
Last Updated: November 9, 2025
Introduction to Hunter Syndrome
Hunter syndrome is an X-linked recessive disorder that belongs to the family of disorders identified as lysosomal storage disorders, and historically as one of the mucopolysaccharidoses, specifically mucopolysaccharidosis type II (MPS II). This disorder was first described in two brothers by the Canadian physician, Charles Hunter in 1917.
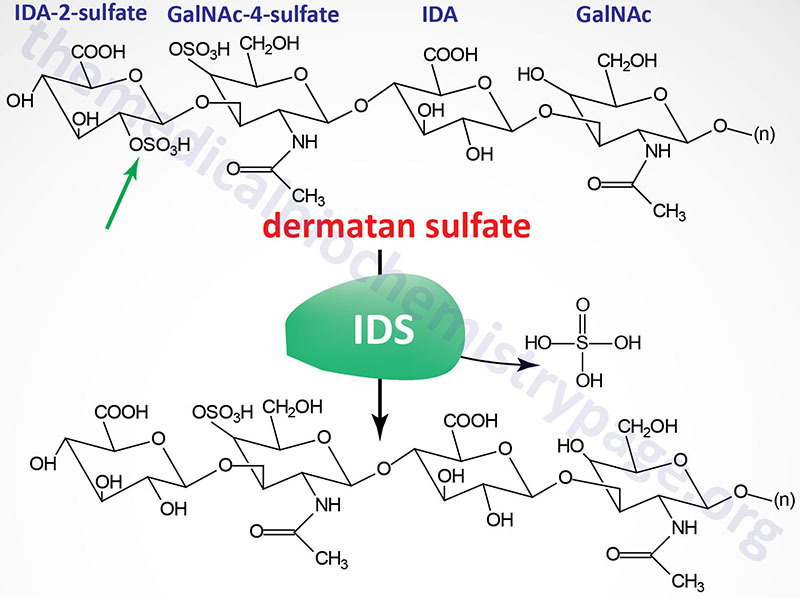
Hunter syndrome is characterized by the lysosomal accumulation of dermatan and heparan sulfates as a consequence of deficiencies in the lysosomal hydrolase, iduronate 2-sulfatase (also called iduronate sulfatase). Iduronate 2-sulfatase is a member of a family of sulfatases, many of which are necessary for the degradation of the glycosaminoglycans.
The reaction catalyzed by iduronate 2-sulfatase is depicted in the Figure. Iduronate 2-sulfatase hydrolyzes sulfates from the 2- position of L-iduronate-2-sulfate, present in dermatan and heparan sulfates. The enzyme was originally referred to as the “Hunter corrective factor”. Hunter syndrome is the only mucopolysaccharidotic disease that is X-linked.
Molecular Biology of Hunter Syndrome
The iduronate 2-sulfatase gene (IDS) is located on the X chromosome at Xq28 spanning 24 kb and encompassing 11 exons that generate three alternatively spliced mRNAs encoding three isoforms (a, b, and c). Removal of the 25 amino acid signal sequence generates the proprotein and finally, an additional 8 amino acids are removed from the N-terminus of the proprotein generating the functional glycoprotein.
As of 2025 a total of 556 pathogenic mutations have been identified in the IDS gene resulting in Hunter syndrome. The majority of Hunter syndrome mutations are missense, nonsense, and frameshift mutations with several small deletions or insertions as well. All patients harboring mutations caused by large deletions or rearrangements in the IDS gene manifest with the severe forms of Hunter syndrome.
The estimated incidence of Hunter syndrome varies from 1/60,000 to 1/150,000. Higher rates of the disorder have been reported within the ethnic Ashkenazi Jewish population.
Clinical Features of Hunter Syndrome
Clinically, Hunter syndrome is divided into severe and mild (attenuated) classifications. The clinical presentation of the severe form of Hunter syndrome has many similarities to those seen in Hurler syndrome without the corneal clouding. In addition, the symptoms of Hunter progress slower than those of Hurler syndrome. The mild form of Hunter syndrome is similar to the Hurler-Scheie syndrome and Scheie syndrome. Hunter syndrome is inherited as an X-linked recessive disorder and affected males do not reproduce. This latter fact precludes the presentation of affected females.
Patients with the severe form of Hunter syndrome present with characteristic clinical features. These include short stature, skeletal deformities, hydrocephalus, joint stiffness, coarse facial features, and intellectual impairment. Onset of symptoms of the severe form are usually seen between 2 and 4 years of age. Typical symptoms include macroglossia, a broad nose, full lips, and large rounded cheeks. As a result of vocal cord enlargement a hoarse, deep voice results. Patients with the severe form of Hunter syndrome develop hepatosplenomegaly, macrocephaly, umbilical hernia, inguinal hernia, hearing loss, and recurrent respiratory infections.
As a consequence of autonomic nervous system involvement many patients suffer from chronic diarrhea. Extensive neurological degeneration precedes death in patients with the severe form of Hunter syndrome with death occurring between 10 and 15 years of age due to coronary and/or pulmonary dysfunction.
Patients with the mild form of Hunter syndrome generally have normal intelligence and survive into adulthood. Some of the symptoms in mild form patients can be as severe as those in the severe form but the progression of deterioration is much slower. Although some patients with the mild form of Hunter can survive into their 50’s or 60’s death often occurs in early adulthood as a consequence of cardiac failure and pulmonary obstruction.
Treatment of Hunter Syndrome
Treatment options for Hunter syndrome are limited and depend upon the time of correct diagnosis since the available treatments cannot reverse, but can only prevent symptoms or worsening of symptoms. Enzyme replacement therapy (ERT) has been approved by the US FDA since 2006 and involves intravenous infusion of the drug called idursulfase. This form of treatment has not undergone controlled studies in patients with severe forms of Hunter syndrome. Nonetheless, some patients with the severe form that have been treated with idursulfase have shown reduction in liver and spleen volumes, increased range of joint motion, and possible improvement in growth velocities. Since the drug cannot cross the blood brain barrier there is no improvement to the cognitive decline in Hunter syndrome patients.
Additional therapeutic options for Hunter syndrome patients include gene therapy or hematopoietic stem cell transplantation.