
Last Updated: February 13, 2026
Introduction to the Urea Cycle Disorders (UCD)
A complete lack of any one of the enzymes of the urea cycle is associated with an significant increase in the likelihood for death shortly after birth. Nonetheless, deficiencies in each of the enzymes of the urea cycle have been identified. In addition, deficiencies in N-acetylglutamate synthase (NAGS), the enzyme necessary for the synthesis of N-acetylglutamate (NAG) which is required for allosteric activation of carbamoylphosphate synthetase 1 (CPS1), have been identified. Collectively, these enzyme deficiencies lead to the urea cycle disorders or UCDs.
The most dramatic presentation of UCD symptoms occurs in neonates between 24 and 48 hours after birth (sometimes symptoms are delayed for several days). Afflicted infants exhibit progressively deteriorating symptoms due to the elevated ammonium levels. Deficiencies in arginase do not lead to symptomatic hyperammonemia as severe or as commonly as in the other UCDs. Deficiencies in carbamoylphosphate synthetase 1 (CPS1), ornithine transcarbamylase (OTC), argininosuccinate synthetase (AS), and argininosuccinate lyase (AL) comprise the common neonatal UCDs.
The Reactions of the Urea Cycle
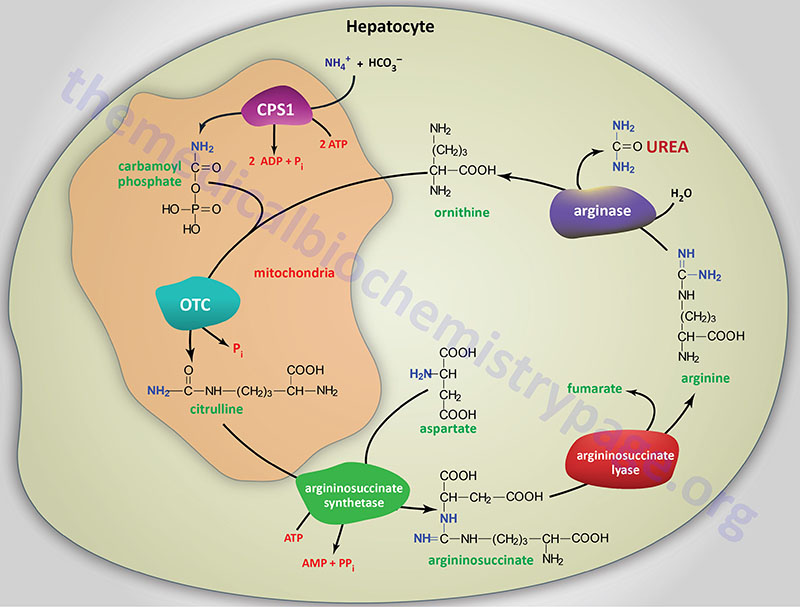
Overview of Urea Cycle Disorders
Severe deficiency or total absence of activity of any of the first four enzymes (CPS1, OTC, AS, AL) in the urea cycle or the cofactor producer (NAGS) results in the accumulation of ammonia and other precursor metabolites during the first few days of life.
Infants with a severe urea cycle disorder are normal at birth but rapidly develop cerebral edema and the related signs of lethargy, anorexia, hyper- or hypoventilation, hypothermia, seizures, neurologic posturing, and coma.
In milder (or partial) deficiencies of these enzymes and in arginase (ARG) deficiency, ammonia accumulation may be triggered by illness or stress at almost any time of life. In these disorders the elevations of plasma ammonia concentration and symptoms are often subtle and the first recognized clinical episode may not occur for months or decades.
Carbamoyl Phosphate Synthetase 1 Deficiency
Carbamoyl phosphate synthetase 1 deficiency (CPSD) is also referred to as Type 1 Hyperammonemia. CPSD is an autosomal recessive disorder representing one of the most severe forms of UCD. The incidence of CPSD in the US is on the order of 1 in every 975,000 live births. Typical symptoms include ammonia intoxication with very high ammonia levels in the blood, and intellectual impairment.
Ornithine Transcarbamylase Deficiency
Ornithine transcarbamylase deficiency (OTCD) is also referred to as Type 2 Hyperammonemia. OTCD is an X-linked disorder and is the most common UCD. The incidence of OTCD in the US is on the order of 1 in 63,000 live births. Symptoms of OTCD include elevated ammonia and amino acids (particularly glutamine) in the blood and increased orotic acid in the urine. Glutamine is also high in the cerebrospinal fluid (CSF) and the urine.
Argininiosuccinate Synthetase Deficiency
Argininosuccinate synthetase deficiency (ASD) is commonly referred to as citrullinemia type 1 (CTLN1). Symptoms of CTLN1 include elevation of ammonia and citrulline in the blood, urine, and CSF. Another form of citrullinemia termed citrullinemia type 2 (CTLN2) results from mutations in the gene encoding the transporter, citrin.
Argininosuccinate Lyase Deficiency
Argininosuccinate lyase (argininosuccinase) deficiency (ALD) is known as argininosuccinic aciduria. This disorder is characterized by high levels of ammonia and argininosuccinate in the blood, CSF and urine. ALD can manifest early as a neonatal UCD or late (around 2 years of age) and is usually a fatal disorder.
Arginase Deficiency
Deficiency in arginase (AD) is also known as hyperargininemia. This disorder is quite rare and manifests with abnormal physical and mental development, as well as the accumulation of ammonia and arginine in the blood and CSF. The amino acids arginine, lysine and ornithine are also elevated in the urine of AD patients.
N-Acetylglutamate Synthase Deficiency
N-acetylglutamate synthase (NAGS) deficiency (NAGSD) represents the rarest form of UCD. NAGS is not directly involved in the reactions of the urea cycle, however, it is critical to the production of the activator of CPS1, N-acetylglutamate. Because of this function of NAGS, a deficiency of this enzyme results in a functional deficiency in CPS1. Laboratory studies and clinical findings are unable to distinguish NAGS from CPS1 deficiency, thus molecular testing is required. Deficiency in NAGS is an extremely rare autosomal recessive disorder. Treatment of NAGS deficiency utilizes an analog of N-acetylglutamate, N-carbamoylglutamate, that effectively activates CPS1.
Differential Diagnosis of the Neonatal Urea Cycle Disorders
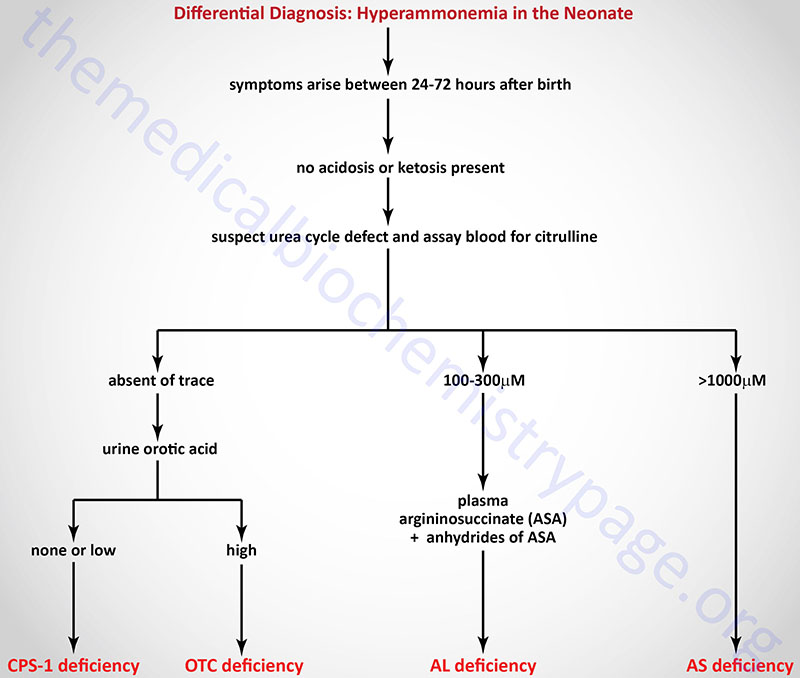
Treatment of Urea Cycle Disorders
In general, the treatment of UCDs has as common elements the reduction of protein in the diet, removal of excess ammonia and replacement of intermediates missing from the urea cycle.
Administration of lactulose (a nonabsorbable disaccharide) results in reduced ammonia production in the gut and, therefore, less ammonium ion is delivered to the portal circulation from the intestines. Bacteria metabolize lactulose to acidic byproducts which then promotes excretion of ammonia in the feces as ammonium ion (NH4+) and interferes with the production of ammonia through reduced catabolism of other nitrogenous compounds. Antibiotics can be administered to kill intestinal ammonia producing bacteria.
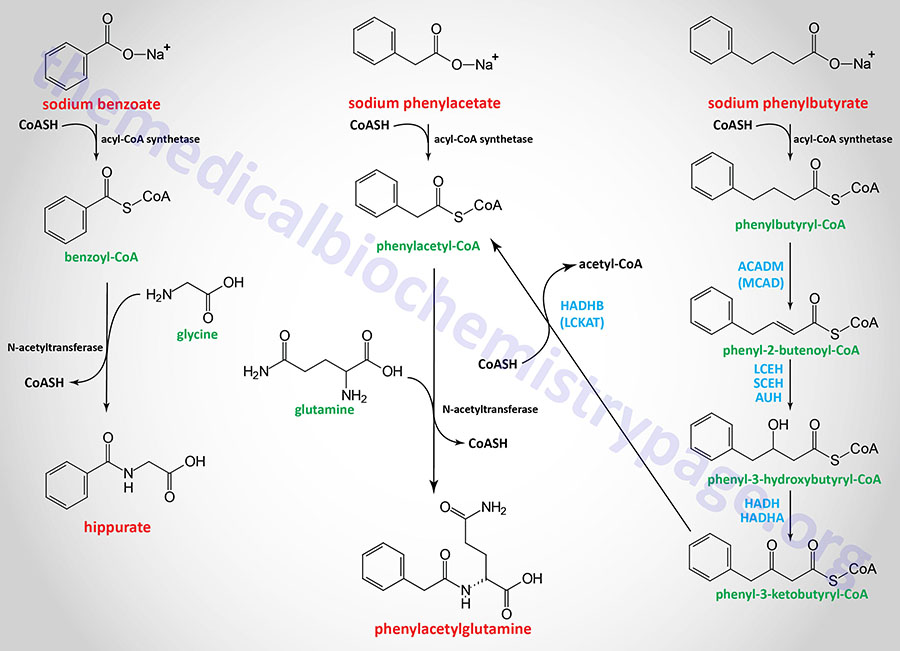
Sodium benzoate and sodium phenylacetate can be administered to covalently bind glycine (forming hippurate) and glutamine (forming phenylacetylglutamine), respectively, see Figure below. These latter compounds, which contain the ammonia nitrogen, are excreted in the feces. Ammunol® is an FDA-approved intravenous solution of 10% sodium benzoate and 10% sodium phenylacetate used in the treatment of the acute hyperammonemia in UCD patients. However, hemodialysis is the only effective means to rapidly reduce the level of circulating ammonia in UCD patients. Buphenyl® and Olpruva®, which are formulations of sodium phenylbutyrate, are FDA-approved oral medications for chronic adjunctive therapy of hyperammonemia in UCD patients. Phenylbutyrate is metabolized to phenylacetate via enzymes of mitochondrial fatty acid oxidation as outlined in the Figure below.
Dietary supplementation with arginine or citrulline can increase the rate of urea production and/or waste nitrogen excretion in certain UCDs. The role of arginine supplementation in UCDs is two fold. The activity of CPS1 is absolutely dependent upon the allosteric activator N-acetylglutamate (NAG) which is synthesized from glutamate and acetyl-CoA (see the Nitrogen Metabolism and the Urea Cycle page for details) via the action of NAG synthase (NAGS). NAGS is itself allosterically activated by arginine, thus, providing arginine in the diet can increase the level of active CPS1 driving more ammonia into carbamoylphosphate. Secondly, the consumption of large amounts of arginine increases the production of ornithine via the arginase catalyzed reaction. This keeps sufficient levels of ornithine available to the urea cycle to ensure that some waste nitrogen can be eliminated as citrulline and argininosuccinate via the OTC and argininosuccinate synthetase catalyzed reactions, respectively.
Citrullinemias not Associated with Urea Cycle
Citrullinemia Type 2 (CTLN2)
Another form of citrullinemia, with sudden onset between the ages of 11 and 79 years, is called citrullinemia type 2 (CTLN2). Manifestations of CTLN2 are recurrent hyperammonemia with neuropsychiatric symptoms including nocturnal delirium, aggression, irritability, hyperactivity, delusions, disorientation, restlessness, drowsiness, loss of memory, flapping tremor, convulsive seizures, and coma. The symptoms of CTLN2 are often provoked by alcohol and sugar intake, as well as by certain medications, and/or surgery.
CTLN2 is caused by deficiency in the gene encoding citrin. Citrin is a mitochondrial solute carrier protein (SLC25A13) involved in the mitochondrial uptake of glutamate and export of aspartate and as such functions in the malate-aspartate shuttle. Deficiencies in SLC25A13 result in mild hyperammonemia and citrullinemia. The clinical course in adults with citrullinemia type 2 is milder than that of CTLN1, possibly distinguishing it from milder late-onset citrullinemia type I. It is not known why CTLN2 is milder and later in onset than CTLN1 and distinguishing between the two forms of citrullinemia is difficult.
Citrin deficiency can manifest in newborns as neonatal intrahepatic cholestasis caused by citrin deficiency (NICCD) and in older children as failure to thrive and dyslipidemia caused by citrin deficiency (FTTDCD). One of the most common characteristics of citrin deficiency is a fondness for protein-rich and/or lipid-rich foods and aversion to carbohydrate-rich foods.
In NICCD children younger than age one year exhibit growth retardation with transient intrahepatic cholestasis, hepatomegaly, diffuse fatty liver and parenchymal cellular infiltration associated with hepatic fibrosis, variable liver dysfunction, hypoproteinemia, decreased coagulation factors, hemolytic anemia, and/or hypoglycemia. NICCD is generally not severe and symptoms may often resolve by one year of age provided appropriate treatment is given. However, some infants will die due to infection and liver cirrhosis.
FTTDCD is usually observed in children around one to two years of age with the aforementioned food preferences being apparent. Some FTTDCD patients have growth retardation, hypoglycemia, hyperlipidemia, pancreatitis, fatty liver, hepatoma, and fatigue. In some cases of FTTDCD, the affected individual will manifest with characteristic CTLN2 symptoms later in life.
Hyperornithinemia, Hyperammonemia, and Homocitrullinuria (HHH) Syndrome
Cytosolic ornithine is transported into the mitochondria in exchange for citrulline transport from the mitochondria to the cytosol. As described in the Nitrogen Metabolism and the Urea Cycle page, this transporter is identified as ornithine translocase 1, ORNT1. ORNT1 is a member of the solute carrier (SLC) family of transporters and as such is encoded by the SLC25A15 gene.
Mutations in the SLC25A15 gene result in the disorder identified as HHH syndrome for hyperornithinemia, hyperammonemia, and homocitrullinuria. HHH syndrome is a heterogeneous disease that manifests with a high degree of clinical variability.
Symptoms of HHH syndrome can range from a mild form (late-onset) that is associated with learning difficulties and mild neurological involvement, to a more severe form (neonatal-onset) that can manifest with hepatic involvement, lethargy, seizures, and coma. Similar to other UCD, an early diagnosis of HHH syndrome has the best chance improve clinical outcomes.
The inability to transport ornithine into the mitochondria is the basis for the hyperornithinemia. Due to defective urea cycle activity, as a result of reduced mitochondrial ornithine, there is the hyperammonemia that is typical of most disorders that affect the functioning of the urea cycle. The homocitrullinuria is thought to be due to the action of OTC combining lysine with carbamoyl phosphate in the absence of citrulline.