
Last Updated: October 30, 2025
Introduction to McArdle Disease
Glycogen storage disease type 5 (GSD5) is an autosomal recessive disorder more commonly known as McArdle disease. This disease was originally described by Brian McArdle in 1951, hence the association of his name with the disease. The disease was seen in a 30-year-old patient who was suffering from muscle weakness, pain, and stiffness following bursts of activity such as during sprinting. It was observed that blood lactate fell in this patient during exercise instead of the normal rise that would normally be seen. This indicated the patient had a defect in the ability to convert muscle glycogen to glucose and ultimately lactate.
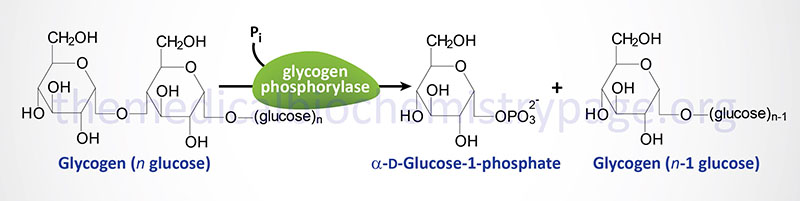
The identification that the deficiency causing these symptoms was the result of a muscle phosphorylase defect was not made until 1959. Glycogen phosphorylase (most commonly just called phosphorylase) exists in multiple tissue-specific isoforms. The muscle, brain, and liver forms are encoded by separate genes. The muscle form is the only isozyme found expressed in mature muscle.
Molecular Biology of McArdle Disease
Humans express three genes encoding proteins with glycogen phosphorylase activity. One gene (PYGL) expresses the hepatic form of the enzyme, a second (PYGM) expresses the muscle form (referred to as myophosphorylase), and the third (PYGB) expresses the brain form.
The gene for muscle phosphorylase (symbol: PYGM) is located on chromosome 11q13.1 and is composed of 20 exons that generate two alternative splice variant mRNAs. These two mRNAs encode PGYM isoform 1 (a protein of 842 amino acids) and PGYM isoform 2 (a protein of 754 amino acids). The muscle isoforms represent about 50% of total phosphorylase in cardiac muscle, 30% of the brain phosphorylase, and is completely absent from the liver.
As of 2019 a total of 147 pathogenic mutations have been identified as causing McArdle disease. The most commonly encountered mutation in the Caucasian population (40%-50% of patients) is a nonsense mutation that changes a C to a T in exon 1 converting an arginine codon at amino acid position 50 to a stop codon (R50X). In a large cohort of Spanish individuals with McArdle disease, 78% of the cases involved the R50X mutation or one of two other mutations where Gly at amino acid 205 was changed to a Ser (G205S) or where Trp at amino acid 797 was changed to an Arg (W797R).
Clinical Features of McArdle Disease
The clinical presentation of McArdle disease is usually seen in young adulthood and is characterized by exercise intolerance and muscle cramps following bursts of muscle activity such as during sprint training and bicycle riding, or other forms of brief strenuous exercise. Attacks of rhabdomyolysis with myoglobinuria frequently accompany the muscle symptoms of McArdle disease. About half of McArdle disease patients will exhibit burgundy colored urine after exercise due to the myoglobinuria. In addition, these patients will present with significant increases in serum creatine kinase.
Although skeletal muscle pathology represents the predominant presentation in McArdle disease patients, other organ system involvement has been identified in numerous patients. These other organ systems are likely to be affected by mutations in the PYGM gene since expression of the gene is known to occur in the retina, specialized cells of the kidney, and in glial cells in the brain. Indeed, the expression of mutant phosphorylase in the retinal pigment epithelium RPE) is likely to account for RPE dystrophy seen in several McArdle disease patients.
Some of the clinical presentation in McArdle disease may be due to disturbances in the normal role of glycogen, not only as a source of glucose for energy, but also as a source glucose in the process of post-translational protein modification. The glucose-1-phosphate released from glycogen, through the action of phosphorylase, serves as the precursor for the role of glucose-6-phosphate in the hexosamine biosynthesis pathway (HBP). The HBP is the pathway for the synthesis of UDP-GlcNAc which can be used for the O-GlcNAcylation of numerous proteins, a critical form of post-translational modification. Thus, defective production of phosphorylase in McArdle disease patients could very well have profound impacts on O-GlcNAcylation pathways not only in muscle but other tissues as well.
Diagnosis of McArdle Disease
Diagnosis of muscle glycogenoses such as McArdle disease was originally made by the observation of a lack of increased blood lactate upon ischemic exercise testing. Using this test it is also that patients experience a large increase in blood ammonia levels. However, the ischemic test is accompanied by muscle cramping and pain and is, therefore, a fairly invasive test. A non-ischemic test that involves an intermittent handgrip contraction has been shown to provide the same blood results as the ischemic test but without the associated cramping and pain. Given the advent of rapid and specific DNA testing, this is the preferred means to assess a patient suspected of having McArdle disease by identifying mutations in the PYGM gene.
In order to distinguish McArdle disease from other muscle defects along the pathway from glycogen to glucose to lactate, an enzymatic evaluation of muscle phosphorylase must be done. Also, molecular analysis for known mutations in the muscle phosphorylase gene can be accomplished using DNA extracted from leukocytes. Muscle biopsy tissue can be analyzed using a histopathological stain for carbohydrate such as Periodic acid-Schiff (PAS). The Schiff reagent is a bleached basic fuschin that reacts with aldehyde groups such as in carbohydrates like glycogen. PAS stains carbohydrates and carbohydrate rich macromolecules a magenta color.
Treatment of McArdle Disease
There is no specific treatment for McArdle disease. Avoidance of intense physical activity is the best course of action. Some patients respond well to the ingestion of sucrose prior to exercise.