
Last Updated: December 2, 2025
Introduction to Fructose
The major natural sources of fructose in the human diet are fruits, honey, and sucrose (a disaccharide of glucose and fructose). Under normal dietary intake the majority of the ingested fructose is metabolized by the enterocytes of the small intestine primarily to glucose which is then delivered to the systemic circulation. In addition to glucose, the carbon atoms from dietary fructose are converted, by intestinal enterocytes, into several other metabolites including glycerate, glutamate, glutamine, alanine, ornithine, and citrulline.
However, diets containing large amounts of sucrose, high fructose corn syrup, or fructose alone, overwhelm the ability of the small intestine to metabolize it all and under these conditions a significant amount of fructose is then metabolized by the liver and to a lesser extent by other organs such as skeletal muscle and the brain.
It should be pointed out that the difference between the amount of fructose available from sucrose obtained from cane or beet sugars is not significantly different than that from high fructose corn syrup. High fructose corn syrup (HFCS) is somewhat improperly identified as being high in fructose giving the impression that it contains a large amount of fructose. However, whereas the fructose content of sucrose is 50% (since it is a pure disaccharide of only glucose and fructose), the content in HFCS is only around 45%–55% and in many cases is actually only 40%–45% fructose.
The reason that corn syrup (which is all glucose to begin with) is labeled as HFCS is because the glucose extracted from corn starch is enzymatically treated to convert some of the glucose to fructose. This is done in order to make the corn syrup sweeter which is why it is particularly popular in the food industry, in addition to the fact that it is cheaper to process than pure sucrose from sugar cane or sugar beet. Therefore, any disorder and/or dysfunction (see below), attributed to the consumption of fructose, can be manifest whether one consumes cane or beet sugar, HFCS, or pure fructose such as in honey and most fruits.
Digestion and Uptake of Dietary Fructose
Although discussed within the context of carbohydrate digestion and absorption in the Digestion and Digestive Processes page, the digestion and absorption of carbohydrates containing fructose will be reviewed here as well. Fructose is obtained in the diet primarily in three forms, as free fructose, in the disaccharide sucrose, or in high fructose corn syrup (HFCS). Sucrose is hydrolyzed into free glucose and free fructose via the action of the intestinal disaccharidase, sucrase-isomaltase. Sucrase-isomaltase also hydrolyzes fructose from HFCS. The fructose is then transported into intestinal enterocytes via the action of the GLUT5 transporter.
The fate of the fructose within intestinal enterocytes depends upon the energy demands of the enterocyte. Under conditions of normal fructose intake the majority is metabolized within the enterocyte for energy production as well as for conversion to fatty acids. Under conditions of excess fructose intake, as is typical in the modern diet of highly processed and sweetened foods, the fructose is transported from the enterocyte into the blood via the actions of the GLUT2 transporter. Following entry into the portal circulation of the liver the fructose is taken up by hepatocytes via GLUT2 transport and metabolized. Approximately 70% of fructose, that enters the blood from the diet, is metabolized by hepatocytes.
Metabolism of Fructose
Fructose Phosphorylation: Hexokinases and Fructokinases
The pathway to utilization of fructose differs in muscle and liver due to the differential distribution of fructose phosphorylating enzymes. Hexokinases are a family of enzymes that phosphorylate hexose sugars, such as glucose or fructose, at the C-6 hydroxyl. In the case of glucose they produce glucose-6-phosphate (G6P) and in the case of fructose they produce fructose-6-phosphate (F6P).
Humans express four genes encoding isozymes of ATP-dependent hexokinase. The encoded enzymes are designated as HK1, HK2, HK3, and HK4. A fifth member of the hexokinase family, identified as hexokinase domain containing 1 (encoded by the HKDC1 gene) is an RNA-binding protein as well as a protein-protein interacting partner.
The HK4 isoform is more commonly referred to as glucokinase and it is encoded by the GCK gene. All five mammalian hexokinases function as monomeric enzymes. Glucokinase is the form of the enzyme found in hepatocytes and pancreatic β-cells. Several of the hexokinases, but not GCK, can phosphorylate various different hexoses including fructose.
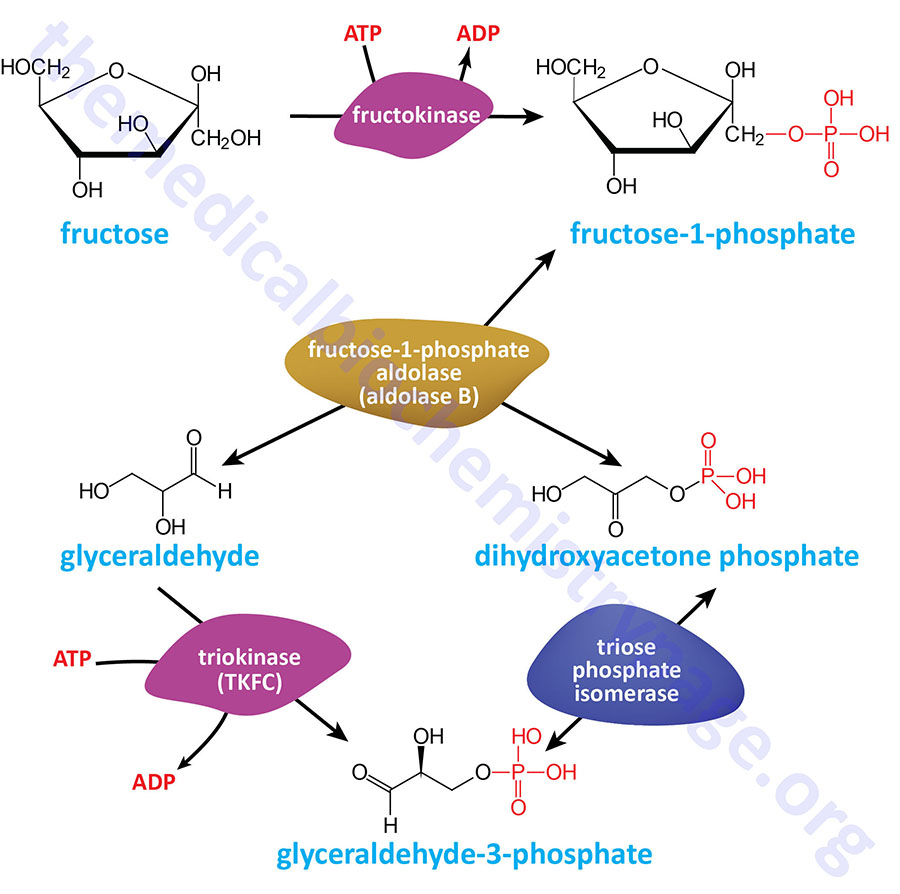
In addition to hexokinases, fructose can be phosphorylated by fructokinases. Fructokinases are formally referred to as ketohexokinases (KHK). There are two forms of KHK in mammals that result from alternative splicing of the KHK gene. These two isoforms are called KHK-A (fructokinase A) and KHK-C (fructokinase C). Fructokinases phosphorylate fructose on the C-1 hydroxyl generating fructose-1-phosphate (F1P).
The KHK gene is located on chromosome 2p23.3 and is composed of 9 exons that generate the two alternatively spliced mRNAs encoding KHK-A and KHK-C.
Fructokinase C (KHK-C) has a very high affinity (low KM) for fructose resulting in rapid phosphorylation with concomitant depletion in ATP. Fructokinase A (KHK-A) has a very low affinity (high KM) for fructose thus, exerting minimal effects on overall fructose metabolism and levels of ATP.
Expression of fructokinase C (KHK-C) is primarily in the intestines, liver, pancreas, and kidney. Expression of fructokinase A (KHK-A) is more ubiquitous and expressed at highest levels in skeletal muscle. Although both fructokinase C (KHK-C) and fructokinase A (KHK-A) can phosphorylate fructose, fructokinase C (KHK-C) is considered to be the primary enzyme involved in fructose metabolism because its’ KM for fructose is very much lower than that of the fructokinase A (KHK-A) isoform. Because of its very high KM for fructose there is some question as to whether or not fructokinase A (KHK-A) actively metabolizes fructose in vivo.
Muscle contains two types of hexokinase (HK1 and HK2), both of which can phosphorylate fructose to fructose-6-phosphate (F6P). The resulting F6P is a direct glycolytic intermediate. However, the affinity of hexokinase for fructose is substantially less than that of fructokinase C (KHK-C).
The liver expresses predominantly glucokinase (hexokinase type IV) which is specific for glucose as its’ substrate. Due to this pattern of expression, there is the requirement for KHK-C in the liver in order to deliver fructose into hepatic glycolysis. Hepatic KHK-C phosphorylates fructose on C–1 yielding fructose-1-phosphate (F1P) which is then hydrolyzed by a specific isoform of aldolase identified as aldolase B.
Functions of HKDC1
Despite being a member of the hexokinase family of enzymes, HKDC1 does not likely participate in the normal process of glycolysis. Analysis of cancer cells demonstrates that overexpression of HKDC1 likely promotes tumorigenesis by affecting lipid metabolism more than glucose metabolism. Recent evidence has also shown that HKDC1 plays a critical role in mitochondrial and lysosomal homeostasis.
The HKDC1 gene is located upstream (5′) of the HK1 gene on chromosome 10q21.1. The HKDC1 gene is composed of 21 exons that encode a 917 amino acid protein that shares 70% amino acid sequence identity with HK1. The existence of HKDC1 was only recently (2007) identified with the use of genome-wide association studies (GWAS) of glucose homeostasis during pregnancy, specifically in the context of gestational diabetes mellitus. Four variants of HKDC1, that were found to be associated with gestational diabetes, were all within separate enhancers that cooperate to regulate expression of the HKDC1 gene. All four polymorphisms cooperate to reduce the expression level of the HKDC1 gene.
Normal expression of the HKDC1 gene is highest in the small intestines but is also differentially expressed in many other tissues. Elevated expression of the HKDC1 gene is found in numerous types of cancer and this is associated with poor prognosis such as is the case for hepatocellular carcinoma (HCC) and squamous cell lung carcinoma (SQCLC).
Expression of the HKDC1 gene has been shown to be regulated by the transcription factor encoded by the TFEB (transcription factor EB, where the EB refers to enhancer box binding) gene. Increased TFEB expression is associated with both lysosomal and mitochondrial stress. Under these latter conditions, the TFEB-induced expression of HKDC1 results in the association of HKDC1 with the mitochondrial outer membrane protein identified as translocase of outer mitochondrial membrane 70, commonly identified as TOM70. TOM70 is encoded by the TOMM70 gene.
Increased expression of HKDC1 is also associated with an increase in the association between mitochondria and lysosomes. These interactions involve the association of HKDC1 with the mitochondrial voltage-dependent anion-selective channels (VDAC), specifically VDAC1. The interaction of HKDC1 with VDAC1 enhances mitochondrial outer membrane contacts with lysosomal transient receptor potential cation channel mucolipin subfamily member 1 (TRPML1). The enhanced membrane contacts between mitochondria and lysosomes (and potentially endoplasmic reticulum) enhances the exchange of Ca2+, ATP, and lipids which contribute to increased lysosomal and mitochondrial homeostasis.
Expression of HKDC1 is significantly upregulated in gastric cancer and the enhanced expression is associated with poor prognosis. Within gastric cancer cells, the HKDC1 protein interacts with the protein, GTPase activating protein (SH3 domain) binding protein 1 (G3BP1) as well as with the mRNA encoding the protein identified as protein kinase, DNA-activated, catalytic subunit (PRKDC). The interaction of HKDC1 with G3BP1 results in enhanced stability of the HKDC1 interaction with the PRKDC mRNA, thereby stabilizing the mRNA. Increased levels of PRKDC are associated with resistance of certain cancers to chemotherapy. The observations that HKDC1 interaction with the PRKDC mRNA results in increased synthesis of PRKDC suggests that targeting the activity of HKDC1 may prove useful in the treatment of certain types of cancers.
Fructose-1-Phosphate Activities
Fructose-1-phosphate (F1P) is not solely an intermediate in the metabolism of fructose in the intestines, liver, pancreas, and kidneys. As discussed in the Glycolysis and the Regulation of Blood Glucose page, within the liver F1P interacts with the glucokinase regulatory protein (GKRP) in the nucleus stimulating the release of glucokinase. The glucokinase can then be transported to the cytosol where it can phosphorylate glucose stimulating its oxidation.
Fructose-1-phosphate has also been shown to function as a signaling molecule within intestinal enterocytes. In the intestines, F1P, generated by fructokinase-mediated metabolism of fructose, inhibits the PKM2 isoform of pyruvate kinase by stimulating the monomeric form of the enzyme. Within intestinal enterocytes monomeric PKM2 interacts with and activates HIF1α. This effect of F1P in the intestines promotes hypoxic cell survival and the elongation of intestinal villi resulting in an increase in surface area which improves the capacity for nutrient uptake.
Aldolase Metabolism of Fructose
Humans express three distinct forms of aldolase; aldolase A, aldolase B, and aldolase C. Aldolase A (fructose-1,6-bisphosphate aldolase) is expressed in most tissues. Aldolase A catalyzes the hydrolysis of fructose-1,6-bisphosphate (F1,6BP) into two 3-carbon products: dihydroxyacetone phosphate (DHAP) and glyceraldehyde-3-phosphate (G3P). The aldolase A reaction proceeds readily in the reverse direction, being utilized for both glycolysis and gluconeogenesis.
Aldolase B is expressed exclusively in the liver, kidney, and small intestine. Aldolase B is unique from the other two isoforms in that it can catalyze the hydrolysis of both fructose-1,6-bisphosphate and fructose-1-phosphate with equal affinity.
Aldolase C is ubiquitously expressed at low levels with the sites of predominant expression being in the brain, specifically in the hippocampus and in Purkinje cells, as well as in smooth muscle cells.
The aldolase A enzyme is encoded by the ALDOA. The ALDOA gene is located on chromosome 16p11.2 spanning 7.5 kb and is composed of 11 exons that generate four alternatively spliced mRNAs that collectively encode protein isoforms of 364 amino acids (isoform 1) and 418 amino acids (isoform 2).
The aldolase B enzyme is encoded by the ALDOB gene. The ALDOB gene is located on chromosome 9q31.1 and is composed of 9 exons that produce a protein of 363 amino acids. In the liver, aldolase B can utilize both F1,6BP and F1P as substrates. Therefore, when presented with F1P the enzyme generates DHAP and glyceraldehyde. The DHAP is converted, by triose phosphate isomerase (TPI), to G3P and enters glycolysis. As indicated below, the glyceraldehyde can be metabolized by several different pathways.
The aldolase C enzyme is encoded by the ALDOC gene. The ALDOC gene is located on chromosome 17q11.2 and is composed of 10 exons that produce a protein of 364 amino acids.
Role of ChREBP in the Metabolism of Fructose
The basic helix-loop-helix/leucine zipper (bHLH/LZ) transcription factor identified as carbohydrate-responsive element-binding protein (ChREBP) was originally identified as being responsive to the levels of glucose in hepatocytes. ChREBP is most highly expressed in the liver but is also abundantly expressed in white adipose tissue (WAT), brown adipose tissue (BAT), pancreatic β-cells, small intestine, kidney, adrenal glands, and brain.
Subsequent to the identification of glucose as an enhancer of ChREBP activity it was found that fructose is a more potent activator than glucose. Fructose-mediated activation of ChREBP involves acetylation of the protein resulting in increased DNA binding to ChREBP target genes. The activation of ChREBP activity by fructose ultimately results in increased metabolism of fructose as well as diversion of its carbon atoms into fatty acid synthesis as detailed in the next section.
As discussed in detail in the Glycolysis and the Regulation of Blood Glucose page, as well as in the Synthesis of Fatty Acids page, ChREBP exists in two isoforms, ChREBP-α and ChREBP-β, as a result of alternative splicing. The ChREBP-β isoform is a more potent transcriptional regulator than is the ChREBP-α isoform. In the intestine, ChREBP has been shown to regulate the expression of the SLC2A5 (GLUT5) gene, the KHK gene, the ALDOB (aldolase B) gene, and the TKFC (triokinase and FMN cyclase) gene. Within the liver ChREBP also activates expression of the KHK, ALDOB, and TKFC genes. ChREBP also promotes metabolism of fructose by the kidneys via activation of expression of the SLC2A5, KHK, ALODB, and TKFC genes.
As discussed in the following section, enhanced expression of the TKFC gene allows for enhanced lipogenesis in response to fructose uptake and metabolism in both the intestines and the liver.
Metabolism of Glyceraldehyde Contributes to Fructose-Mediated Lipogenesis
The glyceraldehyde generated via the action of aldolase B can be metabolized by three distinct routes with the dominant route being the one diverting it into glycolysis. Glyceraldehyde can be phosphorylated to glyceraldehye-3-phosphate by triokinase (also called triose kinase) which is encoded by the TKFC (triokinase and FMN cyclase) gene. The glyceraldehyde-3-phosphate can then directly enter the glycolytic pathway where it eventually enters the TCA cycle in the form of acetyl-CoA.
As the rate of the TCA cycle increases in response to fructose metabolism, the citrate will be transported into the cytosol where ATP-citrate lyase generates acetyl-CoA and oxaloacetate. The acetyl-CoA can be used for the synthesis of fatty acids demonstrating how increased fructose intake and metabolism contributes to lipogenesis. This effect of fructose metabolism is a significant contributor to the development of the metabolic syndrome and metabolic dysfunction-associated fatty liver disease (MAFLD). MAFLD is also referred to as metabolic dysfunction-associated steatotic liver disease, MASLD. MAFLD was formerly referred to as non-alcoholic fatty liver disease, NAFLD.
Glyceraldehyde can also be converted to glycerate (D-glyceric acid) via the action of an aldehyde dehydrogenase (ALDH). The glycerate can then be converted to either of the glycolytic intermediates (3-phosphoglycerate or 2-phosphoglycerate) via the action of glycerate kinase (encoded by the GLYCTK gene). The conversion of glyceraldehyde to glycerate represents a clinically relevant pathway given that the glycerate persists in the blood and has been shown to induce pancreatic islet cell damage contributing to the development of diabetes.
Glyceraldehyde can also be converted to glycerol via an NADP-dependent alcohol dehydrogenase and the glycerol is then phosphorylated by glycerol kinase yielding glycerol-3-phosphate. Glycerol-3-phosphate can then be converted to the glycolytic intermediate, dihydroxyacetone phosphate (DHAP), via the action of glycerol-3-phosphate dehydrogenase. Glycerol-3-phosphate is also the starting point for the synthesis of triglycerides which allows fructose metabolism to contribute to overall lipogenesis in concert with the role of glyceraldehyde-3-phosphate.
Intestinal Fructose Contributes to Hepatic Lipogenesis
When fructose is consumed in excess significant quantities remain unabsorbed and reach the colon where it is metabolized by colonic bacteria into short-chain fatty acids (SCFA) with the predominant SCFA being acetate. The acetate is transported into colonic cells via the action of transporters of the monocarboxylate transporter (MCT) family, primarily MCT1 (encoded by the SLC16A1 gene) and to a lessor extent by MCT2 (encoded by the SLC16A7 gene). Acetate also enters colonic cells through aquaporin-mediated facilitated diffusion. The acetate is then transported into the portal circulation and is taken up into hepatocytes via MCT1-mediated transport.
Within hepatocytes the acetate is converted to acetyl-CoA via the action of the acetyl-CoA synthetase encoded by the ACSS2 (acyl-CoA synthetase short-chain family member 2) gene. The acetyl-CoA then serves as a substrate for the fatty acid synthesizing enzymes, acetyl-CoA carboxylase (ACC) and fatty acid synthase (FAS) resulting in the accumulation of lipids in hepatocytes. The acetyl-CoA is also used for the synthesis of cholesterol, further contributing to de novo lipogenesis.
Fructose Effects on Food Intake
As pointed out in the Glycolysis and the Regulation of Blood Glucose page, glucose is the primary fuel used for energy production in the brain. When glucose is metabolized within the hypothalamus, a signaling pathway is initiated that ultimately results in the suppression of food intake. More detailed information on the role of the hypothalamus in the control of food intake can be found in the Gut-Brain Interrelationships and Control of Feeding Behaviors page. The principal participants in this signaling cascade include AMPK, acetyl-CoA carboxylase (ACC), and the product of the ACC reaction, malonyl-CoA.
The mechanism by which AMPK and ACC are regulated, ultimately resulting in altered levels of malonyl-CoA are detailed in the Synthesis of Fatty Acids page. Briefly, activation of AMPK results in the phosphorylation of ACC resulting in reduced activity of the latter enzyme. Conversely, when AMPK activity falls the phosphorylation state of ACC falls resulting in increased production of malonyl-CoA. When glucose oxidation is increased in the hypothalamus there is a resultant dephosphorylation and inactivation of AMPK and thereby, activation of ACC. The resultant rise in hypothalamic malonyl-CoA is correlated to reduced expression of several orexigenic peptides (e.g. ghrelin, NPY, and AgRP) concomitant with activated expression of several anorexigenic peptides (e.g. α-MSH and CART). These changes in neuropeptide expression result in suppressed food intake while simultaneously increasing overall energy expenditure.
In contrast to the anorexigenic effect of hypothalamic glucose metabolism, the metabolism of fructose in the brain exerts an orexigenic effect. Although the overall mechanisms by which fructose exerts this orexigenic effect are complex, it is due, in part, to the fact that the brain, like the liver, possesses a unique set of sugar transporters and metabolizing enzymes that enables fructose to bypass the PFK-1 catalyzed step of glycolysis. The PFK-1 catalyzed reaction is the rate-limiting step in glycolysis and is critical in the overall regulation of ATP production and consumption. Since hypothalamic fructose metabolism bypasses this important regulatory step its metabolism rapidly depletes ATP in the hypothalamus. When ATP levels fall there is a concomitant rise in AMP which results in activation of AMPK. Activation of AMPK results in phosphorylation and inhibition of ACC which then results in decreased malonyl-CoA levels in the hypothalamus. Therefore, although glucose and fructose utilize the same signaling pathway to control food intake they act in an inverse manner and have reciprocal effects on the level of hypothalamic malonyl-CoA.
Numerous experiments in animals have implicated malonyl-CoA as a key intermediate in the regulation of feeding behavior and overall energy balance initiated via signaling cascades within the hypothalamus. Initial evidence demonstrating this role of malonyl-CoA was the finding that inhibition of fatty acid synthase (FAS) suppressed food intake. FAS is the primary enzyme in the de novo biosynthesis of fatty acids.
The fungal antibiotic, cerulenin, and a related analogue C75, bind to the active site of FAS thereby inhibiting its activity. When these compounds are administered to mice there is a resultant decrease in food intake. Given that inhibition of FAS would be expected to result in an accumulation of its substrate, malonyl-CoA, it was suspected that malonyl-CoA may be a participant in the observed effects. Indeed, intra-cerebrovascular (icv) injection of C75 results in increased levels of hypothalamic malonyl-CoA. Importantly, this effect can be reversed by inhibitors of ACC, the enzyme which synthesizes malonyl-CoA from acetyl- CoA. Further research demonstrated that the anorexigenic effects of FAS inhibitors was due to their ability to rapidly suppress hypothalamic expression of NPY and AgRP, two key orexigenic peptides, while increasing the expression of α-MSH and CART, two key anorexigenic peptides.
Malonyl-CoA levels in the hypothalamus correlate well with nutritional state. Fatty acid synthesis in other tissues, such as the liver and adipose tissue, occurs primarily during energy surplus. Malonyl-CoA, regulates energy metabolism in the liver through two opposing mechanisms. It serves as the substrate for FAS during de novo fatty acid synthesis while at the same time inhibiting mitochondrial fatty acid oxidation through its’ action as an allosteric inhibitor of carnitine palmitoyltransferase 1 (CPT1). Inhibition of CPT1 by malonyl-CoA prevents entry of fatty acids into the mitochondria, thereby inhibiting oxidation. During periods when energy expenditure exceeds intake, such as during fasting, malonyl-CoA levels in the hypothalamus are low. Following food intake there is a rapid rise in hypothalamic malonyl-CoA levels. The changes in hypothalamic malonyl-CoA levels are followed quickly by changes in expression of orexigenic and anorexigenic peptides. In the fasting state NPY and AgRP levels are high, whereas α-MSH and CART levels are low. Upon re-feeding this pattern immediately inverts.
Genetic manipulation of hypothalamic malonyl-CoA levels has also been informative. For example, disruption of FAS gene expression in the hypothalamus results in increased malonyl-CoA levels and decreases in body weight and fat content. Concomitant with disruption of hypothalamic FAS gene expression is an increase in malonyl-CoA concentration. Additionally, the levels of orexigenic peptides decrease while levels of anorexigenic peptides increase leading to suppression of food intake. Conversely, genetic manipulation of the expression of malonyl-CoA decarboxylase (MCD) has the opposite effects to those seen by disruption of FAS expression. MCD functions in opposition to ACC in that it is responsible for the conversion of malonyl-CoA to acetyl-CoA. In fact ACC and MCD are reciprocally regulated to ensure adequate regulation of fatty acid synthesis and fatty acid oxidation.
Overexpression of MCD within the hypothalamus results in increases in both food intake and body weight and also marked increases in body fat content (adiposity). Additionally, when MCD expression is increased within the hypothalamus there is an observed inhibition of the FAS inhibitor (C75)-induced suppression of food intake. These results obtained with forced expression of MCD provide strong evidence that hypothalamic malonyl-CoA acts as an indicator of energy status and participates in the regulation of feeding behavior. In addition, these results indicate that malonyl-CoA, rather than fatty acids, is the effector that regulates energy homeostasis.
Fructose Utilization and Associated Metabolic Dysfunction
Consumption of fructose has been shown to be highly correlated with the development of diabetes, obesity and the metabolic syndrome in addition to being highly correlated with the development of numerous cancers including hepatocellular carcinoma. Consumption of sweetened soft drinks (either with sucrose of HFCS) is associated with an increased risk for obesity in adolescents and for type 2 diabetes in young and middle-aged women. Excess fruit juice (also rich in fructose) is associated with the development of obesity in children.
Studies in mice have shown that the metabolism of fructose by the liver is the major contributor to the development of the metabolic syndrome, despite the fact that intestinal fructose metabolism can contribute to increased serum glucose levels. This was demonstrated by knocking out the KHK gene, which reduces expression of both KHK-C and KHK-A isoforms, specifically in the intestines versus specifically in the liver. When the intestinal expression of the KHK gene was knocked out the protection from the development of fatty liver and the metabolic syndrome results from decreased uptake of fructose. This decreased fructose uptake is the result of an associated decrease in the level of GLUT5 in the intestines as a result of loss of KHK expression. GLUT5 is the fructose specific transporter of the GLUT family.
The combination of a high fat diet combined with increased fructose intake, as is typical of the Western style diet, significantly contributes to the development of the metabolic syndrome and metabolic dysfunction-associated fatty liver disease (MAFLD). MAFLD is the current definition of what was originally defined as non-alcoholic fatty liver disease, NAFLD.
The combination of high fat and fructose results in decreased levels of hepatic fatty acid oxidation which result in the development of fatty liver. The effects of fructose in the liver under these dietary conditions are due to reduction in the level of carnitine palmitoyltransferase 1 (CPT1) which results in reduced fatty acid transport into the mitochondria for oxidation. In addition, fructose included in high fat diets results in the increased acetylation of several enzymes of fatty acid oxidation resulting in a reduced capacity to carry out overall fatty acid oxidation.
One distinction between fructose and glucose metabolism is that the metabolism of fructose results in rapid decreases in ATP and increases in serum uric acid concentration. The increased production of uric acid, as a result of fructose metabolism, is related to the activity of fructokinase (specifically KHK-C). The activity of fructokinase (KHK-C) is different from the other hexokinases by virtue of the fact that it induces transient ATP depletion in the cell. The mechanism is due to the fact that fructokinase (KHK-C) is unregulated and therefore rapidly phosphorylates fructose to fructose-1-phosphate resulting in marked ATP depletion. The activity of fructokinase (KHK-C) is not subject to feed-back inhibition such as is the case for glucose metabolism, thus the ATP depletion is profound. Since the majority of fructose metabolism, beyond the small intestine, occurs in the liver, the effects of this ATP depletion are exerted on numerous important hepatic metabolic processes. ATP depletion also triggers survival responses that include, but are not limited to, increased hunger, increased energy intake, increased thirst, and a reduction in the level of energy expenditure while at rest.
The depletion in ATP is also associated with intracellular phosphate depletion and dramatic increases in AMP generation. Both of the latter stimulate the activity of the purine nucleotide catabolic enzyme, AMP deaminase, increasing degradation of AMP ultimately to uric acid. Humans express three AMP deaminase genes, AMPD1, AMPD2, and AMPD3. Within the context of fructose metabolism it is the AMPD2 encoded enzyme that contributes to enhanced uric acid production.
Elevated serum uric acid is a good predictor for the development of obesity and hypertension. Uric acid is the byproduct of purine nucleotide catabolism. Uric acid contributes to the depletion of ATP by inducing mitochondrial oxidative stress thereby inhibiting ATP production.
Gout is a disorder that is related to excess production of uric acid and deposition of uric acid crystals, particularly in the synovial fluids of the joints. The root cause of gout is in fact hyperuricemia. Therefore, excess consumption of fructose can also result in, and exacerbate, symptoms of gout.
Consumption of fructose by laboratory animals results in their developing several features of metabolic syndrome, including obesity, visceral fat accumulation, fatty liver, and elevated serum levels of insulin and leptin. It is likely that the increase in leptin, following fructose consumption, represents leptin resistance, which could account for the increased food intake observed in fructose-fed animals. All of these phenomena associated with fructose consumption, including hyperuricemia, can be blocked in laboratory animals when both fructokinase C (KHK-C) and fructokinase A (KHK-A) isoforms are eliminated.
The relationship between fructose metabolism-mediated hyperuricemia and development of the metabolic syndrome can also be demonstrated by the fact that treating animals with allopurinol, a drug used to lower uric acid levels in gout patients, partially prevents the fructose-induced metabolic syndrome.
Intestinal Fructose and Hepatocellular Carcinoma
Increased consumption of fructose has been found to be associated with metabolic dysfunction as well as the development of several forms of cancer.
Hepatocellular carcinoma (HCC) is the major form of liver cancer and it is associated with poor outcomes. The incidence of HCC has been shown to be positively correlated with the amount of daily fructose consumption. High dietary fructose intake results in intestinal-barrier deterioration and endotoxemia both of which contribute inflammation-dependent de novo lipogenesis. As a result of increased lipogenesis there is an increased likelihood for the development of non-alcoholic steatohepatitis (NASH), a precursor to HCC. The currently accepted nomenclature for NASH is metabolic dysfunction-associated steatohepatitis (MASH).
Evidence has demonstrated that several genes of fructose metabolism, including the KHK and ALDOB genes, are downregulated in HCC and this downregulation is correlated to negative outcomes. This is significant given that the normal function of these two genes in hepatocytes is associated with induction of apoptosis leading to a reduction in the rate of HCC cell proliferation.
Recent evidence has defined a role for gut microbiota in the metabolism of fructose and how this plays a role in the promotion of cancer in the liver. Gut microbiota metabolize fructose into the short-chain fatty acid (SCFA) acetate. Acetate is then delivered to the liver via the portal circulation where it serves as a precursor for de novo lipogenesis as described above. The acetate is converted to the precursor for fatty acids and cholesterol, acetyl-CoA, via the action of the ACSS2 encoded enzyme and the acetyl-CoA then serves as a substrate for fatty and and cholesterol biosynthesis.
Of significance to the development and progression of HCC, the intestinal-derived acetate contributes to hepatocyte glutamine synthesis via 2-oxoglutarate (α-ketoglutarate) from the TCA cycle. The increased levels of hepatocyte glutamine contribute to UDP-GlcNAc synthesis which drives the O-GlcNAcylation of numerous hepatocyte proteins.
Several proteins that were recently shown to be O-GlcNAcylated as a consequence of high dietary fructose are eEF-1α1 (encoded by the EEF1A1 gene), calpain small subunit 1 (encoded by the CAPNS1 gene), nuclear factor related to KappaB binding protein (encoded by the NFRKB gene), EMSY transcriptional repressor, BRCA2 interacting (encoded by the EMSY gene), and nucleoporin 214 (encoded by the NUP214 gene).
The EMSY gene was given this name because it is the name of the sister of the first author of the original publication describing the protein. The EMSY encoded protein has amino acids at positions 57 to 62 that spell “SISTER” using the single letter amino acid code.
Each of these proteins has been indicated to play a role in proliferation, in particular, the O-GlcNAcylation of the EEF1A1 encoded translation elongation factor has been shown to enhance the proliferation of HCC cells due to its role in protein synthesis.
Role of Fructokinase (Ketohexokinase) in Ethanol Consumption
The metabolism of fructose, initiated via the fructokinase (ketohexokinase, KHK) catalyzed reaction, has implications with respect to ethanol consumption. It has long been known that alcohol consumption induces a preference for carbohydrate (specifically sugars) intake and that sugar intake induces a higher likelihood for alcohol consumption. Indeed, it has been demonstrated that individuals with alcohol use disorder (AUD) are also more likely to have an increased preference for sugar intake, a behavior that is also evident in the children of AUD individuals.
With respect to fructose metabolism, experiments have demonstrated that the consumption of alcohol, and its metabolism in the liver, can lead to glucose diversion into fructose which is a direct contributor to hepatic lipogenesis and the development of hepatic steatosis. The conversion of glucose to fructose involves polyol pathway where glucose is first reduced to sorbitol via the action of the aldose reductase encoded by the AKR1B1 (aldo-keto reductase family 1 member B) gene. Sorbitol can then be oxidized to fructose via the action of sorbitol dehydrogenase encoded by the SORD gene. This pathway is activated in response to ethanol uptake by the liver as a result of the hyperosmolarity of ethanol. Changes in the osmolarity of hepatocytes leads to activation of the AKR1B1 encoded aldose reductase.
As discussed in both the Metabolism of Glyceraldehyde Contributes to Fructose-Mediated Lipogenesis and the Intestinal Fructose Contributes to Hepatic Lipogenesis sections above, fructose metabolism enhances lipogenesis thereby contributing to the development of pathologies associated with excess lipid accumulation such as hepatic steatosis.
Most recently (2025) it has been determined that expression of the KHK gene, that encodes both KHK-A and KHK-C (often abbreviated KHK-A/C) isoforms, is associated with an enhanced preference for ethanol consumption. In knock-out mice, the loss of the KHK gene is associated with a reduced desire for ethanol intake. Mechanistically, the role of KHK in alcohol preference is most likely due to the role of fructose metabolism in the activity levels of aldehyde dehydrogenase (ALDH), the mitochondrial enzyme that metabolizes the toxic intermediate in ethanol metabolism, acetaldehyde. Fructose metabolism in the liver enhances the upregulation of ALDH which in turn results in reduced levels acetaldehyde making consumption of alcohol more tolerable. Conversely, loss of KHK activity prevents increased activity of ALDH thereby causing levels of acetaldehyde to increase. The increased levels of acetaldehyde in turn contribute to negative physiological effects that likely lead to behavioral effects that are associated with a reduced desire to consume alcohol.
Fructose-Mediated Enhancement of Ethanol Metabolism
Two aspects related to the metabolism of fructose contribute to the observation that the rate of hepatic ethanol metabolism is accelerated in the context of fructose metabolism.
The first is that the metabolism of fructose is unregulated, resulting in a dramatic depletion of hepatic ATP and a consequent increase in ADP. The reduced level of ATP will trigger an increase in the rate of mitochondrial oxidative phosphorylation leading to oxidation of the NADH produced during the metabolism of ethanol.
The second is related to the various fates of fructose. Fructose can be reduced to sorbitol with the concomitant oxidation of NADH to NAD+ which is catalyzed by sorbitol dehydrogenase (SORD) of the polyol pathway. In addition, the entry of fructose into the glycolytic pathway will increase the levels of dihydroxyacetone phosphate (DHAP) which in the context of ethanol metabolism, will be driven to glycerol-3-phosphate via the glycerol-3-phosphate dehydrogenase catalyzed reaction which also oxidizes NADH to NAD+.
Fructose metabolism also leads to glyceraldehyde via the aldolase B reaction. The glyceraldehyde can be reduced to glycerol via alcohol dehydrogenase which also oxidizes NADH to NAD+ in the context of this reaction. Glycerol is then phosphorylated by glycerol kinase yielding glycerol-3-phosphate.
The contribution of fructose metabolism to the oxidation of NADH to NAD+ consequently yields a continuous pool of NAD+ for the alcohol dehydrogenase and aldehyde dehydrogenase catalyzed reactions of ethanol metabolism.
Inherited Disorders of Fructose Metabolism
There are three principal disorders that result from inherited mutations in genes encoding enzymes of fructose metabolism. These three disorders are discussed in detail in the Fructose Metabolism Disorders page and include essential fructosuria, hereditary fructose intolerance (HFI), and rructose-1,6-bisphosphatase deficiency.