disorder is left untreated it can lead to severe lethargy, seizures, coma, and eventually death. Metabolic disturbances that are common to the neonatal form of propionic acidemia include ketonuria, hypoglycemia, hyperammonemia, and high anion gap metabolic acidosis.
The late-onset form of propionic acidemia is a milder form of the disorder that presents with developmental regression, chronic vomiting, protein intolerance, failure to thrive, and hypotonia. Occasionally individuals have pathology associated with the basal ganglia that manifest with dystonia and choreoathetosis, and cardiomyopathy.
In patients where there is a suspicion of propionic acidemia analysis of urine organic acids, blood amino acids, and blood acylcarnitine profiles should be performed in order to exclude other metabolic disorders, in particular the methylmalonic acidemias. However, a definitive diagnosis propionic acidemia requires genetic testing for PCCA and PCCB mutations. In addition, DNA testing allows for determination of disease severity.
Treatment of Propionic Acidemia
Treatment of propionic acidemia requires a diet with sufficient levels of amino acids for growth but not in excess of methionine, isoleucine, valine, and threonine. In addition the diet should be restricted in fatty acids with odd numbers of carbon atoms. Supplementation with carnitine prevents loss through the esterification of propionic acid with carnitine. Individuals that experience acute episodic hyperammonemia will require hemodialysis to detoxify the blood.