
Last Updated: December 16, 2025
Introduction to Glutaric Aciduria Type 1 (GA-1)
Glutaric aciduria type 1 (also known as glutaric acidemia type 1) is an autosomal recessive disorder of amino acid metabolism. The disorder is the result of defects in the mitochondrial matrix enzyme, glutaryl-CoA dehydrogenase (GCDH). Glutaryl-CoA dehydrogenase is a member of the acyl-CoA dehydrogenase family of enzymes that include those involved in mitochondrial β-oxidation of fatty acids and as such the enzyme is also known as acyl-CoA dehydrogenase 5 (ACAD5). Glutaryl-CoA dehydrogenase catalyzes the conversion of glutaryl-CoA to crotonyl-CoA in the catabolism of the amino acids lysine, tryptophan, and hydroxylysine.
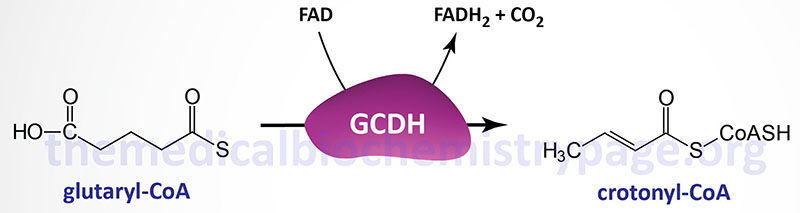
Defects in glutaryl-CoA dehydrogenase result in accumulation of glutaric acid and 3-hydroxyglutaric acid in the blood and subsequently the urine. Glutaric acid is a potent neurotoxin and its accumulation results in the characteristic neural pathology of glutaric aciduria, type 1 which includes gliosis and neuronal loss in the basal ganglia. The damage in the brain manifests with a progressive movement disorder encompassing dysarthria (weakness in muscle used for speech) and choreoathetosis (involuntary twitching or writhing) that usually begins during the first year of life. The frequency of glutaric aciduria type 1 is approximately 1:100,000 worldwide.
Molecular Biology of Glutaric Aciduria Type 1
Glutaryl-CoA dehydrogenase is encoded by the GCDH gene. The GCDH gene is located on chromosome 19p13.13 and is composed of 12 exons that generate two alternatively spliced mRNAs encoding proteins of 438 amino acids (isoform a) and 428 amino acids (isoform b). The isoform b protein is not functional as a glutaryl-CoA dehydrogenase. The isoform a precursor protein undergoes cleavage by mitochondrial processing peptidase complex to form the mature glutaryl-CoA dehydrogenase enzyme. Functional glutaryl-CoA dehydrogenase is a homotetrameric complex.
More than 200 mutations in the GCDH gene have been identified in glutaric aciduria type 1 patients. The majority of mutations in the GCDH gene are missense mutations. In the general population there is no one prevalent mutation, however, certain populations do have high rates of particular mutations. In individuals of German origin, a missense mutation that converts the Arg at position 402 to a Trp (designated R402W), is found in 40% of glutaric aciduria, type 1 cases. In Old Order Amish from Lancaster County, in the US state of Pennsylvania, there is a common missense mutation that converts an Ala at position 412 to a Val (designated A421V). In this Old Order Amish population the carrier frequency of the A412V mutation is estimated to be 10%.
Clinical Features of Glutaric Aciduria Type 1
The clinical spectrum of glutaric aciduria type 1 is generally divided into the infantile-onset form and the late-onset form. The infantile-onset form of glutaric aciduria type 1 is the more common form of the disorder. The late-onset form usually manifests with symptoms appearing after 6 years of age. The designation of infantile- or late-onset is determined by the age at which the first acute encephalopathic crisis occurs. These crises result in the typical complex movement disorders seen in glutaric aciduria type 1 patients resulting from bilateral striatal injury. Although the disorder can be divided into these two categories of pathology, the phenotype can vary widely between affected family members with the same genotype.
Typical of organic acidemias such as glutaric aciduria type 1, there are distinctive metabolic disturbances. The metabolic disturbances in glutaric aciduria type 1 include a high anion gap metabolic ketoacidosis, hyperammonemia, increased protein in CSF, and pancreatic enzyme abnormalities. Alterations in metabolites in the blood include increases in saccharopine (a byproduct of lysine catabolism), glutaric acid, glutaconic acid, and glutarylcarnitine. In contrast, there are reduced levels of serum free carnitine.
In the infantile-onset form of glutaric aciduria type 1 affected children are either normal or experience only mild neurological symptoms. are generally well in the first months of life, or show only mild neurological symptoms. Beginning in the period between 6 and 18 months, depending upon the severity of the mutations in the GCDH gene affected infants will begin to display increasing macrocephaly and severe encephalopathies with metabolic acidosis. The encephalopathies lead to necrosis of the striatum resulting in stroke-like events. These events will ultimately result in a dystonic dyskinetic movement disorders such as dysarthria and choreoathetosis. These encephalopathic crises are often precipitated by infection with fever and dehydration. The lesions that occur in the basal ganglia of glutaric aciduria type 1 patients will result in a variety of neurological, gastroesophogeal, skeletal, and respiratory complications.
Treatment of Glutaric Aciduria Type 1
Treatment of glutaric aciduria type 1 includes protein restriction to reduce the load of the toxic metabolites derived from the catabolism of lysine and tryptophan. In addition, supplementation with carnitine promotes the synthesis and excretion of the less toxic organic acid conjugates. Infants should also be fed a lysine and tryptophan restricted metabolic formula. Common childhood illnesses in glutaric aciduria type 1 patients should be attended to with prompt preventive measures, including parenteral caloric supplementation, to prevent poor outcomes and to reduce morbidity and mortality.