

Last Updated: July 13, 2026
Introduction to MLD
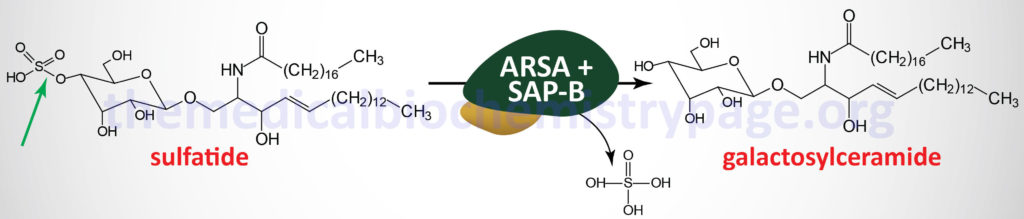
Metachromatic leukodystrophy (MLD) is an autosomal recessive disorder that belongs to the family of disorders identified as lysosomal storage disorders. This disorder is characterized by the lysosomal accumulation of sulfated glycolipids, specifically 3-O-sulfogalactosyl-containing glycolipids (galactocerebroside sulfate), as a consequence of defects in the lysosomal hydrolase, arylsulfatase A (ARSA). Because the desulfurating action of ARSA depends on its interaction with one of the sphingolipid activator proteins, specifically saposin B (see the Gaucher disease page for an explanation of the role of the saposins), defects in saposin B can also lead to MLD, although this latter is a very rare occurrence.
Molecular Biology of MLD
Arylsulfatase A is encoded by the ARSA gene. The ARSA gene is a small gene located on chromosome 22q13.33 spanning only 3.2 kb and is composed of 9 exons that generate six alternatively spliced mRNAs. Four of these mRNAs encode the same 509 amino acid precursor protein (isoform a) and two encode a 423 amino acid precursor protein (isoform b).
As of 2025 there have been a total of 197 pathogenic variants identified in the ARSA gene resulting in MLD. The ultimate severity of the disease is a consequence of how much or how little residual enzyme activity remains in the presence of a given pathogenic variant.
The accessory protein, saposin B, is derived from the processing of the prosaposin protein. Prosaposin is encoded by the PSAP gene that is located on chromosome 10q22.1 and is composed of 15 exons that generate three alternatively spliced mRNAs, each of which encode distinct protein isoforms.
As of 2025 a total of 47 pathogenic variants in the PSAP gene, that encodes prosaposin, have been identified, some of which affect the saposin B protein and as such are associated with metachromatic leukodystrophy.
Clinical Features of MLD
The major site of 3-O-sulfogalactosy-containing glycolipids is the myelin sheaths of central and peripheral neurons. Because of this location the clinical manifestations of MLD are predominately neurological in nature. Histopathologically, MLD is characterized by demyelination of central and peripheral nerves. The accumulation of sulfated glycolipids in the lysosomes results in the characteristic metachromatic staining of the tissues, hence the derivation of the name of this disease.
There is a wide degree of phenotypic variation in MLD and as such the disorder is divided into late infantile, early juvenile, late juvenile and adult forms. The late infantile form presents by the second year of life (usually between 6 months and 4 years) and leads to early mortality. The juvenile forms manifest between 4 years of age and puberty and the adult form becomes clinically overt at any age after puberty. In all forms of MLD mental regression along with disturbances in walking are the earliest physical symptoms observed.
In the childhood forms, quadriparesis, peripheral neuropathy, loss of vision, and loss of speech as well as seizures are the common signs. As the late infantile disease progresses, patients can no longer stand and they show clear signs of mental regression. Patients eventually lose all contact with their surroundings, enter a decerebrate state, and are blind. Death usually occurs within 5 years of the appearance of symptoms of the infantile form of MLD.
In the juvenile forms patients show gradual deterioration in scholastic performance, walking becomes difficult and their speech slurs. There are also emotional and behavioral disturbances in these patients. As this form of the disease progresses cerebellar ataxia and spastic paresis develop. Progressive brainstem involvement results in the necessity for a feeding tube. Most patients will die before the age of 20. There is no specific treatment for MLD.