
Last Updated: October 29, 2025
Identification of Lp(a)
Lipoprotein(a) [Lp(a)] was originally described as a new serum lipoprotein particle by Kare Berg in 1963. Lp(a) is composed of a common LDL nucleus linked to a molecule of apolipoprotein(a) [apo(a); encoded by the LPA gene] by disulfide bonds between a cysteine residue in a Kringle-IV (KIV) type 9 domain in apo(a) and a cysteine residue in apolipoprotein B-100 (apoB-100). When attached to apoB-100 the apo(a) protein surrounds the LDL molecule.
Synthesis of Lp(a) occurs in the liver. The half-life of Lp(a) in the circulation is approximately 3–4 days. Although Lp(a) was described over 40 years ago its precise physiological function remains unclear. However, numerous epidemiological studies have demonstrated that elevated plasma levels of Lp(a) are a significant risk factor for the development of atherosclerotic disease.
The Kringle domains of apo(a) exhibit 75%-85% similarity to the KIV domains of plasminogen. The Kringle domain is a highly glycosylated domain found in numerous proteins and is so-called because the three dimensional structure resembles a looped Danish pastry. Each Kringle domain is composed of approximately 80 amino acid residues and the structure is stabilized by three internal disulfide bonds. There are 10 distinct sub-classes of KIV domains in apo(a) designated KIV1 through KIV10. Apo(a) also contains a Kringle V (KV) domain that resembles the catalytic domain of plasminogen.
Molecular Biology of apo(a)
The apo(a) protein is encoded by the LPA gene. The LPA gene is located on chromosome 6q25.3-q26 and is composed of 39 exons. The apo(a) KIV1 and KIV3 through KIV10 domains are present as single-copy domains. The KIV2 domain is present in a variable number of repeated copies (from 2–43) and constitutes the molecular basis for the highly variable size of Lp(a) in different individuals. The LPA gene is a member of the plasminogen superfamily and given the similarities between apo(a) and plasminogen it has been hypothesized that apo(a) influences the processes of hemostasis.
Apo(a) proteins exhibit a variability in size due to a polymorphism caused by a variable number of the KIV repeats. To date at least seven different isoforms of Lp(a) have been characterized based upon electrophoretic mobilities. These different isoforms are designated F, B, and S1 through S5. The different isoforms are grouped into low molecular weight (LMW) and high molecular weight (HMW) isoforms determined by the number of KIV repeats in the apo(a) protein found in the Lp(a). The level of Lp(a) found in healthy individuals depends upon whether their plasma contains the LMW or HMW isoforms. Individuals with the LMW isoforms have high plasma Lp(a) concentration while those with the HMW isoforms have low concentrations.
Pathology of Lp(a)
When in the circulation Lp(a) particles can be affected by oxidative modification similar to that of the other plasma lipoprotein particles. Lp(a) and oxidized Lp(a) [oxLp(a)] particles interact with macrophages via scavenger receptor uptake leading to cholesterol accumulation and foam cell formation. Indeed, oxLp(a) are phagocytosed more rapidly than other lipoprotein particles and therefore accumulate in the subendothelial space at high levels. This process can lead to progression of atherogenesis, thus accounting for the direct correlation between the plasma level of Lp(a) and coronary artery disease. In addition to oxidation of Lp(a) leading to increased foam cell production, glycation of the particle also may contribute to atherogenesis. In fact, there is a strong correlation in the level of glycated Lp(a) and the severity of hyperglycemia observed in poorly controlled type 2 diabetes.
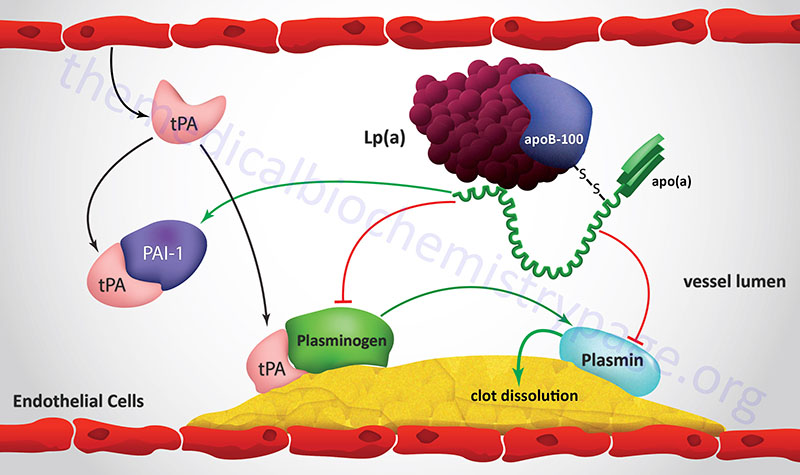
Although the precise physiology of Lp(a) is poorly understood, as indicated above, there is a strong correlation between plasma concentration of Lp(a) and atherogenic events that lead to coronary artery disease. For a discussion of the processes of blood coagulation and the role of plasminogen visit the Blood Coagulation page. Because of the high degree of similarity between apo(a) and plasminogen it is suggested that Lp(a) may contribute to the thrombotic aspects of ischemic heart disease. Lp(a) has been shown to competitively inhibit the binding of plasminogen to its receptor on endothelial cells as well as to its binding sites on fibrinogen and fibrin. This interference of plasminogen binding leads to reduced surface-dependent activation of plasminogen to plasmin.
The normal function of plasmin is to degrade the fibrin clot that forms as a result of vessel injury. Therefore, high plasma concentrations of Lp(a) may represent a source of antifibrinolytic activity. Of significance to the potential for atherogenesis, the antifibrinolytic potential of Lp(a) particles is related to their size. The LMW isoforms of Lp(a) have been shown to have a higher fibrin-binding capacity than the HMW isoforms.
Lp(a) also interferes with other aspects of the normal processes of coagulation in addition to its effects on plasminogen function. Lp(a) stimulates the production of plasminogen activator inhibitor-1 (PAI-1) leading to a reduced ability of t-PA to activate the process of clot dissolution. Increased production of PAI-1 also leads to enhanced proinflammatory events via activation of monocyte adhesion to the vessel wall. Lp(a) has also been shown to modulate platelet activation interfering with the interaction of platelets with exposed collagen fibers in the injured vessel wall.
In addition to the role of Lp(a) in inhibiting plasminogen binding, Lp(a) has been shown to inhibit the release of tissue plasminogen activator (t-PA) from endothelial cells. With reduced release of the enzyme (t-PA) that converts plasminogen to plasmin and interference with plasminogen binding to fibrin clots Lp(a) can exert a significant negative effect on the ability to dissolve blood clots.
In addition to the interactions with plasminogen, leading to enhanced atherogenesis, Lp(a) has been shown to stimulate smooth muscle cell (SMC) growth. This effect of Lp(a) is exerted via an inactivation of transforming growth factor-β (TGF-β). Activated TGF-β inhibits the proliferation and migration of SMC, thus the inhibition of this regulatory effect of TGF-β leads to accelerated blood vessel stenosis with concomitant enhancement of the atherogenic process. Oxidized Lp(a) [oxLp(a)] has also been shown to inhibit nitric oxide-dependent vasodilation which will tend to exacerbate the atherogenic process in hypertensive patients.
Lp(a) also interferes with other aspects of the normal processes of coagulation in addition to its effects on plasminogen function. Lp(a) stimulates the production of plasminogen activator inhibitor-1 (PAI-1) leading to a reduced ability of t-PA to activate the process of clot dissolution. Increased production of PAI-1 also leads to enhanced proinflammatory events via activation of monocyte adhesion to the vessel wall. Lp(a) has also been shown to modulate platelet activation by interfering with the interaction of platelets with exposed collagen fibers in the injured vessel wall. All of the observed effects of Lp(a) on hemostasis result in the persistence of clots which is a significant contributor to atherogenesis and increases the potential for abnormal thrombotic episodes.