
Last Updated: April 2, 2026
Introduction to Mitochondria
The mitochondria are critically vital organelles, second only to the nucleus, whose functions are required for cell viability. The major function of the mitochondria is to generate the high energy molecule ATP, through the utilization of the energy contained in the reduced electron carriers, NADH and FADH2. This vital process is explained in detail in the Oxidative Phosphorylation and Related Mitochondrial Functions page.
Through the process of oxidative phosphorylation the mitochondria interconnects the metabolic processes of carbohydrate, lipid, and amino acid catabolism. In addition, the mitochondria serve as important conduits in the processes of urea synthesis, heme synthesis, and steroidogenesis. Mitochondrial are able to carry out these highly diverse, but interrelated, biochemical processes via the catalytic activity of proteins encoded by both the mitochondrial (mtDNA) and the nuclear genomes. Proteomic analysis has determined that the mitochondrial proteome consists of nearly 1,000 different proteins, all but 13 of which are derived from nuclear genes.
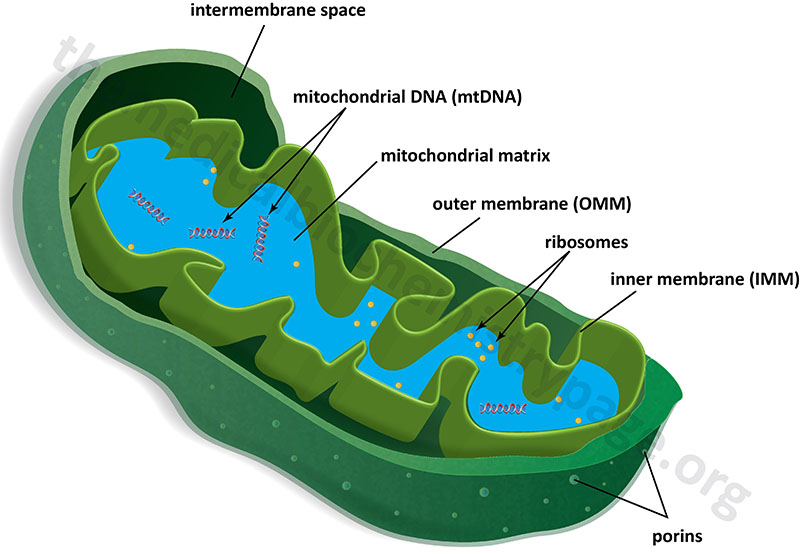
Mitochondria are surrounded by a double-membrane system, consisting of an outer mitochondrial membranes (OMM) and an inner mitochondrial membrane (IMM) separated by an intermembrane space (IMS). The inner membrane forms numerous folds (cristae), which extend into the interior (or matrix) of the organelle. Although depicted as linear molecules in the Figure below, the mitochondrial genome (mtDNA) is a circular double-stranded molecule of DNA.
The outer mitochondrial membrane is in contact with membranes of the endoplasmic reticulum (ER) at sites that are termed ER-mitochondria contact sites, ERMCS. The ERMCS is crucial for exchanging biological molecules between these two organelles. A critical protein in the outer mitochondrial membrane that is involved in forming the ERMCS is mitoguardin-2 (MIGA2). Crucial functions of the ERMCS are the regulation of mitochondrial metabolism and regulation of Ca2+ signaling. The ERMCS plays an important role the maintenance of lipid homeostasis by facilitating local lipid synthesis and lipid transfer between the ER and the mitochondria. The specific role of MIGA2 involves its direct association with glycerophospholipids and liposomes.
With respect to energy generation, pyruvate, derived from carbohydrate oxidation, and fatty acids are imported into the mitochondria from the cytosol and converted to acetyl-CoA in the mitochondrial matrix through the actions of the PDHc and the enzymes of fatty acid oxidation. The resultant acetyl-CoA, and the carbon skeletons of numerous amino acids, are then oxidized via the TCA cycle generating CO2 and the high energy reduced electron carriers, NADH and FADH2. The high-energy electrons from NADH and FADH2 are then transferred, through a series of carriers in the inner mitochondrial membrane, to molecular oxygen (O2) in the process of oxidative phosphorylation.
The energy of the electron transfer is used to drive the synthesis of ATP. Indeed, the inner mitochondrial membrane represents the principal site of ATP generation within the cell. In order to carry out this function the inner mitochondrial membrane has a large surface area as a result of its folding into cristae. In addition, over 70% of the proteins contained within the mitochondria are associated with the inner mitochondrial membrane. These proteins carry out the processes of oxidative phosphorylation and are responsible for the transport of metabolites between the cytosol and the mitochondria.
In the absence of membrane embedded transporters the inner mitochondrial membrane is essentially impermeable to ions and small molecules. This feature of the inner mitochondrial membrane is crucial for the generation of a proton gradient during electron transfer, the energy of which is used for the work of ATP synthesis. The outer mitochondrial membrane, on the other hand, is freely permeable to small molecules principally due to the presence of proteins called porins, which form channels that allow the free diffusion of small molecules. The major porin families are the aquaporins and the voltage-dependent anion channels, VDAC.
Mitochondrial Membrane Lipids
The major lipids of the OMM and the IMM are phospholipids that includes phosphatidylcholines (PC), phosphatidylethanolamines (PE), and cardiolipins (CL). The synthesis of the various mitochondrial phospholipids are detailed in the Synthesis of Phospholipids page. Synthesis of mitochondrial ceramides is discussed in the Sphingolipid Metabolism and the Ceramides page.
Within the OMM phosphatidylcholines are the most abundant lipid followed, in order of abundance, by PE, phosphatidylinositols (PI), CL, phosphatidic acids (PA), and phosphatidylserines (PS). The IMM contains almost exclusively PE, PC, and CL which together constitute over 90% of the lipid in this membrane. The remainder of the IMM lipid content is primarily PI with small amounts of cholesterol and sphingolipids. The IMM has roughly twice the PE content and half the PC content in comparison to their composition in the OMM.
The lipid composition of mitochondrial membranes varies in the different tissues. For example, liver mitochondrial membranes (including the composition of both the OMM and the IMM) are composed of around 50% PC, 35% PE, 20% CL, 5% PI, 1% PS, 1% lysophospholipids (lysoPL), 1% sphingomyelin (SM), and 1% PA. In skeletal muscle mitochondria the lipid composition is around 40% PC, 30% PE, 15% CL, 7% PI, 3% PS, 3% lysophosphatidylcholine (lysoPC), and 2% SM.
In addition to tissue specific differences in overall lipid composition of mitochondrial membranes, there are differences in the distribution of the various lipid types between the OMM and the IMM. For example, in liver mitochondria the OMM is composed of 44–59% PC, 20–35% PE, 5–20% PI, with the remaining phospholipid pool consisting of PS, PA, CL, and lysoPL, whereas the IMM is composed of 38–45% PC, 32–39% PE, 14–23% CL, 2–7% PI, with the remaining consisting mostly of PS, PG, and lysoPL.
The Mitochondrial Genome: mtDNA
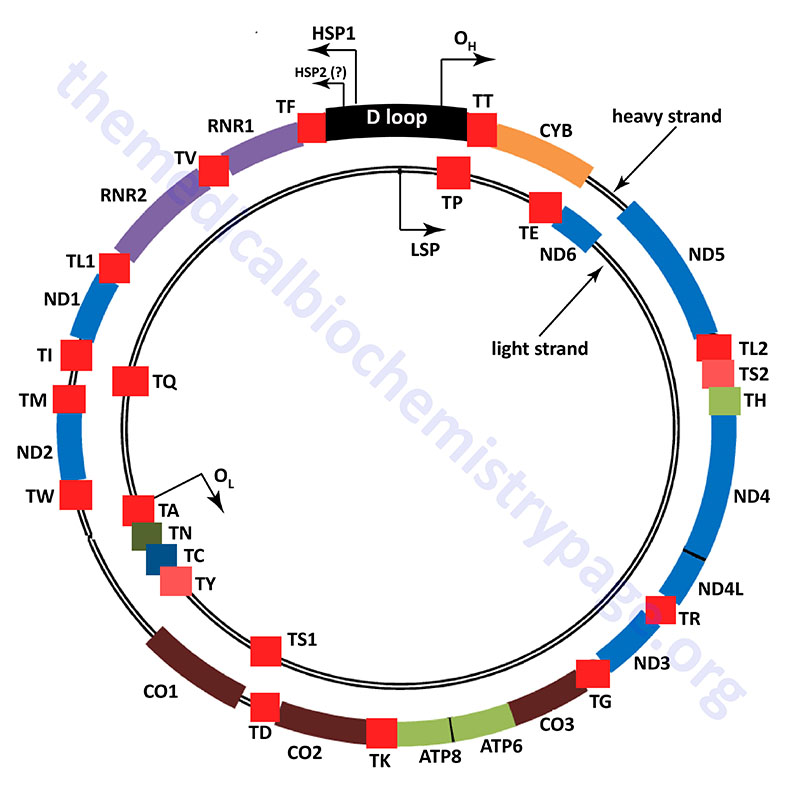
The mitochondrial genome (mtDNA) is a circular double-stranded molecule composed of 16,569 base pairs (bp) of DNA. The two strands of the mtDNA are identified as the heavy strand (H-strand) and the light strand (L-strand). These designations are related to the separation of the two strands in alkaline cesium chloride buoyant density gradients. Due to the phenomenon of mitochondrial heteroplasmy, a single cell in any given tissue, can contain several thousands of copies of mtDNA distributed within the several hundreds of individual mitochondria within that cell.
The mitochondrial genome encompasses a total of 37 genes. All of the mitochondrial genes are intronless. Of the 37 genes in the mtDNA, 13 encode proteins. The remaining mitochondrial proteins, which number in excess of 900, that are necessary for mitochondrial biogenesis and function, are encoded by the nuclear genome. The remaining 24 mtDNA encoded genes include 22 tRNA genes and 2 rRNA genes. The 13 mtDNA encoded proteins all function in the complexes of the oxidative phosphorylation pathway. Only complex II (succinate dehydrogenase) is encoded entirely by nuclear genes. The tRNAs and rRNAs produced by the mtDNA are required for the translation of the 13 mtDNA encoded proteins.
The human mitochondrial genome is organized into nucleoid structures that contain 1–2 copies of the genome. The number of genome copies per nucleoid varies depending on tissue type and the energy demands of the tissue. The function of mitochondrial nucleoids is believed to be the tethering of mtDNA to the inner mitochondrial membrane. This mtDNA tethering aids in organization, distribution and segregation of the DNA. Nucleoids consist of several proteins whose functions are responsible for the maintenance of mtDNA. The primary mtDNA packaging protein is identified as multifunctional mitochondrial transcription factor A (encoded by the TFAM gene: transcription factor A, mitochondrial). The TFAM protein is a member of the high-mobility group (HMG) family of proteins. The function of TFAM is to condense the mtDNA by inducing bending and wrapping of the molecules into compacted nucleoid structures. In addition to its role in mtDNA structure the TFAM encoded protein is critical to the process of transcription from the mitochondrial genome.
Unlike the nuclear genome, where the noncoding DNA is essentially randomly dispersed in the chromosomes, most all of the noncoding DNA in the mitochondrial genome is concentrated in one region referred to as the major noncoding region, or NCR which consists of just over 1 kbp of DNA. Most of the polymorphisms in the human mtDNA sequence are found in the NCR and they are concentrated in three hypervariable sections. Many mtDNA molecules (from 8% to 55% of the total) contain a triple-stranded region which forms what is referred to as the D-loop (displacement loop). This third strand of DNA, consisting of approximately 500 bp, is termed the 7S DNA. The D-loop comprises a large portion of the NCR.
The NCR is also known as the control region due to it harboring important cis-acting DNA elements that control transcription. These transcription initiation start sites are called the heavy- and light-strand promoters, HSP and LSP, respectively. In addition, the NCR contains two origins of replication, the H-strand DNA replication origin (OH) and the L-strand DNA replication origin (OL). The two strands of mtDNA are identified as H-strand and L-strand based upon their differential separation in cesium chloride gradients. The critical role of the NCR in mitochondrial genome maintenance is demonstrated by the fact that all of the partial mtDNA deletions that have been characterized in humans retain the NCR.
Mitochondrial DNA Replication
Like the process of replication of the nuclear genome, replication of the mitochondrial genome requires an adequate pool of dNTPs in addition to all the functional enzymes. The pools of mitochondrial nucleotides are separated from the cytoplasmic pools and they are derived almost exclusively via the pathways of nucleotide salvage. Nucleotide salvage in the mitochondria involves the enzymes thymidine kinase 2 (encoded by the TK2 gene), deoxyguanosine kinase (encoded by the DGUOK gene), as well as the enzymes of the nucleoside monophosphate kinase (NMPK) and nucleoside diphosphate kinase (NDPK) families.
The TK2 gene is located on chromosome 16q21 and is composed of 11 exons that generate seven alternatively spliced mRNAs, three of which encode proteins that lack the mitochondrial import signal and are, therefore, not localized to the mitochondria. The other four TK2 mRNAs encode different sized mitochondrial enzymes with isoform 1 being the largest precursor protein at 265 amino acids. Within the mitochondria TK2 phosphorylates mitochondrial thymidine, deoxycytidine, and deoxyuridine.
The DGUOK gene is located on chromosome 2p13.1 and is composed of 8 exons that generate seven alternatively spliced mRNAs that collectively encode four distinct protein isoforms. Within the mitochondria DGUOK encoded enzymes phosphorylate mitochondrial purine deoxynucleosides. In proliferating cells the pools of nucleotides required for mtDNA replication are imported from the cytosol and are, therefore derived from the cytoplasmic de novo synthesis and salvage pathways.
Mitochondrial DNA is replicated and repaired by the DNA polymerase identified as mtDNA polymerase γ (pol γ) whose subunits are encoded by the nuclear genome. Human pol γ is a heterotrimeric complex consisting of one copy of the 140kDa (p140) catalytic subunit and two copies of the 55kDa (p55) processivity subunit. The p140 catalytic subunit is encoded by the POLG gene. The p55 processivity subunit is encoded by the POLG2 gene.
The POLG gene is located on chromosome 15q26.1 and is composed of 23 exons that generate two alternatively spliced mRNAs, both of which encode the same 1239 amino acid protein.
The POLG2 gene is located on chromosome 17q23.3 and is composed of 11 exons that encode a 485 amino acid protein.
The p140 catalytic subunit of pol γ harbors active sites for 5′ → 3′ DNA polymerase, 3′ → 5′ exonuclease, and 5´-deoxyribose-5-phosphate lyase (5′ dRP lyase) activities. The dRP lyase activity of the POLG encoded enzyme is similar to that of the POLB (pol β) encoded enzyme which is the primary enzyme involved in removing abasic (ribose with no nucleobase attached) sites in nuclear DNA during the course of base excision repair (BER). BER represents the major pathway involved in the repair of nuclear DNA base lesions and it occurs through a series of reactions that result in an abasic site (a 5´-deoxyribose-5-phosphate) where there was a base lesion. The 5´-deoxyribose-5-phosphate (dRP) residue is subsequently removed either through the action of a dRP lyase activity or through displacement by DNA repair polymerase activity. The p55 subunits of the mtDNA polymerase holoenzyme impart high processivity onto the holoenzyme by increasing the overall binding affinity to DNA.
DNA polymerase γ functions in the process of mtDNA replication in concert with numerous additional protein activities. These additional activities include mitochondrial topoisomerase (encoded by the TOP1MT gene), Twinkle mtDNA helicase (encoded by the TWNK gene), mitochondrial RNA polymerase (mtRNAP; encoded by the POLRMT gene), RNaseH1 (encoded by the RNASEH1 gene), mitochondrial single-stranded DNA-binding protein (mtSSB; encoded by the SSBP1 gene), and DNA ligase 3 (encoded by the LIG3 gene). Other activities that have been shown to be critical for the maintenance of the mitochondrial genome include multifunctional mitochondrial transcription factor A (encoded by the TFAM gene), mitochondrial genome maintenance 5′ → 3′ exonuclease 1 (encoded by the MGME1 gene), flap endonuclease 1 which removes 5′ overhanging flaps in DNA and RNA (encoded by the FEN1 gene), and DNA replication helicase/nuclease 2 (encoded by the DNA2 gene). The MGME1, FEN1, and DNA2 encoded proteins have all been implicated in the mtDNA base excision repair (BER) pathways. The DNA2 encoded protein is also involved in mtDNA replication through its ability to stimulate pol γ activity and its co-localization with Twinkle.
Similar to nuclear DNA replication, mtDNA replication requires that pol γ has access to a free 3′-OH of an RNA primer. The primase activity involved in mtDNA replication was originally thought to be solely the function of the mtRNAP protein encoded by the POLRMT gene which is responsible for transcription of the mtDNA (see next section). However, recent experiments have shown that another member of the superfamily of primases called primase-polymerase (PrimPol) is also critical in the process of mtDNA replication and maintenance.
The PrimPol protein is encoded by the PRIMPOL gene. PrimPol is localized to both the nucleus and the mitochondria indicating that it plays a role in DNA maintenance in both compartments. PrimPol is capable of synthesizing both DNA and RNA primers on single-stranded DNA but shows preference for dNTPs as substrates. The PrimPol enzyme is also capable of DNA polymerase activity where it can extend a DNA primer in a template-dependent manner. PrimPol is also capable of carrying out DNA repair functions such as translesion synthesis (TLS) through many different types of DNA lesions, including 8-oxoguanine (8-oxoG) and [6-4] pyrimidine-pyrimidone photoproducts (6-4PP). Of note to human genetic disease, mutation of PRIMPOL is associated with the ocular disorder, high myopia.
Replication of mtDNA is a complex and slow process which takes approximately one hour to synthesize both daughter strands. An asymmetric (also called strand displacement mechanism, SDM) mode of replicating mammalian mtDNA daughter strands is one of the currently accepted modes for mtDNA replication. In the strand displacement model of mtDNA replication, two origins of replication direct the pol γ holoenzyme complex to initiate continuous DNA synthesis. However, this initiation is temporally regulated at the origins. The first step involves synthesis of the daughter strand from the H-strand template which begins at the H-strand origin of replication (OH) located within the NCR. In the context of mtDNA replication the H-strand is referred to as the leading strand and the L-strand is referred to as the lagging strand.
Similar to nuclear DNA replication, initiation of H-strand synthesis by pol γ involves addition of nucleotides to the 3′-end of an existing RNA primer. In human mitochondria the RNA primers occur at very low frequency. As indicated above, primer synthesis involves activities of the POLRMT and PRIMPOL encoded proteins. After initiating synthesis from OH, replication pauses after about 2.5 kb which allows for strand switching. Second strand synthesis, the lagging strand, begins from RNA primers at the OL origin. Although described in rather simplistic terms in this discussion, the process of mtDNA replication is quite complex and beyond the scope of this overview.
Diseases Associated with Mutations in mtDNA Replication Genes
Mutations in the POLG gene are associated with several neurodegenerative disorders that exhibit highly polymorphic pathology. The polymorphism of these disorders relates to the timing of onset, organ system involvement, and symptom presentation. At least 300 pathogenic mutations in POLG have been defined to date.
The POLG disorders are currently divided into at least six major neurodegenerative phenotypes. These six phenotypes include Alpers-Huttenlocher syndrome (AHS), ataxia neuropathy spectrum (ANS), myoclonic epilepsy myopathy sensory ataxia (MEMSA), myocerebrohepatopathy spectrum (MCHS), and the progressive external ophthalmoplegias (PEO) that include the autosomal recessive (arPEO) and autosomal dominant (adPEO) forms.
The POLG encoded protein possesses a polyglutamine (polyQ) repeat that begins at amino acid 43 (encoded by exon 2). Alterations in the polyQ repeat have been shown to be associated with male infertility, testicular cancer, and Parkinsonism.
Alpers-Huttenlocher syndrome (AHS)
The symptoms of Alpers-Huttenlocher syndrome (AHS) become apparent in children between ages 2 and 4. AHS is associated with three characteristic features that includes intractable epilepsy, psychomotor regression, and liver disease. Additional symptoms of AHS include ataxia and neuropathy. The neuropathy can lead to abnormal or absent reflexes and weak muscle tone that becomes progressively worse resulting in the loss of the ability to control muscle movement. Some individuals with AHS lose the ability to walk, sit, or feed themselves.
Ataxia neuropathy spectrum (ANS)
Ataxia neuropathy spectrum (ANS) includes the disorders that were previously called mitochondrial recessive ataxia syndrome (MIRAS) and sensory ataxia neuropathy dysarthria and ophthalmoplegia (SANDO). Individuals afflicted with ANS typically experience ataxia and neuropathy. In ANS the neuropathy is classified as mixed since it involves both sensory and motor deficit. The sensory neuropathy causes numbness, tingling, or pain in the arms and legs, and the motor neuropathy leads to disturbances in muscle movement. Most individuals with ANS also manifest with severe brain dysfunction and suffer from seizures. Additional symptoms of ANS include ophthalmoplegia (weakness of eye muscles), ptosis (drooping eyelids), myoclonus (involuntary twitching), migraines, blindness, depression, and liver disease.
Myoclonic epilepsy myopathy sensory ataxia (MEMSA)
Myoclonic epilepsy myopathy sensory ataxia (MEMSA) is a condition whose symptoms first appear in young adults. MEMSA was originally referred to as spinocerebellar ataxia with epilepsy (SCAE). Symptoms of MEMSA usually begin with the onset of cerebellar ataxia. Recurrent seizures (epilepsy) develop later and often occur in combination with myoclonus. MEMSA individuals may also exhibit encephalopathy and myopathy.
Myocerebrohepatopathy spectrum (MCHS)
Myocerebrohepatopathy spectrum (MCHS), as the name implies, is a disorder associated with muscle, brain, and liver pathologies. The symptoms of MCHS begin to appear in children from a few months of age up to around 3 years. Common symptoms of MCHS include developmental delay, loss of previously acquired motor skills, myopathy, developmental delay or deterioration of intellectual function, and liver disease. Metabolic problems of MCHS include metabolic acidosis due to accumulation of serum lactate and renal tubular acidosis. Children also can experience pancreatitis, recurrent vomiting, and hearing loss.
Progressive external ophthalmoplegia (PEO)
Progressive external ophthalmoplegia (PEO) represents a spectrum of disorders with overlapping signs and symptoms. As the name implies the common pathology is weakness of the eye muscles (ophthalmoplegia). PEO is a late onset disease with manifestations first appearing as bilateral ptosis around 18-40 years of age. The disease is progressive and ultimately involves proximal muscles leading to exercise intolerance and muscle wasting. Additional symptoms include neurological problems such as depression and an avoidance personality as well as hearing loss, cataract formation, hypogonadism, and dysphagia (difficulty swallowing). Mutations in the POLG gene result in the forms of PEO specifically referred to as progressive external ophthalmoplegia with mitochondrial deletions, autosomal recessive 1 (PEOB1) and progressive external ophthalmoplegia with mitochondrial deletions, autosomal dominant 1 (PEOA1).
In addition to the POLG mutations that lead to manifestations of PEO, mutations in the POLG2 gene and the TWNK gene are also associated with PEO.
Specifically the disorder referred to as progressive external ophthalmoplegia with mitochondrial deletions, autosomal dominant 3 (PEOA3) results from mutations in the TWNK gene. The symptoms of PEOA3 include adult onset of ophthalmoplegia, exercise intolerance, proximal muscle weakness, ataxia, peripheral neuropathy, cardiomyopathy, cataracts, depression, and endocrine abnormalities.
Mutations in the POLG2 gene result in progressive external ophthalmoplegia with mitochondrial deletions, autosomal dominant 4 (PEOA4). The symptoms of PEOA4 have a highly variable age of onset from infancy to adulthood. The symptoms of PEOA4 exhibit a range of severity from mild skeletal muscle and external eye muscle weakness to severe multisystem dysfunction characterized by delayed psychomotor development, lactic acidosis, and liver disease.
mtDNA Damage and Repair
Like the DNA in the nuclear genome, mitochondrial DNA (mtDNA) is susceptible to damage. Also, like nuclear DNA, mtDNA that is damaged will undergo repair through mechanisms that are similar to those used to repair nuclear DNA damage. There are certain sequences in the mtDNA, such as the hypervariable segments of the D-loop regulatory region, that are particularly unstable in the natural course of aging. Similar to the effects of nucleotide polymorphisms in the nuclear DNA, single nucleotide variations in mtDNA can be benign or they may promote disease. Lesions in the mtDNA are known to contribute to tissue damage, age-related decline, and to disease.
The number and types of DNA base modifications that have been identified in the nuclear genome are more extensive than those that have been, to date, identified in the mitochondrial genome. Some of the free radical modified bases identified in the nuclear genome that are also found in the mitochondrial genome include 8-oxo-2′-deoxyguanosine (8-oxo-dG), formamidopyrimidines, and 8,5′-cyclo-2′-deoxynucleosides, as well as ribose backbone modifications and DNA strand breaks. Unlike the nuclear genome the removal of 8-oxo-dG, as well as some other DNA base modifications, may not be readily removed from mtDNA.
Mitochondria possess several mechanisms for the repair of damaged DNA including base-excision repair (BER) and nucleotide excision repair (NER) processes. Mitochondria have been shown to possess two methods of BER that includes single-nucleotide base excision repair (SN-BER) and long-patch BER (LP-BER), both of which utilize the same set of proteins used for these mechanisms of BER in the nuclear genome. Mitochondria also carry out single-strand break repair (SSBR), YB-1-mediated mismatch repair, removal of adenine opposite 8-oxo-dG, and nudix hydrolase 1 (encoded by the NUDT1 gene) removal of 8-oxo-dGTP and 8-oxo-2′-dATP. YB-1 refers to Y-box binding protein 1 where the Y-box sequence in DNA is an inverted CCAAT box. The NUDT1 encoded enzyme is also known as oxidized purine nucleoside triphosphate hydrolase. The term nudix is derived from nucleoside diphosphate-linked moiety X. NUDT1 is the mammalian homolog of the bacterial gene, mutT.
Nonhomologous end joining (NHEJ) and homologous recombination activities that are involved in the repair of double-stranded DNA in the nuclear genome have also been shown to be involved in mitochondria. The nuclear genome encoded helicase encoded by the RECQL4 (RecQ like helicase 4) gene has been shown to be involved in double-strand break repairs in the mitochondrial genome.
With respect to BER in the mitochondrial genome the functions of DNA polymerase β, that are involved in nuclear genome BER, are replaced by DNA polymerase γ. Several DNA glycosylases, including the mitochondrial uracil DNA glycosylase encoded by the UNG1 gene, nth like DNA glycosylase (encoded by the NTHL1 gene), the mutY DNA glycosylase (encoded by the MUTYH gene), and the bifunctional glycosylases encoded by the OGG1 (8-oxoguanine DNA glycosylase), NEIL1 (nei like DNA glycosylase 1), and NEIL2 function to repair mtDNA lesions.
The repair of lesions in mtDNA conflicts with the transcription of the mtDNA genes and as such a mechanism of prioritizing the two processes through control of the enzymes involved is operational. When mitochondria are stressed there is activation of a mitochondrial form of the unfolded protein response, UPR. The mitochondrial UPR involves three primary proteins encoded by the ATF4, ATF5, and DDIT3 genes. ATF4 and ATF5 are activating transcription factor 4 and 5, respectively. The DDIT3 encoded protein, DNA damage inducible transcript 3, is also identified as CHOP (C/EBP homologous protein, where C/EBP is CCAAT/enhancer binding protein).
Activation of the mitochondrial UPR results in a reduction in mtDNA damage and this regulatory process is a critical function of the ATF5 protein. Mammalian ATF5 is the homolog of the activating transcription factor associated with stress-1 (atfs-1) gene of the round worm C. elegans. The ATF5 protein strongly interacts with mitochondrial transcription factor A (encoded by the TFAM gene), a mitochondrial transcription factor that is essential for the initiation of mitochondrial transcription. The interaction of the ATF5 and TFAM proteins impedes transcription while simultaneously promoting DNA repair exerted by DNA glycosylases.
Transcription of the Mitochondrial Genome
Transcription of the human mitochondrial genome is the function of the mitochondrial RNA polymerase (mtRNAP) encoded by the nuclear POLRMT gene. The encoded protein functions as a single subunit in conjunction with two transcription initiation factors.
The POLRMT gene is located on chromosome 19p13.3 and is composed of 21 exons that encode a 1230 amino acid precursor protein.
The two transcription initiation factors are identified as mitochondrial transcription factor A (encoded by the TFAM gene) and mitochondrial transcription factor B2 (encoded by the TFB2M gene). A protein related to the TFB2M encoded protein is identified as mitochondrial transcription factor B1 which is encoded by the TFB1M gene.
The TFB1M encoded protein possesses two activities. The primary function of TFB1M is SAM-dependent adenine dimethyltransferase activity that methylates the conserved stem-loop of the mitochondrial 12S rRNA. This function of the TFB1M encoded protein controls mitochondrial protein synthesis. Although the TFB1M protein has been shown to interact with TFAM and POLRMT in vitro, there is no in vivo evidence to indicate that mitochondrial transcription regulation is a function of this protein.
The TFAM gene is located on chromosome 10q21.1 and is composed of 9 exons that generate two alternatively spliced mRNAs that encode two distinct protein isoforms.
The TFB2M gene is located on chromosome 1q44 and is composed of 8 exons that encode a 396 amino acid protein.
Mitochondrial RNA polymerase is capable of promoter recognition on its own but requires the activities of the TFAM and TFB2M encoded proteins in order for the promoter region to be melted into single strands. The L-strand of the mtDNA contains a single transcription promoter identified as LSP. The H-strand is proposed to have two transcription promoters identified as HSP1 and HSP2. However, experimental evidence for the so-called HSP2 promoter has not been demonstrated. The sequences of the LSP and of HSP1 are close together at one end of the D-loop region. This juxtaposition allows the mtRNAP, TFAM, and TFB2M complexes to initiate transcription of both strands under identical circumstances. TFAM binds to mitochondrial DNA between 14–35 bp upstream of the LSP transcription start site. The binding of TFAM promotes the assembly of the transcriptional initiation complex by recruiting POLRMT and TFB2M to the site. The primary function of TFB2M is to melt the promoter and to stabilize the open promoter complex.
Transcription from the LSP yields a single polycistronic RNA that encodes subunit 6 of the NADH dehydrogenase and eight tRNAs. Transcription of the H-strand of mtDNA also yields a polycistronic mRNA that in this case encodes 12 polypeptides, two rRNAs, and the rest (14) of the tRNAs. The transcription start site, directed by the HSP1 promoter, is at the phenylalanine tRNA (tRNAPhe). H-strand transcription is also the function of the mtRNAP, TFAM, and TFB2M proteins.
Following their transcription, the L-strand and H-strand polycistronic RNAs are processed into individual tRNA, rRNA, and mRNA molecules. However, there are two bicistronic mt-mRNAs, one which encodes the MTND4 and MTND4L proteins and the other that encodes the MTATP6 and MTATP8 proteins. All of the mitochondrial mRNA molecules, excluding the MTND6 mRNA, are polyadenylated (approximately 50 adenine residues are added) similar to the post-transcriptional processessing of nuclear genome encoded mRNAs. However, polyadenylation of mt-mRNA is carried out by the mitochondrial polyadenylate polymerase (mtPAP) encoded by the MTPAP gene.
One unique feature of the polyadenylation of mt-mRNAs is that in 7 of these mRNAs the addition of the polyA tail completes the UAA stop codon. Like their cytoplasmic counterparts, mt-tRNAs are modified at various nucleotides and have the CCA trinucleotide added to their 3′-ends. In all mt-mRNAs the translational start codon is found at or within a few nucleotides of the 5′-end.
The mechanisms that control mtDNA transcriptional initiation are not fully understood but may include sensing of metabolic demands such as ATP levels and the influences of hormones. Transcription factors that have been found in the mitochondria include the thyroid hormone receptor, RXR, and p53. Although detected in the mitochondria, and the existence of appropriate response elements in mtDNA, there is as yet, no direct evidence that these transcription factors are necessary nor required for mtDNA transcription. There have been reports that p53 interacts with pol γ in the mitochondria in response to DNA damage.
Control over the termination of mitochondrial transcription was originally clearly demonstrated for transcription that initiates at the HSP1. Termination of the HSP1 transcription unit was shown to be mediated by the protein identified as mitochondrial transcription termination factor 1, mTERF1, which is encoded by the MTERF1 gene. The mTERF1 protein binds in a sequence-specific manner to a 28 bp element that resides at the 3′-end of the tRNALeu gene. mTERF1 can also lead to termination of transcription initiated at the LSP.
In addition to the MTERF1 gene humans express three additional related genes identified MTERF2, MTERF3, and MTERF4 all of whose encoded proteins localized to the mitochondria. The MTERF2 and MTERF3 encoded proteins have been shown to be involved in transcription of mtDNA through interaction with the promoter region of the mtDNA. The MTERF4 encoded protein is involved in mitochondrial translation. The MTERF4 protein binds to the 7S, 12S, and 16S mitochondrial rRNAs and targets the NSUN4 (NOP2/Sun RNA methyltransferase family member 4) encoded enzyme to the mitochondrial large ribosomal subunit.
Translation of Mitochondrial Genome-Derived RNAs
Mitochondrial protein synthesis machinery is referred to as the mitoribosome and like the translational machinery for nuclear encoded genes consists of two distinct subunits. The small ribosomal subunit (mt-SSU) is a 28S complex and the large subunit (mt-LSU) is a 39S complex. Each subunit contains a single rRNA with the mtDNA encoded 12S rRNA in the mt-SSU and the mtDN encoded 16S rRNA in the mt-LSU. The standard nomenclature for mitoribosomal proteins is to use the designation MRP (mitochondrial ribosomal protein) followed by L for large subunit proteins and S for small subunit proteins similar to the cytosolic ribosomal protein nomenclature.
The protein content of the mt-LSU has been more rigorously studied with current estimates of at least 51 different proteins, with at least 8 of these derived from the mtDNA. The current estimates for protein content of the mt-SSU is approximately 30 different proteins.
The protein content of the mitoribosome consists of proteins that are orthologues of the original eubacterial proteins, demonstrating a clear bacterial origin of the mitochondria, and these proteins are referred to as the old group proteins. The new group proteins represent the mitoribosomal proteins that were non-ribosomal proteins but via evolutionary processes became associated with the mitoribosome. Many of these new group proteins were originally identified from studies that were unrelated to work designed to define the composition and functions of the mitoribosome but were only subsequently shown to be mitoribosomal proteins. Examples include several proteins that were identified as functioning in pathways of apoptosis such as programmed cell death protein 9 (originally designated PDCD9) which is now identified as MRPS30 and the protein originally identified as death associated protein 3 (DAP3) which is now referred to as MRPS29.
A certain level of structural conservation exits between the mitoribosome and the cytosolic ribosomes in that there is a typical A-site and a P-site. Evidence is unclear as to whether or not the mitoribosome contains an E-site such as that found in cytosolic ribosomes. One clear distinction related to the function of the mitoribosome is that it is tasked with synthesizing solely the hydrophobic proteins localized to the inner mitochondrial membrane (IMM) and the insertion of these proteins into the IMM occurs co-translationally.
The 22 tRNA genes of the mitochondrial genome represent the entire set of tRNAs required for the translation of mitochondrial mRNAs (mt-mRNA). This compares to the set of nuclear genome encoded tRNA genes which numbers at least 30. The mitochondrial tRNAs exhibit a greater degree of wobble in the anticodon-codon interactions whereby a 5′-U in the anticodon can base pair with any of the four nucleotides at the 3′ position of the codon. The mitochondrial genome translation codons also have some significant differences compared to the nuclear genome codons. A subset of methionine tRNA (met-tRNAmet) becomes formylated, similar to formylation of the initiator fmet-tRNAmet of bacterial translation initiation, and it is this pool of fmet-tRNAmet that serves to initiation translation of mt-mRNAs. The formylation reaction is catalyzed by mitochondrial methionyl-tRNA formyltransferase which is encoded by MTFMT gene.
During translation of mt-mRNAs the initiator fmet-tRNAmet can recognize AUG, AUA, or AUU as the translational start codon for incorporation of methionine, whereas AUA and AUU encode Ile (I) in nuclear genome encoded mRNAs. In addition to alternative start codons, in mt-mRNA the codon UGA is used for Trp (W) incorporation whereas this is a stop codon for nuclear genome encoded mRNAs. Also the codons AGA and AGG, which encode Arg (R) in nuclear genome encoded mRNAs, play a role in translational termination in two mt-mRNAs.
Similar to translational initiation in the cytosol, mitochondrial protein synthesis requires initiation factors. However, whereas cytosolic translation initiation is controlled by numerous proteins, mitochondrial protein synthesis initiation is controlled by just two proteins, identified as mitochondrial initiation factors 2 and 3 (mIF2 and mIF3). The role of mIF2 (which is encoded by the MTIF2 gene), as a dimeric complex with GTP, is to recruit the fmet-tRNAmet to the mt-SSU. Prior to engagement with fmet-tRNAmet:mIF2:GTP the mt-SSU is bound with mIF3 to prohibit premature initiation. When a positive interaction occurs between fmet-tRNAmet:mIF2:GTP and a start codon the mt-LSU is recruited to the complex and protein synthesis can begin.
Elongation of mitochondrial protein synthesis involves the entry of subsequent amino acids into the A-site of the mitoribosome. Each amino acid is carried to the ribosome charged to an appropriate tRNA as for cytosolic translational elongation. In mitochondrial translation the aminoacyl-tRNA is aided to the A-site via interaction with the mitochondrial elongation factor identified as mtEF-Tu (encoded by the TUFM gene) which is complexed with GTP. Following interaction of the aminoacyl-tRNA with the appropriate codon, the GTP is hydrolyzed and the mtEF-Tu:GDP is released. Regeneration of functional mtEF-Tu:GTP is catalyzed by the guanine nucleotide exchange factor identified as mtEF-Ts (encoded by the TSFM gene).
Peptide bond formation is carried out by the peptidyltransferase center of the mt-LSU similar to peptide bond formation in cytosolic ribosomes. Following peptide bond formation the free tRNA residing in the P-site is removed through a translocation process that is similar to that occurring in cytosolic ribosomes. The elongation factor mtEF-G1 (encoded by the GFM1 gene) catalyzes the release of the tRNA and movement of the mitoribosome such that the peptidyl-tRNA resides in the P-side and a free A-site resides over the next codon.
Translational termination of all 13 mitochondrial proteins occurs following recognition of an appropriate stop codon by the termination factor identified as mtRF1a which is encoded by the MTRFL1 (mitochondrial translational release factor 1 like) gene. This gene is so-called since it was shown to be similar to a protein encoded by the MTRF1 ( mitochondrial translation release factor 1) gene that was originally thought to be the translational release factor for mitochondrial translation. The predicted activity of the MTRF1 encoded protein was based upon its amino acid similarity to bacterial translational release factors, however, it was subsequently found to have no function in mitochondrial translational termination.
In nine of the monocistronic human mt-mRNAs the stop codon UAA is utilized for termination and in the two bicistronic mt-mRNAs the stop codon UAG is utilized. In the mt-mRNA that encodes the MTCO1 protein the stop codon utilized is AGA and in the mt-mRNA that encodes the MTND6 protein the stop codon is AGG. However, the actual termination process from these two mt-mRNAs involves a -1 (minus one) frameshift such that the U which precedes the AGA and the AGG results in the recognition of a UAG codon as the actual stop codon. This process is facilitated by a separate release factor encoded by the MRPL58. The MRPL58 encoded protein was originally identified in certain colon cancers and was given the name immature colon carcinoma transcript 1 (ICT1).
Mitochondrial Import of Nuclear Genome-Derived Proteins
The mitochondrial proteins that are encoded by the nuclear genome are translated in the cytoplasm and then these proteins are imported into the mitochondria by specialized recognition and transport proteins and complexes. Mitochondrial protein import and localization is controlled by specific amino acid sequences in the precursor proteins. These sequences not only target the proteins to the mitochondria but also ensure that individual proteins are properly distributed into the four mitochondrial compartments: outer mitochondrial membrane (OMM), intermembrane space (IMS), inner mitochondrial membrane (IMM), and matrix.
Like all but the mitochondrial genome encoded proteins, mitochondrial proteins are synthesized in the cytosol. Proteins destined for the mitochondria posses a presequence akin to the leader peptide of ER targeted proteins. Transfer of these proteins to their appropriate location in the mitochondria requires specific chaperones and transport complexes. At the level of the outer mitochondrial membrane the transport process involves a complex called the translocase of outer mitochondrial membrane, TOM. The TOM is composed of several proteins that are receptors for mitochondrially targeted proteins and that compose the channel itself of the TOM.
The TOM complexes are localized to specialized domains of the outer mitochondrial membrane that are closely associated with openings in the inner mitochondrial membrane that are referred to as cristae junctions. The channel of the TOM complex is formed from the Tom40, Tom22, and Tom7 proteins while the preprotein receptors are Tom20 and Tom70. Accessory subunits of the TOM that are required for assembly and stabilization of the TOM are Tom5 and Tom6. The human Tom proteins are encoded by genes with the designation TOMM. As an example the Tom40 protein is encoded by the TOMM40 gene which is located on chromosome 19q13.32 and is composed of 10 exons that three alternatively spliced mRNAs all of which encode the same 361 amino acid protein.
Mitochondrial precursor proteins that contain an appropriate presequence are initially recognized by Tom20 and then they interact with Tom22 prior to import. Other mitochondrial proteins that are hydrophobic precursors contain an integral targeting sequences that is recognized by Tom70. Whether recognized by Tom20 or Tom70, both classes of mitochondrial protein are imported through the Tom40 channel. Many proteins that are embedded in the outer mitochondrial membrane such as Tom20 and Tom70 are referred to as signal-anchored proteins. These proteins are embedded in the outer membrane via sequences in the N-terminus and have their C-terminal domains extended into the cytosol. Other classes of outer membrane anchored proteins have a C-terminal transmembrane domain and are referred to as tail-anchored proteins. The proteins of the Bcl-2 family of apoptosis regulating proteins are members of the mitochondrial tail-anchored family of proteins.
Proteins of the intermembrane space (IMS) are generally small soluble proteins that are imported via the mitochondrial IMS import and assembly (MIA) pathway. The MIA pathway is unique in that it couples the process of protein import to the folding and oxidation of the imported protein leading to the formation of internal disulfide bonds in the process. The introduction of the disulfide bonds in IMS proteins is catalyzed by a protein identified as Mia40 which acts as the chaperone and the sulfhydryl oxidase identified as growth factor, augmenter of liver regeneration (encoded by the GFER gene; is the human homolog of the yeast Erv1 enzyme). The human Mia40 protein is encoded by the coiled-coil-helix-coiled-coil-helix domain containing 4 (CHCHD4) gene.
Transport of mitochondrial proteins to the inner membrane and the mitochondrial matrix involves complexes identified as translocase of inner mitochondrial membrane, TIM. Like the TOM the TIM complexes are composed of central channel forming proteins. The TIM that is responsible for matrix targeted proteins contains the Tim23 (encoded by the TIMM23 gene) protein which is the primary channel protein, along with Tim17. Proteins that are transported via the Tim23 mediated TIM contain an amphipathic helix at their N-termini that, that like TOM transport, is referred to as a presequence. The TIM23 complex contains several additional proteins such as Tim50 which regulates channel opening and Tim 21 which regulates docking functions. Matrix proteins that contain the appropriate presequence have it proteolytically removed during the transport process.
Mitochondrial inner membrane proteins also contain a TIM recognized presequence. However, multipass integral inner membrane proteins are inserted into the membrane via the TIM complex identified as TIM22. The central channel of the TIM22 complex is generated from the Tim22 protein encoded by the TIMM22 gene. The Tim22 protein is referred to as the carrier translocase. Like the TIM23 complex, the TIM22 complex is composed of several additional subunits such as Tim12, Tim18, and Tim54. The functionally most significant proteins inserted into the inner mitochondrial membrane via the TIM22 complex are the metabolite transporters such as the dicarboxylate transporter encoded by the SLC25A10 gene.
Complexes of Mitochondrial Oxidative Phosphorylation
Electron flow through the mitochondrial electron transport assembly is carried out through several enzyme complexes identified as complexes I, II, III, and IV. The ATP synthase complex is sometimes also called complex V. Electrons enter the transport chain primarily from cytosolic NADH to mitochondrial NADH but can also be supplied by succinate (to mitochondrial FADH2) or by the glycerol phosphate shuttle via mitochondrial FADH2.
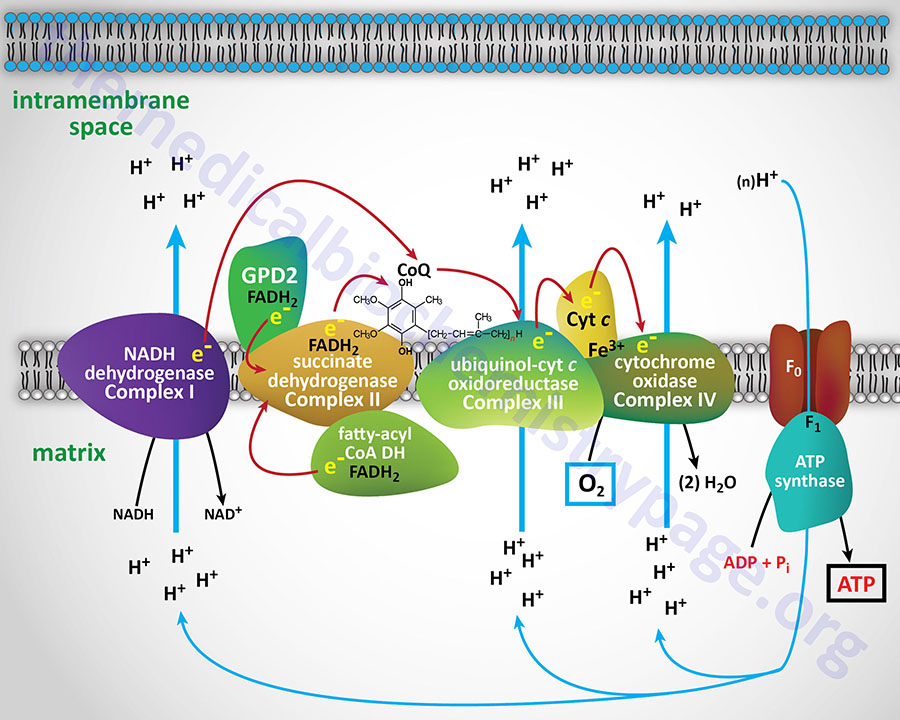
With the exception of NADH, succinate, and CoQ, all of the components of the pathway are integral proteins of the inner mitochondrial membrane whose cofactors undergo redox reactions. NADH and succinate are soluble in the mitochondrial matrix, while CoQ is a small, mobile carrier that transfers electrons between the primary dehydrogenases of complexes I and II and cytochrome b. Although possessing mobility, CoQ is also restricted to the membrane phase because of its hydrophobic character.
As shown in the Figure above, the mitochondrial electron transport proteins are clustered into multiprotein complexes known as Complexes I, II, III, and IV. Within complex I the seven protein subunits that comprise the NADH dehydrogenase activity are all encoded by the mitochondrial genome. The remaining subunits are all encoded within the nuclear genome. All of the subunits of complex II are encoded by the nuclear genome. The cytochrome b of complex III is the only mitochondrial genome encoded subunit of this complex. Within complex IV the three subunits comprising cytochrome c oxidase are each encoded by the mitochondrial genome with the remaining subunits encoded within the nuclear genome. All but two of the subunits of ATP synthase are encoded by the nuclear genome while the ATP synthase 6 and ATP synthase 8 subunits are encoded by the mitochondrial genome.
In addition to the core protein subunits of each of the complexes of oxidative phosphorylation, there are numerous assembly factors required to ensure correct formation of each complex. The importance of the assembly factors in functional formation of these complexes can be demonstrated by the mitochondrial encephalomyopathies that result due to mutations in several genes. For example, GRACILE syndrome (Growth Retardation, Amino aciduria, Cholestasis, Iron overload, Lactic acidosis, and Early death) is caused by mutations in the BCS1L gene which is required for proper assembly of complex III. The BCS1L (BCS1-like) gene is so-called because it is the human homolog of the yeast bsc1 gene. The BCS1L encoded protein is a member of the AAA family of ATPases.
Complex I
Complex I, also known as NADH:CoQ oxidoreductase (also NADH-ubiquinone oxidoreductase or NADH dehydrogenase), is composed of NADH dehydrogenase with FMN as cofactor, plus non-heme-iron proteins having at least one iron sulfur center. Complex I is responsible for transferring electrons from NADH to CoQ. The ΔEo′ for the latter transfer is 0.42V, corresponding to a ΔG′ of –19 kcal/mol of electrons transferred. With its highly exergonic free energy change, the flow of electrons through complex I is more than adequate to drive ATP synthesis.
There are a total of 45 protein subunits in mammalian complex I. Of the 45 proteins, 38 are derived from the nuclear genome and seven are derived from the mitochondrial genome (mtDNA). The organization of complex I is such that a portion of the complex is embedded in the inner mitochondrial membrane and the rest of the complex protrudes into the mitochondrial matrix. Complex I consists of 14 core subunit proteins, of which seven are derived from the mitochondrial genome, and 30 supernumerary (or accessory) proteins, all of which are encoded by the nuclear genome. Along with the core and supernumerary subunits, complex I contains one FMN moiety and eight iron-sulfur (Fe-S) clusters. In addition to the protein subunits of complex I, there are at least nine known complex assembly factor proteins required for the proper assembly of complex I. Although nine genes encoding complex I assembly factors have been identified via mutations linked to human diseases, evidence suggests that there may be as many as 25 assembly factors required for functional assembly of complex I. The nine characterized assembly factor encoding genes are NDUFAF1, NDUFAF2, NDUFAF3, NDUFAF4, NDUFAF5, NDUFAF6, FOXRED1, NUBPL, and ACAD9 genes.
The dehydrogenase activity of complex I is responsible for the oxidation of NADH to NAD+ and contains the NDUFV1, NDUFV2, and NDUFS1 subunits (where the ND designation refers to NADH Dehydrogenase). The hydrogenase activity of the complex transfers electrons to ubiquinone (coenzyme Q) and is composed of the NDUFS2, NDUFS3, NDUFS7, and NDUFS8 subunits. The proton translocation module of complex I (referred to as the core) contains all seven of the mtDNA encoded proteins.
Table of Mammalian Complex I Core Subunits
| Gene | Location | Functions / Comments |
| MT-ND1 | mtDNA | protein identified as ND1; |
| MT-ND2 | mtDNA | protein identified as ND2; |
| MT-ND3 | mtDNA | protein identified as ND3; |
| MT-ND4 | mtDNA | protein identified as ND4; |
| MT-ND4L | mtDNA | protein identified as ND4L; |
| MT-ND5 | mtDNA | protein identified as ND5; |
| MT-ND-6 | mtDNA | protein identified as ND6; |
| NDUFS1 | 2q33.3 | gene composed of 20 exons; largest subunit of the NAHD dehydrogenase activity of complex I; |
| NDUFS2 | 1q23.3 | gene composed of 16 exons; constitutes one of the subunits of the Fe-S fraction of the complex |
| NDUFS3 | 11p11.2 | gene composed of 7 exons; constitutes one of the subunits of the Fe-S fraction of the complex; mutations in gene result in one form of Leigh syndrome |
| NDUFS7 | 19p13.3 | gene composed of 8 exons; mutations in gene result in one form of Leigh syndrome |
| NDUFS8 | 11q13.2 | gene composed of 8 exons; binds two Fe-S clusters in the NADH dehydrogenase activity; mutations in gene result in one form of Leigh syndrome |
| NDUFV1 | 11q13.2 | gene composed of 10 exons; carries the NADH-binding site, the FMN-binding site, and Fe-S-binding sites |
| NDUFV2 | 18p11.22 | gene composed of 9 exons; |
Deficiencies in complex I proteins are the most common causes of mitochondrial diseases in humans and these disorders encompass a wide degree of heterogeneity in pathological presentation. The infantile presenting complex I disorders include Leigh syndrome, fatal infantile lactic acidosis (FILA), neonatal cardiomyopathy, leukoencephalopathy, pure myopathy, and combined hepatopathy and tubulopathy. Due to the composition of complex I being the result of both mtDNA and nuclear DNA encoded proteins, disorders that are associated with defects in complex I proteins are divided accordingly. The complexity and heterogeneity of complex I disorders can be emphasized by the constellation of disorders termed MELAS, mitochondrial encephalomyopathy, lactic acidosis, and stroke-like episodes. MELAS is not the result of a single gene defect represents a constellation of related disorders that can result from defects in several mtDNA encoded proteins of complex I.
Complex II
Complex II which is most commonly called succinate dehydrogenase (the TCA cycle enzyme) is also known as succinate:CoQ oxidoreductase or succinate:ubiquinone oxidoreductase). Complex II is composed of four protein subunits and all four subunits are integral membrane proteins in the inner mitochondrial membrane. All four subunits of complex II are encoded by the nuclear genome and the genes are SDHA, SDHB, SDHC, and SDHD. The ΔEo′ for electron flow through complex II is about 0.05 V, corresponding to a ΔG′ of –2.3 kcal/mol of electrons transferred, which is insufficient to drive ATP synthesis. The difference in free energy, of electron flow through complexes I and II, accounts for the fact that a pair of electrons originating from NADH and passing to oxygen supports production of 2.5 (more commonly reported as 3) equivalents of ATP, while two electrons from succinate support the production of only 1.5 (more commonly reported as 2) equivalents of ATP.
Complex III
Reduced CoQ (CoQH2) diffuses in the lipid phase of the membrane and donates its electrons to complex III, whose principal components are the heme proteins known as cytochromes b (encoded by the MT-CYB gene) and c1 (encoded by the CYC1 gene) and a non-heme-iron protein, known as the Rieske iron sulfur protein (encoded by the UQCRFS1 gene). Complex III is known as ubiquinol-cytochrome c oxidoreductase or as CoQ-cytochrome reductase and often as the cytochrome bc1 complex. Complex III is composed of 11 protein subunits, all of which, excepting cytochrome b, are encoded by the nuclear genome. The MT-CYB, CYC1, and UQCRFS1 encoded proteins constitute the electron transfer center of complex III. In contrast to the heme of hemoglobin and myoglobin, the heme iron of all cytochromes participate in the cyclic redox reactions of electron transport, alternating between the oxidized (Fe3+) and reduced (Fe2+) forms. The electron carrier from complex III to complex IV is the smallest of the cytochromes, cytochrome c (molecular weight 12,000; encoded by the CYCS gene). The release of cytochrome c from the mitochondria to the cytosol is the major trigger of the mitochondrially-induced apoptosis pathway.
Another important protein in the functioning of complex III is the assembly factor encoded by the BCS1L gene (BCS1-like: the human homolog of a yeast gene identified as BCS1). Mutations in the BCS1L gene result in the lethal disorder called GRACILE syndrome (GRACILE: Growth Retardation, Aminoacidurina, Cholestasis, Iron overload, Lactic acidosis, Early death). Another disorder that results from mutations in the BCS1L gene and the MT-CYB gene is known as Björnstad syndrome.
Complex IV
Complex IV, also known as cytochrome c oxidase (COX), contains the hemeproteins known as cytochrome a and cytochrome a3, as well as copper-containing proteins in which the copper undergoes a transition from Cu+ to Cu2+ during the transfer of electrons through the complex to molecular oxygen. Complex IV is composed of a total of 13 protein subunits. Oxygen is the final electron acceptor, with water being the final product of oxygen reduction. The catalytic core of complex IV is composed of the mitochondrial genome (mtDNA) encoded proteins MT-CO1 and MT-CO2 as well as cytochrome a and cytochrome a3. Another mtDNA encoded protein, MT-CO3, is a structural protein in the complex.
The other 10 subunits of complex IV are encoded by the nuclear genome and are identified as COX genes (COX4, COX5A, COX5B, COX6A, COX6B, COX6C, COX7A, COX7B, COX7C, and COX8). The COX6A and COX7A genes encode tissue-specific isoforms (COX6A1 and COX6A2; COX7A1 and COX7A2) with the H (heart)-type being expressed in heart and skeletal muscle (COX6A2 and COX7A1) and the L (liver)-type being expressed in non-muscle tissues (COX6A1 and COX7A2). There two isoforms of the COX4 protein encoded by the COX4I1 and COX4I2 genes. There is a testis-specific isoform of COX6B (identified as COX6B2). Mutations in the COX genes are the most common causes of defects in oxidative-phosphorylation.
ATP Synthase (Complex V)
The process of mitochondrial ATP synthesis occurs via the activities of the ATP synthase complex, often called complex V of oxidative phosphorylation. ATP synthase is a multiple subunit complex that forms two functional domains identified as the Fo and the F1 domains. The F1 domain resides in the matrix of the mitochondria and the Fo domain is located in the inner mitochondrial membrane. The Fo complex is composed of a ring structure called the c-ring that consists of at least eight copies of the same protein (the C proteins).
Additional components of the Fo complex (one copy each) are the subunits identified as A(a), B1(b1), D(d), F6, and O (commonly identified as oligomycin sensitivity-conferring protein, OSCP) that collectively form the peripheral stalk of the complex. Additional protein subunits of the F0 complex include the E(e), F2(f), G(g), and A6L proteins that each span the inner mitochondrial membrane. All of the nuclear genes that encode components of the ATP synthase complex are members of the ATPase 5 family.
There are three genes in humans that encode c-ring C proteins. The ATP5G1 gene encodes the C1 protein, the ATP5G2 gene encodes the C2 protein, and the ATP5G3 gene encodes the C3 protein. The B1 subunit is encoded by the ATP5F1 gene. The D subunit is encoded by the ATP5H gene. The F6 subunit is encoded by the ATP5J gene. The O subunit (OSCP) is encoded by the ATP5O gene. The E subunit is encoded by the ATP5I gene. The F2 subunit (sometimes identified as the f subunit) is encoded by the ATP5J2 gene. The G subunit is encoded by the ATP5L gene. Another gene, ATP5L2, encodes a G subunit related protein identified as G2. Two of the subunits of the Fo complex are encoded by mitochondrial genes, the A and A6L subunits. The A subunit is encoded by the MT-ATP6 gene and the A6L subunit is encoded by the MT-ATP8 gene. The F1 complex is composed of three copies of both the α and β subunits and one copy of the γ, δ, and ε subunits. The α subunit is encoded by the ATP5A1 gene. The β subunit is encoded by the ATP5B gene. The γ subunit is encoded by the ATP5C1 gene. The δ subunit is encoded by the ATP5D gene. The ε subunit is encoded by the ATP5E gene.
In addition to the Fo/F1 core complex proteins there are several regulatory, assembly, and associated proteins that are fundamental to the synthesis of ATP by ATP synthase. During the normal process of mitochondrial respiration (oxidative phosphorylation) there is a high proton-motive force (strong proton gradient across the inner mitochondrial membrane) that favors ATP synthesis. Under conditions of compromised mitochondrial function, the proton-motive force falls below the threshold required for ATP synthesis and the activity of ATP synthase can actually reverse such that it hydrolyzes ATP to pump protons back into the inner membrane space. Since depletion of ATP can result in cell death there must be a mechanism to prevent excess reverse activity of ATP synthase.
The regulatory function is carried out by the associated protein identified as inhibitor protein, IF1. The function of IF1 to inhibit ATP synthase activity operates in a pH-dependent manner, being most active at a pH below 6.5. The activity of IF1 protects cells during periods of ischemia where O2 levels fall leading to a reduced proton gradient. IF1 functions as a homodimer which binds to two ATP synthase complexes through interactions with the F1 β and γ subunits.
Another important ATP synthase regulatory protein is called factor B. Factor B interacts with the Fo complex on the matrix side of the inner mitochondrial membrane. The function of factor B is to block leaking of protons across the inner mitochondrial membrane. The ATP synthase assembly factors ATP11 and ATP12 are required to protect the hydrophobic surfaces of the unassembled α and γ subunits of the F1 complex, preventing them from aggregating within the mitochondrial matrix. Another ATP synthase associated protein is called TMEM70 (transmembrane protein 70) which maintains the normal activity level of ATP synthase.
Mitochondrial Supercomplexes
Mitochondria can form what are referred to as supercomplexes which consist of the assembly of complexes I and III and in some cases complex IV. The formation of these supercomplexes promotes highly efficient electron transport by creating pools of coenzyme Q and cytochrome c. The formation of supercomplexes allows for functionally relevant reprogramming of metabolic processes such as glycolysis, glutathione metabolism, and pyrimidine nucleotide nucleotide synthesis. The most significant function of mitochondrial supercomplexes is in the regulation of flux of metabolites into and out of the TCA cycle.
The physiological significance of supercomplex formation has been demonstrated by the fact the formation of supercomplexes increases in skeletal muscle in response to exercise. In contrast, supercomplex assembly is impaired in individuals with diabetes.
The nuclear genome encoded protein, cytochrome c oxidase subunit 7A2 like, which is encoded by the COX7A2L gene, is currrently the only identified supercomplex assembly factor. The COX7A2L encoded protein is also identified as COX7RP and also as SCAF1 (supercomplex assembly factor 1). The COX7A2L encoded protein is structurally similar to the complex IV protein COX7A, specifically to the non-muscle isoform identified as COX7A2, hence the identity of the protein as COX7A2L.
COX7A2L is required for the formation of two supercomplexes that have been termed N-respirasome and Q-respirasome. The N-respirasome is composed of complexes I, III, and IV. The Q-respirasome is composed of complexes III and IV. Whereas, the COX7A2L protein is required for supercomplex assembly, it plays no role in the stability of the individual complexes, I, III, and IV.
The formation of supercomplexes allows for enhanced production of ATP, such as in skeletal muscle in response to exercise, as well as for the suppression of reactive oxygen species (ROS) production. Recent studies have shown that the ability of cells to generate mitochondrial supercopmplexes enhances exercise endurance and, at least in mice, results in suppression of cellular senescence, a key characteristic of aging cells.
Mitochondrial Dynamics: Fusion and Fission
Mitochondria are dynamic organelles that undergo a constant state of flux that increases and decreases the number and size of mitochondria in any given cell under a wide array of metabolic and physiologic conditions. Mitochondrial flux involves the processes defined as fusion and fission. Both fusion and fission are required to fine-tune fundamental cellular processes such as the generation of ATP and reactive oxygen species, ROS. Reactive oxygen species include super oxide anion (O2•–), hydrogen peroxide (H2O2), hydroxyl radical (•OH), ozone (O3), singlet oxygen (1O2), and peroxy radicals (RO• and RO2•).
Mitochondria are intimately coupled to the membranes of the endoplasmic reticulum (ER) at specialized regions of membrane adherence termed mitochondria-associated membranes. These mitochondria-associated membranes are crucial to the modulation of mitochondrial and overall cellular calcium homeostasis. The intimate association between the mitochondria and the ER, via the mitochondria-associated membranes, allows for increases in mitochondrial calcium levels that are necessary for the regulation of the metabolic processes of this organelle. Within the mitochondria, the generation of ATP and ROS, as well as the regulation of calcium homeostasis, exerts important controls on oxygen sensing, cell-cycle progression, mitophagy, and apoptosis.
Mitochondrial Fusion
As the term fusion implies, mitochondrial fusion refers to the process where two originally distinct mitochondria merge (fuse) into one. The process of mitochondrial fusion involves integration of both the inner (IMM) and outer mitochondrial membranes (OMM) from both of the originally distinct mitochondria into the IMM and OMM of the new single mitochondria. The physiological and metabolic significance of mitochondrial fusion is that it allows for the exchange of contents between mitochondria, allowing defective mitochondria to acquire mtDNA and essential components of the oxidative phosphorylation pathway.
Mitochondrial fusion in human cells (as well as other mammals) is mediated by the proteins identified as mitofusin 1, mitofusin 2, and optic atrophy 1.
Mitofusin 1 is encoded by the MFN1 gene which is located on chromosome 3q26.33 and is composed of 19 exons that encode a protein of 741 amino acids.
Mitofusin 2 is encoded by the MFN2 gene which is located on chromosome 1p36.22 and is composed of 20 exons that generates two alternatively spliced mRNAs, both of which encode the same 757 amino acid proteins. Mutations in the MFN2 gene are associated with Charcot-Marie-Tooth disease type 2A2 (CMT2A2) and hereditary motor and sensory neuropathy VI.
The optic atrophy 1 protein is encoded by the OPA1 gene. The OPA1 gene is located on chromosome 3q29 and is composed of 32 exons that generate 10 alternatively spliced mRNAs. The optic atrophy 1 encoding mRNA that was originally characterized was composed of 29 exons that encoded a preproprotein of 960 amino acids.
Certain mutations in the OPA1 gene result in the autosomal dominant disorder called optic atrophy type 1 which is a form of optic neuropathy that leads to progressive bilateral degeneration of the optic nerves. In other situations heterozygous mutations in the OPA1 gene are known to be associated with pathology distinct from those of the optic nerve including ataxia, sensorineural deafness, chronic progressive external ophthalmoplegia, axonal sensory-motor polyneuropathy, and mitochondrial myopathy.
Both mitofusin proteins are dynamin-related GTPases whose functions are responsible for fusion of the mitochondrial outer membranes. In addition to mitochondrial localization, mitofusin 2 is localized to the endoplasmic reticulum (ER) where it promotes ER–mitochondrial tethering resulting in enhanced mitochondrial calcium uptake. The OPA1 protein is also a dynamin-related GTPase but it is localized to the inner mitochondrial membrane and there it is involved in the fusion of these membranes.
The mitofusins are large proteins that span the outer mitochondrial membrane twice resulting in both the N- and C-termini presented to the cytoplasm. Mitofusins form both homo-oligomeric and hetero-oligomeric complexes between two mitochondria. Once the mitofusin proteins form these complexes the hydrolysis of GTP enables mitochondrial membrane fusion.
However, prior to the fusion process the cardiolipin in the membrane is hydrolyzed to phosphatidic acid through the action of phospholipase D (PLD). The OPA1 protein is tethered to the inner mitochondrial membrane facing the intermembrane space. From here OPA1-mediated hydrolysis of GTP promotes fusion of the inner mitochondrial membranes, a function that also requires the activity of mitofusin 1.
Mitochondrial Fission
Mitochondrial fission is the process whereby a single mitochondrion is divided into two distinct mitochondria. This is a critical process that occurs primarily during the cell cycle to ensure that the resulting daughter cells contain the same, or very similar, numbers of mitochondria as the mother cell. Mitochondrial fission is also important for mitochondrial replication and the removal of damaged mitochondria through the process of mitophagy (mitochondrial autophagy).
The process of mitochondrial fission requires that the cytosolic protein, dynamin-related protein 1 (Drp1), be recruited to the outer mitochondrial membrane. As its name implies Drp1 is a dynamin-related GTPase. Dynamin-related protein 1 is encoded by the DNM1L (dynamin like 1) gene which is located on chromosome 12p11.21 and is composed of 21 exons that generate eight alternatively spliced mRNAs. which encode distinct protein isoforms where isoform 1 is the largest at 736 amino acids. In addition to mitochondrial fission Drp1 is involved in peroxisomal fission. Mutations in the DNM1L gene result in the autosomal dominant disorder called encephalopathy due to defective mitochondrial and peroxisomal fission 1 (EMPF1).
Recruitment of Drp1 to the outer mitochondrial membrane involves several integral membrane proteins of the outer mitochondrial membrane. These proteins are mitochondrial fission factor (Mff), mitochondrial fission protein 1 (Fis1), and two mitochondrial dynamics (MiD) proteins identified by their molecular weight, MiD49 and MiD51. Mitochondrial fission factor is encoded by the MFF gene and mutations in this gene are the cause of autosomal recessive encephalopathy due to defective mitochondrial and peroxisomal fission 2 (EMPF2).
Once interaction with the outer mitochondrial membrane is initiated Drp1 proteins multimerize forming a structure that wraps completely around the mitochondrial surface. The energy of GTP hydrolysis is used to pinch the mitochondrion into two smaller mitochondria. The dynamin 2 protein (encoded by the DNM2 gene) plays a role in the fission process by promoting further constriction of the mitochondrion following the action of Drp1. The ability of Drp1 to promote mitochondrial fission is controlled by its state of phosphorylation at Ser 616 ans Ser 637.
Phosphorylation of Drp1 at Ser 616 by cyclin dependent kinase 1/cyclin B (CDK1/cyclin B) promotes mitochondrial fission during mitosis. PKA-mediated phosphorylation of Drp1 at Ser 637 interferes with its translocation to mitochondrial fission sites and promotes Drp1 detachment from the mitochondrion. On the other hand the calcium-sensitive phosphatase, calcineurin, mediates dephosphorylation of Ser 637 in Drp1 resulting in the recruitment of Drp1 to the mitochondria, thereby promoting mitochondrial fission.
Another important modulator of Drp1 activity is its state of O-GlcNAcylation. Attachment of GlcNAc to Thr 585 and 586 results in reduced ability to phosphorylate Ser 637 and promotes translocation of Drp1 from the cytosol to the mitochondrial membrane.
Mitochondrial Signal Transduction Processes
Mitochondria are responsible for far more than just being the major producers of ATP energy. Mitochondria are capable of sensing and responding to endogenous and environmental inputs that result in signal transduction processes emanating from the mitochondria to influence the functions of other organelles and to systemically regulate physiological processes. Mitochondrial signal transduction influences metabolic, biochemical, neuroendocrine, and other local or systemic processes that ultimately enhance organismal adaptation. A term has been coined to collectively describe the processes of mitochondrial signal transduction: mitochondrial information processing system (MIPS)
Within the context of the MIPS there is sensing, integration, and signaling. Mitochondria are responsive to metabolic and hormonal inputs that lead to an alteration in biochemical and morphological processes in the mitochondria. These inputs include, but are not limited to, nutrient levels, both peptide and steroid hormones, biochemical metabolites, gases such as O2 and NO, ions, and ATP/ADP and NAD(P)+/NAD(P)H ratios. The sensing of these various inputs are then integrated into the overall functioning of the mitochondria ultimately resulting in changes to mitochondrial signaling (outputs). Mitochondrial outputs include, but are not limited to, biochemical metabolites, reactive oxygen species (ROS), ions, ATP, steroid hormones, DNA and RNA, lipids, gases, peptides, and heat.
Mitochondrial sensing involves numerous processes such as G-protein coupled receptors (GPCR), nuclear receptors, and ion and metabolite channels. With respect to GPCR, these are found in the outer mitochondrial membrane (OMM). GPCR identified in the OMM include the purine (ATP) receptor P2Y5, the cannabinoid receptor 1 (CB1), the angiotensin receptors (AT1R and AT2R), and the melatonin receptor (MT1). Canonical nuclear receptors that are found within the matrix of the mitochondria include the glucocorticoid receptor (GR), the thyroid (T3) hormone receptor (TR), the estrogen receptor beta (ERβ), and the androgen receptor (AR). Ion and metabolite channels are found in the inner mitochondrial membrane (IMM).
Activation of the AT2R in the mitochondria is associated with increased production of nitric oxide (NO) while also decreasing complex I-driven O2 consumption capacity. Activation of the mitochondrial MT1 receptor has been shown to inhibit the release of cytochrome c and protects from downstream ischemic effects. Activation of the CB1 receptor results in reduced complex I activity and overall mitochondrial respiration.
Glucocorticoid response elements (GRE) are found in the D loop of the mtDNA as well as in several mtDNA genes including those encoding the 12S rRNA, the tRNALeu(UUR), the COX I, and the COX III genes. The mitochondrial AR is involved in regulating mtDNA levels and mtDNA transcription and translation.
Mitochondrial Calcium and Metabolic Regulation
Mitochondria have long been known to be a significant organelle for the storage of calcium, Ca2+. Calcium uptake by mitochondria occurs at the outer mitochondrial membrane (OMM) via the transporters of the voltage-dependent anion transport (VDAC) family, most likely VDAC1. The VDAC proteins are members of the mitochondrial porin family.
Calcium transport into the matrix of the mitochondria occurs via the action of the inner mitochondrial membrane (IMM) localized transporter identified as mitochondrial calcium uniporter, MCU. The MCU is actually a complex of several proteins in addition to the actual pore-forming subunit identified as MCU (also designated mtCU). The complex includes many regulatory proteins that includes an inhibitor identified as MCUb, the essential MCU regulator (EMRE), and three EF-hand proteins identified as mitochondrial Ca2+ uptake 1 (MICU1), MICU2, and MICU3. In addition, the protein identified as MCU regulator 1 (MCUR1) serves as a scaffold that connects MCU and EMRE.
The mitochondrial calcium uniporter protein is encoded by the MCU gene. The MCU gene is located on chromosome 11q22.1 and is composed of 12 exons that generates three alternatively spliced mRNAs, each of which encode a distinct protein isoform.
The essential MCU regulator (EMRE) is encoded by the SMDT1 (single-pass membrane protein with aspartate rich tail 1) gene. The SMDT1 gene is located on chromosome 22q13.2 and is composed of 4 exons that encode a 107 amino acid precursor protein.
The MICU1 gene is located on chromosome 10q22.1 and is composed of 17 exons that generate four alternatively spliced mRNAs, each of which encode a distinct protein isoform.
The MICU2 gene is located on chromosome 13q12.11 and is composed of 15exons that encode a 434 amino acid protein.
The MICU3 gene is located on chromosome 8p22 and is composed of 22 exons that generates 11 alternatively spliced mRNAs, each of which encode distinct protein isoforms. The isoform 1 protein is the longest an is composed of 530 amino acids.
The MICU1, MICU2, and MICU3 proteins located at the IMM and each form dimers. When Ca2+ concentration is low they are unoccupied and block the MCU pore. When Ca2+ levels rise, these EF-hand proteins bind Ca2+ which alters their conformation allowing the pore to function in its role of Ca2+ transport into the matrix. The primary contributors to increased Ca2+ near the mitochondria are the inositol-1,4,5-trisphosphate (IP3; Ins-1,4,5-P3) receptors in the endoplasmic reticulum (ER) and the calcium release channels, the ryanodine receptors (RyR) which are prevalent in cardiac and skeletal muscle sarcoplasmic reticulum (SR). The tethering of ER and SR membranes with the mitochondria allows for rapid localized increases in Ca2+ to alter the metabolic activity of the mitochondria.
Efflux of Ca2+ from the matrix of the mitochondria is primarily the function of the IMM-localized mitochondrial Ca2+ efflux exchanger, NCLX (Na+-Ca2+-Li+ exchanger). The NCLX transporter carries out electrogenic transport of four Na+ ions into the mitochondria in exchange for one Ca2+ ion and one K+ ion out of the mitochondria.
The NCLX transporter is a member of the SLC superfamily of transporters and as such is encoded by the SLC8B1 gene. The SLC8B1 gene is located on chromosome 12q24.13 and is composed of 16 exons that generate three alternatively spliced mRNAs that collectively encode two distinct protein isoforms.
In addition to NCLX, evidence indicates that there is also a Na+-independent transporter involved in mitochondrial Ca2+ efflux. This Ca2+ efflux transporter, also localized to the IMM, is an antiporter that import protons (H+) in exchange for Ca2+ efflux. The transporter is identified as transmembrane BAX inhibitor motif containing protein 5 (TMBIM5). The human TMBIM5 protein is encoded by the GHITH (growth hormone inducible transmembrane protein) gene.
When Ca2+ is transported out of the mitochondria it can be taken back up by the ER and SR through the action of the Ca2+ re-uptake transporters of the SERCA (sarco/endoplasmic reticulum Ca2+-ATPase) family. SERCA transporters are members of the P-type ATPases and humans express three genes in the family, ATP2A1 (encoding SERCA1), ATP2A2 (encoding SERCA2), and ATP2A3 (encoding SERCA3). SERCA1 predominates in cardiac fast twitch muscle fibers. SERCA2 predominates in cardiac slow twitch fibers. SERCA3 is ubiquitously expressed.
Within the context of mitochondrial metabolism, the uptake of Ca2+ is necessary for the function of the pyruvate dehydrogenase complex (PDHc), the TCA cycle, and the electron transport chain of oxidative phosphorylation. Calcium activation of the PDHc occurs primarily as a result of the activation of PDH phosphatase which then dephosphorylates the PDHc increasing its activity, thereby providing increased acetyl-CoA delivery to the TCA cycle. Within skeletal muscle mitochondria Ca2+ inhibits the PDHc kinase, PDK4. Inhibition of PDK4 prevents the PDHc from being phosphorylated allowing the complex to remain active at oxidizing pyruvate to acetyl-CoA.
The activity of two of the dehydrogenases of the TCA cycle is regulated by Ca2+ ions. These include isocitrate dehydrogenase (ICDH) and the 2-oxoglutarate (α-ketoglutarate) dehydrogenase complex (OGDH). Both ICDH and OGDH are directly activated by Ca2+ ions that function by lowering the Km for substrate.
All three of the dehydrogenases that are affected by mitochondrial Ca2+ generate NADH which drives the electron transport chain. With respect to oxidative phosphorylation and the production of ATP, complex III of the electron transport chain and the ATP synthase (complex V) are activated by mitochondrial Ca2+.
When the amount of intramitochondrial Ca2+ exceeds a threshold the ion mitochondrial permeability transition pore (PTP) opens allowing for release of the accumulated Ca2+ along with several smaller mitochondrial proteins and cofactors. The significance of the Ca2+ triggered opening of the PTP has been demonstrated to be an underlying cause of diseases associated with mitochondrial Ca2+ overload as well as the cause of cell death in ischemia and reperfusion injury.
Mutations in either of the two EF-hand subunits of the MCU complex encoded by the MICU1 and MICU2 genes have been identified in humans. These mutations are associated with muscle fatigue, myopathy, and neurodevelopmental disorders. The syndrome associated with mutations in the MICU1 gene is referred to as myopathy with extrapyramidal signs, MPXPS. A nonsense mutation in the MICU2 gene (W14X) represents a variant of unknown significance (VUS) identified in two siblings of Arab kindred who exhibited impaired intellectual development.
Mitochondria and the Integrated Stress Response
Mitochondrial stress results in numerous changes to cellular metabolism, the redox environment, and the capacity for correct protein folding. The changes that occur in response to mitochondrial stress are coordinated with what is collectively referred to as the integrated stress response, ISR. When considering the role of mitochondrial stress in the ISR, it is often referred to by the acronym: ISRmt.
The ISR functions to generate conditions in the cell that reduce the vulnerability to future stress. Various conditions activate the ISR including, but is not limited to, amino acid deprivation, iron deprivation, increased reactive oxygen species (ROS), and accumulation of unfolded proteins. With respect to protein folding, the role of mitochondrial processes in response to stress is referred to as the mitochondrial unfolded protein response (mtUPR; also designated UPRmt).
Numerous signals are sensed to assess the health of mitochondria. These include mitochondrial membrane potential, calcium ions, reactive oxygen species (ROS), mitochondrial protein import, various metabolites, and the level of protease activity. The overall changes that occur in response to mild mitochondrial stress have been termed mitohormesis.
A major contributor to the integrated responses that cells undertake to stress and mitochondrial dysfunction is the phosphorylation of the alpha subunit of the heterotrimeric translation initiation factor, eIF-2. Human cells express four genes that encode kinases that phosphorylate eIF-2α as described in detail the Protein Synthesis (Translation): Processes and Regulation page. One of the eIF-2α kinases is the heme regulated inhibitor (HRI). The protein is also called the heme controlled repressor (HCR) or the heme controlled inhibitor (HCI). The gene encoding HRI is identified as EIF2AK1.
When mitochondria are stressed, primarily due to increased reactive oxygen species (ROS), or when they sense nuclear DNA damage, a mitochondria-localized protease known as OMA1 zinc metallopeptidase, or simply as OMA1, is activated. The term OMA1 refers to Overlapping activity with the M-AAA protease A1. M-AAA protease A1 is a member of the Mitochondrial ATPases Associated with Any diverse cellular activities (AAA) protease family.
Mitochondrial membrane-localized AAA-type proteases are defined as m-AAA (where “m” refers to matrix) and i-AAA (where “i” refers to intermembrane space). The m-AAA protease is oriented in the inner mitochondrial membrane such that the catalytic domain is in the matrix. The i-AAA protease is oriented in the inner mitochondrial membrane such that the catalytic domain is in the inter-membrane space. OMA1 interacts with m-AAA and i-AAA in a complex that also contains the membrane localized proteins, PHB2 (prohibitin 2 which is encoded by the PHB2 gene) and SLP2 (stomatin-like protein 2 which is encoded by the STOML2 gene).
One of the substrates of OMA1 is the protein DAP3 binding cell death enhancer 1 which is encoded by the DELE1 gene. DELE is derived from DAP3-binding, Enhancer of Ligand-mediated Endopeptidase. The DELE1 encoded protein is a signaling peptide that is critical to the activation of the ISR.
When OMA1 cleaves DELE1, a short form results identified as DELE1s (also designated S-DELE1). DELE1s is transported out of the mitochondria to the cytosol where it interacts with HRI. The interaction of DELE1s with HRI activates the kinase which in turn phosphorylates eIF-2α. The phosphorylation of eIF-2α results in the inhibition of global protein synthesis and the activation of the translation of specific proteins required to initiate the ISR. As a result of eIF-2α phosphorylation translation of the transcription factor, activating transcription factor 4 (ATF4) is enhanced. ATF4 is the master transcriptional regulator of the ISR.
In addition to its role in mitochondrial stress-related activation of the ISR, the OMA1 protein also functions in mitochondrial apoptosis (through cleavage of the OPA1 protein), mitochondrial autophagy (mitophagy; through cleavage of the PINK1 protein), and mitochondrial necroptosis (through cleavage of the PGAM5 protein). As discussed in the Mitochondrial Fusion section above, OPA1 is a mitochondrial dynamin-like GTPase encoded by the OPA1 gene. The OPA1 encoded protein can induce mitochondrial outer membrane permeabilization. PINK1 (PTEN INduced putative Kinase 1; where PTEN stands for Phosphatase and TENsin homolog) is a Ser/Thr kinase that is critical to the maintenance of mitochondrial health by protecting mitochondria from damage through the process of mitophagy. The PGAM5 (phosphoglycerate mutase 5) protein is a phosphatase that is involved in mitochondrial health. PGAM5 removes phosphate from its target substrates which can promote mitochondrial fusion and biogenesis thereby contributing to cell survival or the result can promote mitochondrial fission and apoptosis leading to programmed cell death.
Dysfunction in mitochondrial oxidative phosphorylation results in an increase in the NADH to NAD+ ratio. Increased NADH lead to depletion of aspartate (Asp) production, and consequently asparagine (Asn) as well since Asp serves as a predominant precursor for the generation of Asn. These effects represent nutrient (amino acid) deprivation, an event that in turn activates another eIF-2α kinase, GCN2. The activation of GCN2 leads to further enhancement in the level of ATF4 activity.
The activation of ATF4 leads to increased transcription of target genes that includes FGF21, GDF15, MTHFD2, and ATF5. Increased expression of the FGF21 gene leads to enhanced systemic glucose uptake and increased hepatic lipid metabolism. GDF15 is involved in central control of feeding behaviors. Increased expression of the MTHFD2 gene (encoding the mitochondrial NAD+/NADP+-dependent enzyme that possesses 5,10-methylene-THF dehydrogenase and 5,10-methenyl-THF cyclohydrolase activities) enhances the generation of the mitochondrial pool of tetrahydrofolate (THF) derivatives. Enhancement of the THF pool allows for increased nucleotide biosynthesis, a process that is required in the context of DNA damage repair.
Another member of the ATF family of transcription factors, ATF5, is also critical to the role of the mitochondria in the ISR. ATF5 is normally localized to the mitochondria but under conditions of mitochondrial protein folding stress, increased levels of reactive oxygen species (ROS), or electron transfer chain dysfunction, the ATF5 protein begins to accumulate in the cytosol and is transported into the nucleus where is activates genes in the mtUPR. Genes encoding proteins of the heat shock protein family of chaperones, such as HSPD1 (encoding hsp60), HSPE1 (encoding chaperonin 10 which is a co-chaperone of hsp60), and HSPA9 (encoding a mitochondrial hsp70 family protein) are some of the targets of ATF5 transcriptional activity. Additional genes that are regulated by ATF5 are the ATF5 gene itself and the LONP1 gene which encodes a mitochondrial protease.
As indicated in the mtDNA Damage and Repair section above, the protein encoded by the DDIT3 gene is also important in the overall ISR. The DDIT3 (DNA damage inducible transcript 3) encoded protein is also identified as CHOP (C/EBP homologous protein, where C/EBP is CCAAT/enhancer binding protein). Activation of ATF4 and ATF5 results in increased transcription of the DDIT3 gene. The DDIT3 encoded protein (CHOP) functions as a dominant-negative factor by forming heterodimers with other proteins of the C/EBP family, thereby inhibiting their functions. The primary activity of CHOP is activation of apoptosis.
When cells sense mitochondrial dysfunction a specific transcriptional program is initiated that encompasses genes that promote mitochondrial protein homeostasis. This process is referred to as the mitochondrial unfolded protein response, UPRmt, which constitutes a portion of the overall involvement of the mitochondria in the ISR.
Under conditions of excessive reactive oxygen species (ROS) generation mitochondria will contribute to the UPR via the activation of the co-chaperone of the Hsp40 family encoded by the DNAJA1 gene. The activation of the DNAJA1 encoded protein induces its interaction with Hsp70 family chaperones. In the context of the UPR, that interaction of DNAJA1 with Hsp70 results in the release of transcription factor encoded by the HSF1 (heat shock transcription factor 1) gene. Upon its release, HSF1 migrates to the nucleus where it activates the expression of genes whose encoded proteins are involved in the UPR, many of which are mitochondrial chaperones such as the HSPA9 (mitochondrial Hsp70 family member), HSPD1 (Hsp60 family member), and HSPE1 (Hsp10 family member) genes.
Mitochondrial Autophagy (Mitophagy)
Autophagy (from Greek for “self-eating”) represents the highly regulated pathways by which cellular components can be degraded and recycled as a means to support cell survival during times of energy stress and nutrient starvation. Three primary pathways of autophagy have been characterized.
Microautophgy represents the pathway whereby the lysosome directly engulfs a small portion of the cytosol. Chaperone-mediated autophagy (CMA) represents the pathway by which chaperones target selective substrates for degradation in the lysosome.
Macroautophagy (most commonly just referred to as autophagy) is the pathway by which cytoplasmic organelles are sequestered within membrane compartments referred to as pre-autophagosomes or phagophores (also referred to as isolation membranes, IM).
In yeast the initial autophagy-associated structure is referred to as the pre-autophagosomal structure, PAS. The sources of the membrane material from which the phagophore forms is quite varied. Evidence demonstrates that membrane from the endosomes, the endoplasmic reticulum (ER), the Golgi, the ER-Golgi intermediate complex (ERGIC), the mitochondria, and ER-mitochondrial contact sites are sources of the phagophore membranes.
The newly formed membrane compartments fuse at their edges to form a double membrane vesicular structure called the autophagosome. The autophagosome is, therefore, the membrane enclosed structure which contains cytosolic constituents and subcellular organelles destined for degradation. The autophagosome ultimately fuses with the lysosome allowing its contents to be degraded. Although the induction of the various pathways of autophagy include both physiological and pathological processes, the response of cells to nutrient starvation represents the canonical form of (macro)autophagy. Dysfunction in the processes of autophagy can have severe clinical consequences. The etiology of numerous diseases and disorders such as Crohn disease, many cancers, diabetes, and several neurodegenerative disorders is associated with defects in components of autophagy such as members of the autophagy related family of proteins.
Table of Membrane Trafficking Proteins in Autophagosome Formation
List is not intended to be a complete catalog of all membrane trafficking proteins involved in autophagy
| Protein Name | Gene(s) | Functions / Comment |
| Rab1 family | RAB1A, RAB1B, RAB1C | member of the RAB family of monomeric G-proteins; belongs to RAS superfamily; involved in phagophore biogenesis from ER exit sites (ERES) which are membrane domains involved in the protein secretory pathway |
| Rab5 family | RAB5A, RAB5B, RAB5C | member of the RAB family of monomeric G-proteins; belongs to RAS superfamily; associated with early endosomes; involved in the process of induction of autophagy |
| Rab7 family | RAB7A, RAB7B | member of the RAB family of monomeric G-proteins; belongs to RAS superfamily; associated with lysosomes and late endosomes; involved in the fusion of autophagosomes with the lysosomes |
| Rab11 family | RAB11A, RAB11B | member of the RAB family of monomeric G-proteins; belongs to RAS superfamily; associated with recycling endosomes; involved in the transport of endosomal membrane to the phagophore |
| Rab25 | RAB25 | member of the RAB family of monomeric G-proteins; belongs to RAS superfamily; associated with recycling endosomes; involved in the regulation of autophagosome maturation |
| Rab32 | RAB32 | member of the RAB family of monomeric G-proteins; belongs to RAS superfamily; associated with the ER; involved in autophagosome formation |
| synaptosome associated protein 29 | SNAP29 | involved in the fusion between autophagosome membranes and lysosomal membranes |
| sorting nexin 18 | SNX18 | associated with recycling endosomes; participant in endocytosis and intracellular vesicle trafficking; involved in formation of the autophagosome |
| syntaxin 7 | STX7 | involved in trafficking membranes from the plasma membrane to early endosomes along with STX8; involved in formation of the autophagosome |
| syntaxin 8 | STX8 | involved in trafficking membranes from the plasma membrane to early endosomes along with STX7; involved in formation of the autophagosome |
| syntaxin 17 | STX17 | a member of the SNARE (SNAP REceptor, where SNAP is Soluble N-ethylmaleimide-sensitive factor Attachment Protein) family of membrane fusion proteins; the SNARE family proteins are required for membrane fusion processes exocytotic and endocytotic pathways; involved in the control of autophagosome membrane fusion with the lysosome membrane |
| vesicle associated membrane protein 3 | VAMP3 | a member of the SNARE family; involved in vesicular transport from the late endosomes to the trans-Golgi network (TGN); associated with the ATG12-ATG5-ATG16L1 conjugate fusion with the phagophore membranes |
| vesicle associated membrane protein 7 | VAMP7 | involved in the targeting/fusion of transport vesicles to their target membrane during protein transport to endosomes and lysosomes; role in ATG16L1 association with membranes forming the phagophore |
| vesicle associated membrane protein 8 | VAMP8 | a member of the SNARE family; interacts with STX17; involved in the control of autophagosome membrane fusion with the lysosome membrane |
In addition to the more global processes of autophagy there are several selective processes that include autophagic removal of mitochondria (called mitophagy and described in more detail below), peroxisomes (termed pexophagy), endoplasmic reticulum (called reticulophagy), and ribosomes (called ribophagy). Part of the nucleus can also be removed via specialized autophagic processes referred to as nucleophagy. There are two stages of nucleophagy termed piecemeal microautophagy of the nucleus (PMN) which is the early stage process, and late nucleophagy (LN) which occurs following prolonged nutrient deprivation. The process referred to as xenophagy involves the autophagic elimination of invading pathogens such as bacteria and viruses.
The complete process of autophagy induction, organelle turnover, and recycling of the required proteins of autophagy is exceedingly complex and beyond the scope of this discussion. However, an overview of the basics of the regulation of induction of autophagy and the major protein activities of (macro)autophagy will be carried out. Numerous genes that encode proteins that regulate the processes of autophagy are identified by the ATG acronym which refers to AuTophaGy related gene. Not all proteins in the large family of proteins referred to as the autophagy related family are designated by the ATG nomenclature. Humans express a total of 33 genes in the autophagy related protein family, several of which are described in the following Table. In addition to these 33 proteins of the autophagy related family, there are numerous other proteins required to activate and regulate the overall processes of autophagy. Indeed, several proteins that regulate autophagy also play important roles in the regulation of apoptosis.
Table of Mammalian Autophagy Related Protein Family
| Protein Name | Gene | Functions / Comment |
| autophagy related 2A | ATG2A | required for the formation of the autophagosome via its role in the regulation of lipid droplet formation and dispersion; functions along with the ATG2A, WIPI1, and WIPI2 encoded proteins to forma critical functional complexes of autophagy |
| autophagy related 2B | ATG2B | required for the formation of the autophagosome via its role in the regulation of lipid droplet formation and dispersion; functions along with the ATG2B, WIPI1, and WIPI2 encoded proteins to form a critical functional complexes of autophagy |
| autophagy related 3 | ATG3 | functions as a ubiquitin conjugating E2-like activity; covalently attaches phosphatidylethanolamine (PE) to the C-terminus of the human ATG8 family member proteins: GABARAP, GABARAPL1, GABARAPL2, and MAP1LC3A |
| autophagy related 4A | ATG4A | functions as a cysteine protease; cleaves the C-terminus of ATG8 family member ubiquitin-like modifier proteins exposing a terminal glycine residue |
| autophagy related 4B | ATG4B | functions as a cysteine protease; cleaves the C-terminus of ATG8 family member ubiquitin-like modifier proteins exposing a terminal glycine residue |
| autophagy related 4C | ATG4C | functions as a cysteine protease; cleaves the C-terminus of ATG8 family member ubiquitin-like modifier proteins exposing a terminal glycine residue |
| autophagy related 4D | ATG4D | functions as a cysteine protease; cleaves the C-terminus of ATG8 family member ubiquitin-like modifier proteins exposing a terminal glycine residue |
| autophagy related 5 | ATG5 | is conjugated with ATG12 via the ubiquitin conjugating E2-like activity of ATG10 |
| autophagy related 7 | ATG7 | functions as a ubiquitin conjugating E1-like activity to activate ATG12 |
| autophagy related 9A | ATG9A | multipass membrane protein that is thought to assist in brining membrane to the developing phagophore |
| autophagy related 9B | ATG9B | multipass membrane protein that is thought to assist in brining membrane to the developing phagophore |
| autophagy related 10 | ATG10 | functions as a ubiquitin conjugating E2-like activity to transfer ATG12 to ATG5 forming a complex |
| autophagy related 12 | ATG12 | activated by ATG7 then transferred to ATG5 via the ubiquitin conjugating E2-like activity of ATG10 |
| autophagy related 13 | ATG13 | involved in the complex formed from the ULK1, ULK2, RB1CC1, and ATG101 encoded proteins; required to stabilize ATG101 |
| autophagy related 14 | ATG14 | also known as ATG14 like (ATG14L); component of the beclin 1 complex; involved in the association of the ATG12-ATG5-ATG16L1 complex and the ATG8-PE conjugate to the phagophore |
| autophagy related 16 like 1 | ATG16L1 | forms a ubiquitin conjugating E3-like activity when conjugated with ATG12 and ATG5 |
| autophagy related 16 like 2 | ATG16L2 | forms a ubiquitin conjugating E3-like activity when conjugated with ATG12 and ATG5 |
| autophagy related 101 | ATG101 | interacts with and stabilizes, in a reciprocal manner, ATG13; involved in the formation of the ULK1/ULK2 autophagy initiating complex |
| beclin 1 | BCN1 | mammalian homolog of yeast ATG6 (which is also known as Vps30); functions in a complex with several proteins that includes ATG14; activity regulated by ULK1/ULK2; activity also regulated by AMPK; activity inhibited by the pro-survival proteins BCL-2 and BCL-XL; |
| RB1 induced coiled-coil 1 | RB1CC1 | gene name derived from fact that the encoded protein enhances retinoblastoma 1 (RB1) gene expression in cancer cells; commonly referred to as FIP200 (focal adhesion kinase family interacting protein of 200 kDa); interacts with the ULK1 kinase; is the human ortholog of the yeast ATG17 gene |
| UNC-51-like autophagy activating kinase 1 | ULK1 | represents one of two human homologs of the yeast ATG1 gene (often identified as ATG1A); critical regulator of autophagy induction; activity regulated by the mTOR complex 1 (mTORC1) |
| UNC-51-like autophagy activating kinase 2 | ULK2 | represents one of two human homologs of the yeast ATG1 gene (often identified as ATG1B); critical regulator of autophagy induction; activity regulated by the mTOR complex 1 (mTORC1) |
| WD repeat domain, phosphoinositide interacting 1 | WIPI1 | represents one of two human homologs of the yeast ATG18 gene; often designated ATG18A; functions along with the ATG2A, ATG2B, and WIPI2 encoded proteins to form a critical functional complex of autophagy |
| WD repeat domain, phosphoinositide interacting 2 | WIPI2 | represents one of two human homologs of the yeast ATG18 gene; often designated ATG21 or ATG18B; functions along with the ATG2A, ATG2B, and WIPI1 encoded proteins to form a critical functional complex of autophagy |
A central regulator of the signaling pathways that trigger autophagy is the Ser/Thr kinase mTOR (mechanistic target of rapamycin) which is the principal component of both the mTOR complex 1 (mTORC1) and mTORC2 protein complexes. The mTORC1 is the major mTOR-containing complex that regulates cellular responses to growth factor signaling, nutrient deprivation, and various forms of cellular stress.
When mTOR is activated by a variety of different growth factor receptors autophagy is suppressed. Upstream signaling proteins that stimulate the activity of mTOR include the Ser/Thr kinases, PKB/AKT, PI3K, and several members of the MAPK family of kinases. Under conditions of energy depletion, such as nutrient starvation, the master metabolic regulatory kinase, AMPK, is activated which phosphorylates and inhibits mTOR, thereby promoting autophagy induction. The tumor suppressor p53 can also activate autophagy under conditions of genotoxic stress through repression of mTOR activity.
Within the context of autophagy there are two related downstream kinases whose activities are regulated by activated mTORC1. These Ser/Thr kinases are known as UNC-51-like autophagy activating kinases -1, and -2, which are encoded by the ULK1 and ULK2 genes. Humans express a third related gene, UKL3, whose encoded kinase is primarily involved in the regulation of the activity of the sonic hedgehog (SHH) protein. However, evidence does indicate that ULK3 can function in some capacity in the regulation of autophagy.
The UNC genes represent a large family of genes identified in the round worm C. elegans that, when mutated, result in uncoordinated motor activity. The human ULK1 protein functions similarly to the yeast ATG1 protein. ULK1 (or its close family member ULK2) interacts with several other proteins to form a large complex that is required for the formation of an autophagosome.
The other proteins that complex with ULK1/ULK2 include ATG13, ATG101, and RB1CC1 (FIP200/ATG17). As indicated, under conditions of nutrient deprivation AMPK is activated, thereby phosphorylating and inhibiting the mTORC1. When the mTORC1 is active it phosphorylates ULK1 on a regulatory Ser residue (S757) thereby, inhibiting the activation of ULK1. Thus, AMPK-mediated inhibition of mTORC1 activity allows ULK1 to induce autophagy. AMPK also directly phosphorylates ULK1 on two different Ser residues (S317 and S777) which results in activation of the autophagy induction properties of ULK1.
In addition to the role of AMPK in the regulation of ULK1/ULK2 activity via its effects on mTORC1 activity, this master metabolic regulatory kinase also regulates the autophagy complex downstream of ULK1/ULK2 at the level of the beclin 1 complex, also called the class III PI3K complex.
The beclin 1 complex consists of several proteins including, but not limited to beclin 1 (the mammalian homolog of yeast ATG6), ATG14 (often referred to as ATG14-like, ATG14L), ultraviolet irradiation resistance-associated gene (UVRAG), AMBRA1 (autophagy and beclin 1 regulator 1), RUBICON (RUN and cysteine-rich domain containing beclin 1 interacting protein), and SH3 domain containing GRB2 like endophilin B1 (encoded by the SH3GLB1 gene; also known as BIF-1).
The primary function of the activation of the beclin 1 complex is to activate the class III PI3K encoded by the PIK3C3 gene. PIK3C3 encodes a p110 catalytic subunit which forms a heterodimer with the p150 regulatory subunit encoded by the PIK3R4 gene. Activation of the class III PI3K results in the production of phosphatidylinositol 3-phosphate (PI3P) at the site of phagophore formation.
One important role of PI3P is the recruitment of the WIPI proteins (human homologs of the yeast ATG18 protein) to the phagophore membrane. In addition, PI3P may recruit other downstream effector proteins of the autophagy process such as the ATG5-ATG12-ATG16L1 complex.
The function of the beclin 1 protein within the complex can be inhibited via interaction with the pro-survival (anti-apoptotic) proteins BCL-2 and BCL-XL (see Mitochondrial Apoptosis section the Protein, Organelle, and Cell Turnover page). BCL-2 and BCL-XL bind to the BH3 domain of beclin 1 thereby inhibiting its activity. The interaction of beclin 1 with either of the two pro-survival proteins can be inhibited by phosphorylation of beclin 1 or BCL-2 or by the ubiquitylation of beclin 1. Therefore, it is clear that the beclin 1 complex plays an important role in promoting mitochondrial apoptosis as well.
Mitochondria localized beclin 1 induces the translocation of the BAX protein which forms a outer mitochondrial membrane (OMM) pore complex allowing pro-apoptotic factors, such as cytochrome c to be released setting off the intrinsic apoptotic cascade.
The nucleation of membrane into the initial pre-autophagosome (phagophore) requires several ATG proteins and lipids in what is referred to as the conjugation step of autophagosome formation. Phosphatidylethanolamine (PE) is known to be a critical lipid component of the conjugation process and it is believed that the PI3P derived via the action of the class III PI3K is also an important lipid. Phagophore formation begins when ATG12 is activated via the ubiquitin conjugating E1-like activity of ATG7. Activated ATG12 is then transferred to ATG5 via the ubiquitin conjugating E2-like activity of ATG10. The ATG12-ATG5 conjugate then interacts with ATG16L1 forming an ATG12-ATG5-ATG16L1 complex that possesses ubiquitin conjugating E3-like activity. The ATG12-ATG5-ATG16L1 complex is functionally coupled to proteins of the human ATG8 family. The ATG8 proteins are considered members of the ubiquitin-like modifier family of proteins.
In yeast there is but a single ATG8 protein involved in autophagy, however, in humans there are at least seven ATG8 family members. The human ATG8 family proteins are GABA type A receptor-associated protein (encoded by the GABARAP gene; considered ATG8A), GABA type A receptor-associated protein like 1 (encoded by the GABARAPL1 gene; considered ATG8B), GABA type A receptor-associated protein like 2 (encoded by the GABARAPL2 gene; considered ATG8C), microtubule associated protein 1 light chain 3 alpha (encoded by the MAP1LC3A gene; considered ATG8E; also called simply LC3), microtubule associated protein 1 light chain 3 beta (encoded by the MAP1LC3B gene; considered ATG8F), microtubule associated protein 1 light chain 3 beta2 (encoded by the MAP1LC3B2 gene; considered ATG8G), and microtubule associated protein 1 light chain 3 gamma (encoded by the MAP1LC3C gene; considered ATG8J).
The ATG8 family proteins are cleaved at their C-termini via the action of the several human ATG4 family cysteine proteases. The cleavage results in the exposure of a C-terminal glycine residue. In the process of protein ubiquitylation (described in detail in the Protein, Organelle, and Cell Turnover page) the exposed C-terminal glycine of ubiquitin would be the site of attachment to protein substrate. However, in the case of the ATG8 family proteins they are attached to phosphatidylethanolamine, PE. The attachment of ATG8 family proteins to PE is catalyzed by the E3-like activity of the ATG12-ATG5-ATG16L1 conjugate in concert with the E2-like activity of ATG3. The ATG12-ATG5-ATG16L1 conjugate, along with the associated ATG8 family protein covalently attached to PE, are recruited to the membranes of the forming phagophore via the function of the ATG14 protein which is in a large complex with the beclin 1 protein.
The recognition of the “cargo” that is to be engulfed by the autophagosome is carried out be specific proteins termed autophagy receptors. All autophagy receptors characterized to date possess both an ATG8E (LC3) interacting region (more commonly referred to as an LC3-interacting region, LIR) and a ubiquitin-binding domain, UBD. The selective autophagy receptor called sequestosome 1 (encoded by the SQSTM1 gene) was the first member of the family characterized. The SQSTM1 encoded protein is also known as nucleoporin 62 (p62). There are five well characterized autophagy receptors which includes SQSTM1, NBR1 (Neighbor of BRCA1 gene 1; encoded by the NBR1 gene), NDP52 [Nuclear Domain 10 Protein 52; encoded by the calcium binding and coiled-coil domain 2 (CALCOCO2) gene], OPTN (optineurin; encoded by the OPTN gene), and BNIP3-like [BCL-2 interacting protein 3 (BNIP3) like; encoded by the BNIP3L gene; BNIP3 and BNIP3L are members of the pro-apoptotic BH3-domain only BCL-2 related protein family). BNIP3L is also called NIX (NIP3-like protein X).
When the formation of the autophagosome is complete it fuses with endosomes which then fuses with lysosomes to form autolysosomes. When the fusion with the lysosomes takes place, the contents of the autophagosome are degraded by the resident hydrolases. The resulting amino acids and other basic cellular constituents are reused by the cell. When intracellular amino acids rise high enough through the actions of the autophagosome and subsequent lysosomal protein degradation they will reactivate the mTORC1 resulting in suppression of autophagy.
Mitochondrial function is critical for metabolic homeostasis and the production of energy. As mitochondria age they become progressively damaged primarily from the accumulated effects of the reactive oxygen species (ROS) that are inherently generated in this organelle during the process of electron transport. The progressive ROS damage leads to release of mitochondrial components, some of which, such as cytochrome c, can trigger the intrinsic apoptotic program.
Another mitochondrial damage response pathway is the autophagic pathway termed mitophagy. Mild oxidative stress triggers mitophagy and equally significant is that ROS accumulation is required for hypoxia-induced mitophagy as well as for nutrient deprivation-induced autophagy. Thus, ROS limits its own production via stimulation of mitophagy, thereby, preventing cellular damage. Although mitophagy involves several proteins of the primary (macro)autophagy pathway, the induction of mitophagy occurs via a pathway that is distinct in its protein effectors. The ultimate role of mitophagy is to remove damaged or surplus mitochondria from the cell so as to ensure cell survival. However, if the damage is significant the intrinsic apoptotic program may be initiated. Defects in the mechanisms that ensure mitochondrial viability, such as mitophagy, are associated with numerous disease states, in particular neurodegenerative diseases such as Parkinson disease (PD).
Parkinson disease is characterized by death of dopaminergic neurons in the substantia nigra which is located in the mesencephalon. Although substantia nigra neuron degeneration represents the primary pathological feature of PD, there are multiple neuron types and neuronal circuits that become disrupted in the course of the disease. The dopaminergic neurons in the substantia nigra possess dense axonal arborizations and because of their continuous pacemaking activity they are under high mitochondrial stress. It is this intrinsic property of dopaminergic neurons that makes then exceptionally sensitive to defects in mitochondrial quality control mechanisms and the resultant mitochondrial dysfunction.
Familiar (inherited) forms of PD do not represent a major proportion of cases, however, study of patients with these forms has been instrumental in identifying proteins involved in the pathways of mitophagy. In patients with early-onset autosomal recessive forms of PD, two genes, whose encoded proteins are critical for mitochondrial quality control, were identified.
One gene, identified as PINK1 (PTEN INduced putative Kinase 1; where PTEN stands for Phosphatase and TENsin homolog) is a Ser/Thr kinase and the other gene, identified as PARK2 (parkin RBR E3 ubiquitin protein ligase) is a ubiquitin conjugating E3 ligase. The PARK2 encoded protein is commonly called Parkin. The Parkin protein is a member of one of the three E3 ligase families referred to as RBR (Ring-Between-Ring) family. During mitochondrial stress the PINK1 protein is localized to the mitochondrial outer membrane (OMM), whereas the Parkin protein is normally localized to the cytosol.
Using genetic studies in fruit flies has been determined that PINK1 functions to recruit Parkin from the cytosol to the damaged mitochondrial membrane and to activate its E3 ligase activity. The PINK1 and Parkin mediated pathway represents the most well characterized inducer of mitophagy.
PINK1, which a nuclear encoded protein, is imported into healthy mitochondria by the translocase of outer membrane (TOM) and translocase of inner membrane (TIM) complexes which are described in detail in the Protein Targeting page. The N-terminal transmembrane domain of PINK1 is inserted into the inner mitochondrial membrane (IMM) and subsequently cleaved by the serine protease encoded by the PARL (presenilin associated rhomboid like) gene. The cleaved C-terminal fragment of PINK1 is transported back to the cytosol and degraded via the N-end rule proteasomal pathway (described in the section on Protein Acetylation in the Protein Modifications page). Within the cytosol the C-terminal fragment of PINK1 can also bind to Parkin, thereby, preventing its recruitment to the OMM.
This process of PINK1 synthesis, mitochondrial import, proteolysis, release from the mitochondria, and subsequent N-end rule proteasomal degradation keeps the steady state level of PARK1 in the mitochondria low which is a signal of mitochondrial health. Under conditions where PINK1 activity accumulates mitophagy is stimulated. Mitochondrial stabilization and accumulation of PINK1 can occur due to the over expression of misfolded proteins in the mitochondrial matrix. This most likely occurs due to impaired protein import at the TIM complex.
When PARK1 import into the IMM, or when the IMM-associated proteases that hydrolyze the N-terminus of PARK1 are impaired, the protein autophosphorylates and dimerizes in the OMM at the TOM complex. Under these conditions the kinase domain of PARK1 faces the cytosol. In the unstimulated state, Parkin remains in a closed conformation unable to attach ubiquitin to an internal Cys residue (C431) which is the typical intermediate step in the activity of ubiquitin conjugating E3 ligases.
When PINK1 dimerizes in the OMM, and is activated by autophosphorylation, it can phosphorylate Parkin (on S65), as well as ubiquitin, which is required for the full E3 ligase activity of Parkin. The phosphorylated ubiquitin serves as a Parkin receptor on the mitochondria. When activated, Parkin ubiquitylates numerous substrates such as the mitochondrial voltage-dependent anion channels (encoded by the VDAC1, VDAC2, and VDAC3 genes), the mitochondrial RHO family protein 1 identified as MIRO1 (also identified as RHOT1), and the mitochondrial fission protein dynamin related protein-1 (encoded by the DRP1 gene). In addition to MIRO1, mitochondria contain a second RHO family member identified as MIRO2 (also identified as RHOT2). The MIRO1 and MIRO2 proteins serve as adaptors that allow the mitochondria to interact with the kinesin and dynein/dynactin motor proteins of the cell. Another mitochondrial protein that links mitochondria to the motor proteins is homolog of the Drosophila melanogaster Milton gene which is encoded by the TRAK1 (trafficking kinesin protein 1) gene.
As described above, autophagosome cargo selection is determined, in part, by recognition of ubiquitylated proteins via the ubiquitin-binding domains (UBD) of autophagy receptors. The autophagy receptors, NDP52 and OPTN are critical activators of mitophagy. These two autophagy receptors recruit components of the autophagosome, such as the ULK1 complex and the Pi3P-binding proteins WIPI1 and WIPI2, to the mitochondrial membrane, thereby, stimulating mitophagy.
Control of mitophagy can be exerted by the removal of ubiquitin from Parkin substrates. As for the deubiquitylation process described in the Protein, Organelle, and Cell Turnover page, removal of ubiquitin from Parkin substrates is catalyzed by several deubiquitylating enzymes, DUBs. The DUBs encoded by the USP15, USP30, and USP35 (ubiquitin specific peptidase) genes are known to limit the extent of mitophagy by removal of ubiquitin from Parkin substrates. USP30 is likely to be the major DUB controlling mitophagy since it is tethered to the OMM with its catalytic domain facing the cytoplasm.
Mitochondrial Dysfunction and Diabetes
Well established data demonstrate that mitochondrial dysfunction, particularly as it relates to the processes of oxidative phosphorylation, is contributory to the development of encephalomyopathy, mitochondrial myopathy, and several age-related disorders that include neurodegenerative diseases, the metabolic syndrome, and diabetes. Indeed, with respect to diabetes, several mitochondrial diseases manifest with diabetic complications such as mitochondrial encephalomyopathy with lactic acidosis and stroke-like episodes (MELAS) and maternally inherited diabetes and deafness (MIDD).
Normal biogenesis of mitochondria is triggered in response to changes in the ATP/ADP ratio and to activation of AMPK which in turn results in increased expression of PPARγ co-activator 1α (PGC-1α) and nuclear respiratory factor-1 (NRF1). PGC-1α is a master transcriptional co-activator of numerous genes involved in mitochondrial biogenesis. NRF1 is a transcription factor that regulates the expression of mitochondrial transcription factor A (TFAM, for transcription factor A, mitochondrial; also designated mtTFA) which is a nuclear transcription factor essential for replication, maintenance, and transcription of the mitochondrial genome (mtDNA). NRF1 also controls the expression of nuclear genes required for mitochondrial respiration and heme biosynthesis. Evidence has shown that both PGC-1α and NRF1 expression levels are lower in diabetic patients as well as in non-diabetic subjects from families with type 2 diabetes. The expression of NRF1 is highest in skeletal muscle which is also the tissue that accounts for the largest percentage of glucose disposal in the body and, therefore, is the tissue that is most responsible for the hyperglycemia resulting from impaired insulin signaling.
Mitochondrial dysfunction results in increased production of ROS which activates stress responses leading to increased activity of MAPK and JNK. Both of these serine/threonine kinases phosphorylate IRS1 and IRS2 resulting in decreased signaling downstream of the insulin receptor. Inhibited IRS1 and IRS2 activity results in decreased activation of PI3K. PI3K activation is involved in the translocation of GLUT4 to the plasma membrane resulting in increased glucose uptake. Therefore, inhibition of PI3K results in reduced glucose uptake in skeletal muscle and adipose tissue. Mitochondrial dysfunction results in a reduction in the level of enzymes involved in β-oxidation leading to increases in intramyocellular lipid content. Indeed, skeletal muscle metabolism of lipids has been shown to be impaired in type 2 diabetics. An increased delivery of fatty acids to skeletal muscle, as well as diminished mitochondrial oxidation, results in increased intracellular content of fatty acid metabolites such as diacylglycerol (DAG), fatty acyl-CoAs, and ceramides. These metabolites of fatty acids are all known to induce the activity of protein kinase C isoforms (PKCβ and PKCδ) that phosphorylate IRS1 and IRS2 on serine residues resulting in impaired insulin signaling downstream of the insulin receptor.
Because skeletal muscle consumes the largest amount of serum glucose, mitochondrial dysfunction in this tissue will have the greatest impact on glucose disposal. However, adipose tissue also plays an important role in glucose homeostasis and mitochondrial dysfunction in this tissue has been shown to result in impaired glucose homeostasis resulting in diabetes. For example, when animals are treated with inhibitors of mitochondrial oxidation insulin-stimulated glucose uptake in adipose tissue is significantly impaired. Adipose tissue secretes a number of proteins classified as adipokines. Adiponectin is an adipokine that promotes insulin-sensitivity in insulin-responsive tissues, such as skeletal muscle. When plasma levels of adiponectin are measured in obese or type 2 diabetic subjects it is found to be significantly lower than in age and sex matched control subjects that are of normal weight or that do not have diabetes. In animal studies, the enhancement of adipocyte mitochondrial biogenesis results in increased adiponectin release from adipose tissue. Conversely, expression of adiponectin expression is decreased in adipocytes with mitochondrial dysfunction.
Given that impaired mitochondrial function is clearly associated with obesity and type 2 diabetes, it is not surprising that there is great interest in the use of pharmacology to augment mitochondrial function in the treatment of these disorders. Of significance is the fact that the thiazolidinedione (TZD) class of drugs used to treat the hyperglycemia of type 2 diabetes activate PPARγ which in turn increases the level of activity of PGC-1α. Although the TZDs were first marketed due to their ability to improve insulin sensitivity, they have since been shown to increase mitochondrial functions both in vitro and in vivo. Antioxidants have also been shown to enhance mitochondrial function by reducing the production of ROS.
Resveratrol (found in grape skins and red wine) is a potent antioxidant whose activity is, in part, due to its ability to activate the deacetylase SIRT1. Activated SIRT1 deacetylates PGC-1α resulting in increased transcriptional activity and thus, enhanced mitochondrial biogenesis.
Obesity-Induced Mitochondrial Dysfunction
Recent evidence has shown that a monomeric G-protein of the RAS family, specifically RalA, plays a significant role in the mitochondrial dysfunction that is prevalent in obesity. Expression of the gene (RALA; RAS like proto-oncogene A) encoding the RalA protein, as well as the activity of RalA, are both increased in white adipose tissue under conditions of high fat diet-induced obesity.
Under conditions of normal diet, when insulin activates its receptor in white adipocytes, RalA activity is enhanced. Following insulin activation, RalA participates in the mobilization of membrane vesicles containing GLUT4 to the plasma membrane. The activation of RalA by insulin results as a consequence of the phosphorylation and inhibition of the the GTPase activating complex (RalGAP) that normally serves to reduce the activity of RalA. In addition, insulin enhances the localization of the guanine nucleotide exchange factor (GEF) for RalA (Ral guanine nucleotide dissociation stimulator like 2; encoded by the RGL2 gene) to the RalA complex allowing for enhanced recycling of GTP for GDP, keeping RalA active. However, under conditions of high fat diets, the activity of RalA becomes contributory to obesity.
In mice, the targeted deletion of RalA in white adipocytes results in the attenuation of high fat diet-induced obesity. This effect is the result of increased mitochondrial fatty acid oxidation. Under conditions of a high fat diet there is an increase in mitochondrial fission in white adipocytes. RalA is involved in this process through its interaction with, and activation of, the phosphatase, PP2A.
With respect to mitochondrial biogenesis, the activity of PP2A leads to the removal of an inhibitory phosphorylation the protein Drpl1 which is encoded by the DNM1L gene. As detailed above, the Drpl1 protein functions in both mitochondrial fission and peroxisomal fission. PKA-mediated phosphorylation of Drp1 at Ser 637 interferes with its translocation to mitochondrial fission sites and promotes Drp1 detachment from the mitochondrion. This inhibitory phosphorylation is the target for PP2A. Therefore, the activation of PP2A by RalA results in an increase in mitochondrial fission and ultimately mitochondrial fragmentation.
Mitochondrial Disorders
Mitochondrial diseases are some of the most common inherited disorders in metabolism. This large class of disorders can be the result of mutations in genes of the nuclear genome as well as genes of the mitochondrial genome. A hallmark of the mitochondrial diseases is that, depending on the particular disease, symptom manifestation can occur at any age and can cause symptoms in any organ system. Most mitochondrial proteins are nuclear-encoded and so it is not surprising that the vast majority of mitochondrial diseases are the result of mutations in genes from the nuclear genome. Mutations in 228 nuclear genes and all 13 mitochondrial mRNA genes are associated with rare monogenic syndromes in which mitochondrial dysfunction is central to the resulting pathology of the disease. The most common causes of mitochondrial disorders in humans are mutations in the genes that encoded the proteins of complex I of the oxidative phosphorylation pathway.
Although many mitochondrial disorders result from monogenic mutation, many reflect changes in the normal processes of mitochondrial biogenesis triggered by changes in the cellular milieu. As discussed in the previous section, disordered mitochondrial dynamics contributes to the pathogenesis of type 2 diabetes. It is also important to note that numerous conditions, that are not classically considered to involve mitochondria, such as cancers, cardiovascular diseases, and neurodegenerative diseases are impacted by alterations in overall mitochondrial dynamics.
Many of the disorders that result from defects in mitochondrial biogenesis and/or function have multiple genetic causes such that manifestation can result from maternal inheritance, X-linked inheritance, autosomal recessive inheritance, or autosomal dominant inheritance, or they can be the result of sporadic somatic mutation. Several of the more commonly occurring mitochondrial disorders result from not one single gene defect but can result from mutations or deletions in multiple different genes, each of which result in similar pathologies. Leigh syndrome represents this latter class of mitochondrial disorder given that the general pathology of Leigh syndrome has been shown to result from mutations in at least 16 different genes that includes both mtDNA genes and nuclear genome genes. Several defined mitochondrial disorders have been shown to result from mutations in the same gene leading to potential diagnostic complications. For example mitochondrial encephalopathy, lactic acidosis, stroke-like episodes (MELAS) and myoclonic epilepsy with ragged-red fibers (MERRF) are both associated with mutations in the mtDNA encoded genes MT-TK, MT-TL1, MT-TH, MT-TS1, and MT-ND5.
Mitochondrial diseases can be caused by mutations in genes required for mtDNA replication as described in the section above as well as genes required for mtDNA maintenance. Mitochondrial diseases can also result from mutations in genes that encode proteins of the complexes of the oxidative phosphorylation machinery or genes that encode proteins required for proper assembly of these complexes. Mutations in several of the genes required to synthesize coenzyme Q (ubiquinone) also result in mitochondrial disorders whose symptoms overlap with those of the common disorders such as Leigh syndrome. Mutations in genes encoding proteins required for mitochondrial protein synthesis also cause pathologies resulting from defects in mitochondrial function.
Table of Common Mitochondrial Disorders
| Disorder | Gene(s) | Pathology / Comments |
| mitochondrial encephalopathy, lactic acidosis, stroke-like episodes: MELAS | MT-TL1, MT-TQ, MT-TH, MT-TK, MT-TC, MT-TS1, MT-ND1, MT-ND5, MT-ND6, MT-TS2 | all mutations causing MELAS are in mtDNA encoded genes; inheritance is strictly maternal; most common cause (80% of cases) of MELAS is inheritance of an A → G transition mutation in the MT-TL1 gene which is a mtDNA gene that encodes a Leu (L) tRNA that recognizes the codons UUA and UUG; symptoms usually appear in early childhood following a period of normal growth; earliest symptoms include muscle weakness and pain, recurrent headaches, loss of appetite, vomiting, and seizures; common pathology is lactic acidosis resulting in vomiting, abdominal pain, extreme fatigue, muscle weakness, and difficulty breathing; most individuals with MELAS experience stroke-like episodes beginning before age 40. |
| myoclonic epilepsy with ragged-red fibers: MERRF | MT-TK, MT-TL1, MT-TH, MT-TS1, MT-TS2, MT-TF, MT-ND5 | all mutations causing MERRF are in mtDNA encoded genes; inheritance is strictly maternal; most common cause (80% of cases) of MERRF is inheritance of mutations in the MT-TK gene which is a mtDNA gene that encodes a Lys (K) tRNA; symptoms of MERRF begin to appear in childhood or early adolescence; MERRF is characterized by myoclonus (muscle twitching), muscle weakness (myopathy), and progressive stiffness (spasticity); additional common symptoms of MERRF are seizures, ataxia, peripheral neuropathy, optic neuropathy, hearing loss, and dementia; many MERRF patients are of short stature |
| Leber hereditary optic neuropathy: LHON | MT-ND1, MT-ND2, MT-ND4, MT-ND4L, MT-ND5, MT-ND6, MT-CYB, MT-CO3, MT-ATP6, | symptoms of LHON usually present in midlife (mean age of onset 27–35 years); symptoms are predominantly restricted to the visual system; symptoms include acute or subacute central vision loss that initially manifests with blurring and clouding of vision ultimately leading to blindness; mutations in the mtDNA that cause LHON can act autonomously or in association with each other in the manifestation of the pathology of LHON; males manifest symptoms more often than females; often individuals who harbor LHON mutations never experience symptoms with more than 50% of males and 85% of females remaining symptom free |
| neuropathy, ataxia, and retinitis pigmentosa: NARP | MT-ATP6 | MT-ATP6 encodes the A subunit of the F0 protein of the mitochondrial ATP synthase complex (often called complex V) of oxidative phosphorylation; symptoms usually begin to appear in childhood; major symptoms of NARP include numbness, tingling, or pain in the arms and legs, ataxia, and vision loss |
| Alpers-Huttenlocher syndrome: AS | POLG | see the section covering mtDNA replication |
| Leigh syndrome: LS | BCS1L, NDUFAF2, NDUFA10, NDUFS4, SDHA, NDUFA2, NDUFAF6, NDUFS3, NDUFS8, FOXRED1, NDUFA9, NDUFA12, NDUFS7, COX10, COX15, SURF1; MT-ATP6 | mutations in more than 80 genes (mostly nuclear genome) have been associated with the common pathologies of Leigh syndrome; the most common mtDNA mutations are in the MT-ATP6 gene; MT-ATP6 encodes the A subunit of the F0 protein of the mitochondrial ATP synthase complex (often called complex V) of oxidative phosphorylation; Leigh syndrome (subacute necrotizing encephalomyelopathy) is characterized by progressive psychomotor regression; death usually occurs by 2-3 years of age and is most often the result of respiratory failure; early onset symptoms include vomiting, diarrhea, dysphagia (difficulty swallowing) all of which result in growth impairment and failure to thrive; muscle problems include hypotonia and dystonia; neurological problems include ataxia and peripheral neuropathy |
| progressive external ophthalmoplegia: PEO | POLG, TWNK, RRM2B, SLC25A4, MT-TL1; large mtDNA deletions | mutations in these genes lead to large deletions in the mtDNA in muscle cells; also large deletions in mtDNA are associated with PEO; symptoms usually begin to appear between 18 and 40 years of age; symptoms first manifest with drooping eyelids (ptosis) progressing to weakness and paralysis (myopathy) of the muscles that move the eyes; see the section covering mtDNA replication |
| mitochondrial neurogastrointestinal encephalopathy: MNGIE | TYMP | the TYMP gene encodes thymidine phosphorylase; functions to promote angiogenesis (was originally identified as platelet-derived endothelial cell growth factor: ECGF1); mutations in TYMP allow accumulation of thymidine which negatively affects mtDNA maintenance; most individuals begin to experience symptoms by age 20 but they may occur earlier or later in some individuals; gastrointestinal pathology is the most common symptom of MNGIE which is characterized by reduced gastric motility leading to dysphagia (trouble swallowing), nausea and vomiting following food intake |
| mitochondrial DNA depletion syndrome: MDS | POLG, TWNK, TK2, DGUOK, SUCLA2, SUCLG1, RRM2B, MPV17, TYMP | represents a constellation of disorders that are associated with depletion of mitochondrial DNA due to mutations in nuclear genome genes; due to mutations in different genes different patients can exhibit a wide range of pathologies and a wide range of age of onset; mutations in POLG, TWNK, DGUOK, and MPV17 result in pathologies that affect the brain and the liver; mutations in SUCLA2, SUCLG1, and RRM2B cause pathologies in the brain and skeletal muscles; mutations in TK2 are associated primarily with skeletal muscle pathology; the disorder originally called Alpers syndrome is now included in the categorization of MDS |
| Kearns-Sayre syndrome: KSS | mtDNA deletions | the mtDNA deletions observed in KSS range from 1.3–8 kbp and can vary in position; the most common deletion (35% of patients) is a 4997 bp deletion of nucleotide 8469–13,147 (encompassing 12 genes); symptoms of KSS usually appear before age 20 and most commonly affect the eyes; characteristic symptoms include chronic progressive external ophthalmoplegia (PEO) which represents weakness or paralysis of the eye muscles impairing eye movement and causing drooping eyelids (ptosis); KSS may also manifest with muscle weakness in the limbs, deafness, renal pathology, and dementia |
| Pearson syndrome: PS | mtDNA deletions | infantile onset, exocrine pancreatic insufficiency, sideroblastic anemia and bone marrow failure, tubular and liver dysfunction, encephalopathy; children who survive will develop classic Kearns-Sayre syndrome |