
Last Updated: March 25, 2026
Introduction to NAGS Deficiency
N-acetylglutamate synthase deficiency (NAGSD) is most commonly the result of the inheritance of mutations in the NAGS gene which encodes N-acetylglutamate synthase. NAGSD manifests in two forms identified as neonatal onset and as adult onset. Inherited NAGSD is an extremely rare autosomal recessive disorder with a frequency estimated to be 1 in 2,000,000 live births.
N-acetylglutamate synthase catalyzes the formation of N-acetyglutamate which is the obligate activator of the urea cycle enzyme, carbamoyl phosphate synthetase 1 (CPS1).
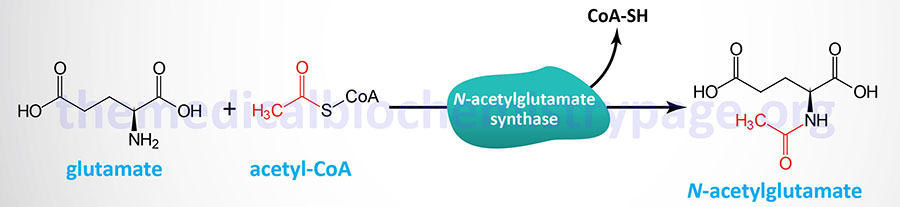
Because of the role of N-acetylglutamate in the activation of CPS1, NAGSD represents a disorder that is associated with a secondary defect in the processes of the urea cycle. The symptoms of inherited NAGSD mimic those of CPS1 deficiency, CPSD and include elevated plasma ammonia, glutamine, and alanine, reduced or absent citrulline, normal urinary orotate, and respiratory alkalosis.
Deficiency of NAGS activity can also result through secondary inhibition. Secondary inhibition of NAGS activity occurs in a number of conditions associated with the accumulation of short fatty acids (SCFA) as in the various organic acidemias. Accumulation of of the CoA derivatives of SCFA is thought to inhibit NAGS activity through either competition with acyl-CoA or through reductions in the overall coenzyme A pool, or both. In the case of the disorders identified as propionic acidemia and isovaleric acidemia, the accumulating propionyl-CoA and isovaleryl-CoA, respectively, is associated with inhibition of NAGS activity.
Molecular Biology of NAGS Deficiency
The N-acetylglutamate synthase gene (NAGS) is located on chromosome 17q21.31 and is composed of 7 exons that encode a 534 amino acid mitochondria-localized protein. The mitochondrial targeting sequence reside in the N-terminal region of the protein. The major tissues of expression of the NAGS gene are the liver, as expected for its role in the urea cycle, and the intestines.
The functional NAGS enzyme is a homotrimer but higher order multimers have been shown to form in the presence of the allosteric activator of the enzyme, arginine. The presence of arginine results in from three to fivefold increases in the level of NAGS activity.
Only a very few mutations have thus far been identified in the NAGS gene associated with inherited NAGSD. These mutations include missense, nonsense, frameshift, and splice site mutations. Missense mutations are the most common and are most often associated with the neonatal onset form of NAGSD.
Clinical Features of NAGS Deficiency
Clinically, inherited cases of NAGS deficiency can be divided into neonatal onset and late onset forms.
Neonatal cases often present with poor feeding or feeding intolerance, irritability, vomiting, lethargy, hypertonia and/or hypotonia, encephalopathy, respiratory distress, hepatomegaly, seizures, and tachypnea.
Late onset NAGS deficiency shows variability in age of onset and disease severity that could be attributed to a number of genetic and environmental factors. The common symptoms associated with late-onset NAGSD include vomiting, confusion or disorientation, ataxia, lethargy, decreased level of consciousness, seizures, and hypotonia. The variability in presenting symptoms in the late-onset form of NAGSD can be due to the amounts and activities of individual proteins, to differences in cellular and tissue organization, as well as differences in the type of diet consumed by affected patients. Indeed, in many cases of late-onset NAGSD affected individuals exhibited a history of avoidance of high protein foods.
Treatment of NAGS Deficiency
As is typical in other UCD, acute episodes of hyperammonemia can be treated with intravenous administration of Ammunol® and with oral Buphenyl® for chronic adjunctive therapy of hyperammonemia. The mechanism of action of these compounds is detailed in the Urea Cycle Disorders: Overview page.
The treatment of NAGSD also includes the administration of the N-carbamoylglutamate which is an analog of N-acetylglutamate (NAG) and activates CPS1 similarly to NAG.