
Last updated: October 28, 2025
Introduction to Maple Syrup Urine Disease: MSUD
Maple syrup urine disease (MSUD), also called branched-chain aminoaciduria, is so called because the urine of affected individuals smells like maple syrup or burnt sugar. The cause of the odor in the urine of MSUD patients is a catabolic metabolite of leucine identified as sotolone [3-hydroxy-4,5-dimethyl-2(5H)-furanone]. This compound is also found in the seeds of the herb, fenugreek (Trigonella foenum-graecum) which are used as a spice.
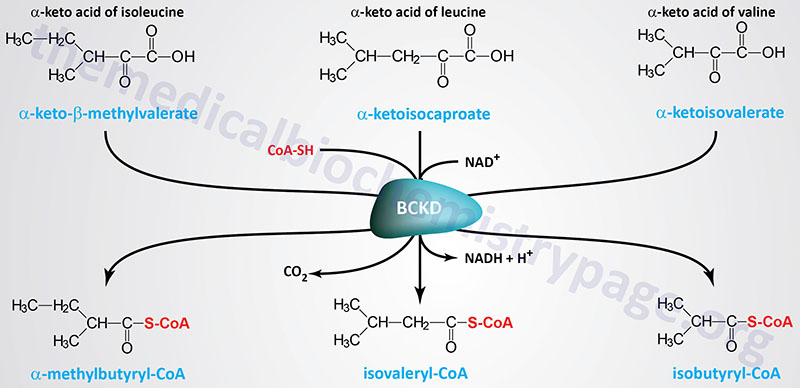
MSUD is an autosomal recessive disorder that results from a deficiency in the enzyme, branched-chain α-keto acid dehydrogenase (BCKD), that is involved in the catabolism of the branched-chain amino acids (BCAA), leucine, isoleucine, and valine. A deficiency in BCKD leads to an accumulation of those three amino acids and their corresponding branched-chain α-keto acids (BCKA). MSUD is associated with a worldwide frequency of approximately 1 in 185,000 persons. In certain inbred populations such as the Old Order Mennonites the incidence of MSUD is extremely high on the order of 1 in every 175 births.
The BCAA represent approximately 35%–40% of the essential amino acids in skeletal muscle. Following the ingestion of protein the BCAA represent about 60% of the increase in amino acids in the blood. In skeletal muscle the BCAA represent a significant source of carbon atom as an alternate to lipid and carbohydrate as a source of energy. The BCAA are also actively metabolized for energy in the heart, kidneys, brain and adipose tissue. In the liver, oxidation of the BCAA provides significant carbon for the production of the ketone bodies used by the brain during periods of fasting.
Branched-Chain Keto Acid Dehydrogenase Complex
Molecular Biology of Maple Syrup Urine Disease
The BCKD complex is a multimeric mitochondrial enzyme composed of three catalytic subunits identified as E1, E2, and E3. The E1 portion of the complex is a heterotetrameric thiamine pyrophosphate (TPP)-dependent decarboxylase with a subunit structure of α2β2. The E2 portion is a dihydrolipoamide branched-chain transacylase composed of 24 lipoic acid-containing polypeptides. The E3 portion is a homodimeric flavoprotein identified as dihydrolipoamide dehydrogenase, DLD.
The E1α subunit is encoded by the BCKDHA (branched chain keto acid dehydrogenase E1, alpha polypeptide) gene. The BCKDHA gene is located on chromosome 19q13.2, spans 55 kb and contains 9 exons that generate two alternatively spliced mRNAs that encode alpha subunit isoform 1 (445 amino acids) and alpha subunit isoform 2 (444 amino acids).
The E1β subunit is encoded by the BCKDHB (branched chain keto acid dehydrogenase E1 subunit beta) gene. The BCKDHB gene is located on chromosome 6q14.1 and is composed of 20 exons that generate three alternatively spliced mRNAs that encode two different protein isoforms (392 amino acid isoform 1 and 322 amino acid isoform 2).
The E2 subunit is encoded by the DBT gene. The DBT gene is located on chromosome 1p21.2, spans 68 kb and contains 15 exons that generate two alternatively spliced mRNAs that collectively encode proteins of 482 amino acids (isoform 1) and 301 amino acids (isoform 2). The biological function, if any, of the DBT isoform 2 protein has yet to be estab,ished.
The E3 subunit is encoded by the DLD gene. The DLD gene is located on chromosome 7q31.1, spans 20 kb and contains 14 exons that generate four alternatively spliced mRNAs, each of which encode a distinct protein isoform. The DLD gene encodes the same dihydrolipoamide dehydrogenase subunits found in the PDH and the 2-oxoglutarate (α-ketoglutarate) dehydrogenase complexes.
The activity of BCKD is regulated by two additional subunits, a kinase and a phosphatase that reversibly phosphorylate/dephosphorylate, respectively, the complex.
The BCKD kinase is encoded by the BCKDK gene which is located on chromosome 16p11.2 and is composed of 13 exons that generate three alternatively spliced mRNAs encoding three distinct isoforms of the enzyme. The kinase phosphorylates the E1α subunit rendering the enzyme inactive. Gain-of-function mutations in the BCKDK gene are associated with a mild form of MSUD.
The phosphatase that removes the inhibitory phosphate is encoded by the PPM1K (protein phosphatase, Mg2+/Mn2+ dependent 1K) gene. The PPM1K gene is located on chromosome 4q22.1 and is composed of 11 exons that encode a protein of 372 amino acids.
The genetic heterogeneity in MSUD patients can be explained by the complexity in the structure of BCKD. There are four molecular phenotypes of MSUD based upon the affected locus:
- Type IA is due to mutations in the E1α gene.
- Type IB is due to mutations in the E1β gene.
- Type II is due to mutations in the E2 gene.
- Type III is due to mutations in the E3 gene.
To date a total of 63 mutations have been identified in the four genes of the catalytic portion of BCKD.
Clinical Classifications of MSUD
Based upon the overall clinical presentation as well as a particular patients response to thiamine administration, MSUD patients can be divided into five phenotypic classifications.
Classic MSUD
Classic MSUD is defined by neonatal onset of encephalopathy and is the most severe form of the disorder. The levels of the BCAA, especially leucine, and their α-keto acids, are dramatically elevated in the blood, urine, and cerebrospinal fluid of afflicted infants.
The primary cause of the encephalopathy and neurodegeneration in MSUD infants is the high levels of the α-keto acid of leucine, α-ketoisocaproic acid, in the brain. This compound triggers apoptosis in neurons and glial cells by interfering with mitochondrial energy metabolism. The presence of alloisoleucine in the fluids is highly diagnostic of MSUD.
The level of BCKD activity in classic MSUD patients is less than 2% of normal. Affected infants appear normal at birth but symptoms develop rapidly, appearing by 4 to 7 days after birth. The first distinctive signs are lethargy and little interest in feeding. As the disease progresses infants will exhibit weight loss and progressive neurological deterioration. Neurological signs will alternate from hypo- to hypertonia and extension of the arms resembling decerebrate posturing. At this time the characteristic burnt sugar or maple syrup odor to the urine is apparent. If left untreated infants will develop seizures, lapse into a coma and die. The prognosis for untreated infants is poor with death occurring within several months of birth due to complications of metabolic crisis and neurological deterioration.
Intermediate MSUD
Intermediate MSUD is distinguished from classic in that patients do not experience the severity of classic MSUD in the neonatal period. Infants will have persistent elevation in BCAAs in body fluids as well as neurological impairment. The level of BCKD in intermediate MSUD individuals ranges from 3% to 30% of normal. Many intermediate MSUD patients do not experience the acute metabolic decompensation present in classic MSUD.
Intermittent MSUD
In patients with the intermittent form of MSUD the activity of BCKD ranges from 5% to 50% of normal. These individuals will show normal early development with normal intelligence. During periods when patients are asymptomatic their fluid levels of BCAAs will be normal. These patients are however, at risk for acute metabolic decompensation during periods of stress. Initial symptoms of the intermittent form of MSUD usually appear between 5 months and 2 years of age in association with an infection.
Thiamine-responsive MSUD
There is a similar course of progress in the symptoms of thiamine-responsive MSUD patients to that seen in intermediate MSUD patients. Plasma BCAA levels are around 5 times normal and alloisoleucine is characteristically detectable in these patients. Administration of thiamine and consumption of a low protein diet results in a reduction of BCAA levels to normal. Withdrawal of thiamine treatment results in a rapid rebound in the elevation of plasma BCAA concentration. These responses to thiamine are the reason for this classification of MSUD. There is a wide range of heterogeneity in thiamine-responsive MSUD patients and thiamine treatment alone is insufficient to result in lower levels of BCAAs.
Dihydrolipoyl dehydrogenase (E3)-deficient MSUD
As the name of this classification implies, this form of MSUD is due to a deficiency in the E3 component of the BCKD complex. This form of MSUD is very rare with only 20 reported cases. The symptoms of the E3-deficient form are similar to those of intermediate MSUD, but there is an accompanying severe lactic acidosis. Infants with the E3-deficient form of MSUD are relatively normal for the first few months of life. Persistent lactic acidosis will be seen to develop between 2 and 6 months of age.
Treatment of Maple Syrup Urine Disease
The treatment of classic, intermediate, intermittent, and thiamine-responsive MSUD involves lifelong therapy to maintain an diet with sufficient calories and protein for growth but does not contribute to enhanced branched-chain amino acid (BCAA) catabolism. In addition, there must be immediate medical intervention for metabolic decompensation. Various techniques have been used to reduce plasma leucine levels including hemodialysis or hemofiltration, the latter being a process where the blood is removed, filtered, and then returned to the body.
Management of an MSUD patient’s diet remains a constant balancing act between giving enough protein and BCAA to provide for normal growth and development while simultaneously ensuring that the patient’s metabolism remains in a therapeutic. Because α-ketoisocaproic acid (the α-ketoacid of leucine) is particularly toxic to the brain it is particularly important to limit the amount of leucine in the diet. MSUD children must be regularly monitored to ensure that their diet is adequate and that amino acid levels remain within acceptable normal ranges.
Because there is a form of MSUD referred to as thiamine-responsive MSUD, it is often recommended that all MSUD patients be given a trial of thiamine therapy to determine whether or not they have the thiamine-responsive form. However, no individual with MSUD can be successfully treated with thiamine alone.
Liver transplantation has been shown to be successful in curing classic MSUD patients of their disease. Following the transplant these patients remain symptom-free and are able to consume a normal diet. The limitation to transplantation is the availability of a donor liver and the high cost of this procedure.