
Last Updated: October 29, 2025
Introduction to the Congenital Adrenal Hyperplasias
The congenital adrenal hyperplasias (CAH) are a group of inherited disorders that result from mutations in one of several genes involved in adrenal cortical steroid hormone synthesis. In the virilizing forms of CAH the mutations result in impairment of cortisol production and the consequent accumulation of steroid intermediates proximal to the defective enzyme. In the virilizing forms of CAH there is increased ACTH secretion which leads to elevated synthesis of adrenal androgens. In addition, there is adrenal cortical hyperplasia, the symptom that imparts the name to these disorders. All forms of CAH are inherited in an autosomal recessive manner.
There are two common and at least four rare forms of virilizing CAH. The common forms are caused by mutations in the genes encoding either 21-hydroxylase (CYP21A2; also identified as just CYP21 or CYP21B) or 11β-hydroxylase (CYP11B1). The majority of CAH cases (90-95%) are the result of defects in CYP21A2 with a frequency of between 1:13,000 and 1:15,000 live births worldwide. The four rare forms of virilizing CAH result from mutations in the genes encoding 3β-hydroxysteroid dehydrogenase (HSD3B2), placental aromatase (CYP19A1), P450-oxidoreductase (POR), or steroidogenic acute regulatory protein (StAR).
The typical signs of virilizing CAH are reflective of the androgen excess as well as the mineralocorticoid and glucocorticoid deficiencies. In general, the degree to which a given mutation results in reduction of enzyme activity is correlated to the level of glucocorticoid and mineralocorticoid deficiency.
An additional form of CAH is caused by mutations that affect either the 17β-hydroxylase, 17,20-lyase, or both activities, encoded in the CYP17A1 gene. Unlike the virilizing forms of CAH, individuals harboring CYP17A1 mutations, that result in severe loss of enzyme activity, have absent sex steroid hormone production accompanied by hypertension resulting from mineralocorticoid excess.
Pathophysiology of the CAH
Mutations in the gene encoding the 21-hydroxylase (CYP21A2) result in decreased cortisol production. The decrease in circulating cortisol results in a disturbance in the normal cortisol-mediated hypothalamic–pituitary–adrenal (HPA) feedback loop. The loss of the effects of cortisol on the hypothalamus results in increased hypothalamic production of corticotropin-releasing hormone (CRH; also termed corticotropin-releasing factor, CRF). The loss of the effects of cortisol on the pituitary result in increased production of ACTH. Low levels of cortisol in the adrenals during development result in adrenomedullary dysplasia and a consequent deficiency in epinephrine production. Mutations in the CYP21A2 gene that result in the salt-wasting form of CAH result in the largest degree of epinephrine deficiency.
Despite there being no direct influence of ACTH on the renin–angiotensin–aldosterone system (RAAS), the volume depletion due to aldosterone insufficiency serves as an additional stimulus for ACTH production. The added ACTH production is due to indirect stimulation of hypothalamic vasopressin synthesis. Vasopressin, which is co-secreted with CRH, functions synergistically with CRH to augment ACTH production and secretion.
ACTH binds to the melanocortin type 2 receptor (MC2R) and activates adrenocortical steroidogenesis. ACTH also functions as an adrenal trophic factor such that the excess ACTH that results from loss of cortisol production results in adrenocortical hyperplasia in addition to the uninhibited synthesis of adrenal androgens and androgen precursors. The major androgen precursor, 17-hydroxyprogesterone, is a diagnostic marker for CAH.
The hormonal imbalances of CAH, including cortisol deficiency, androgen excess, and varying degrees of aldosterone
and epinephrine deficiency leads to the phenotypic presentation of the disorder. The degree of symptoms in a given CAH patient is determined by the level of enzyme impairment along with lifetime hormonal control.
CYP21A2 Deficiency
The reactions catalyzed by the 21-hydroxylase enzyme, encoded by the CYP21A2 gene, include the conversion of 17-hydroxyprogesterone (also designated 17α-hydroxyprogesterone) to 11-deoxycortisol and the conversion of progesterone to 11-deoxycorticosterone. The 21-hydroxylase enzyme possesses two different substrate binding sites, one for 17-hydroxyprogesterone and one for progesterone. The synthesis of 11-deoxycortisol occurs within adrenal cortical cells of the zona fasciculata while synthesis of 11-deoxycorticosterone occurs in adrenal cortical cells of both the zona glomerulosa and the zona fasciculata.
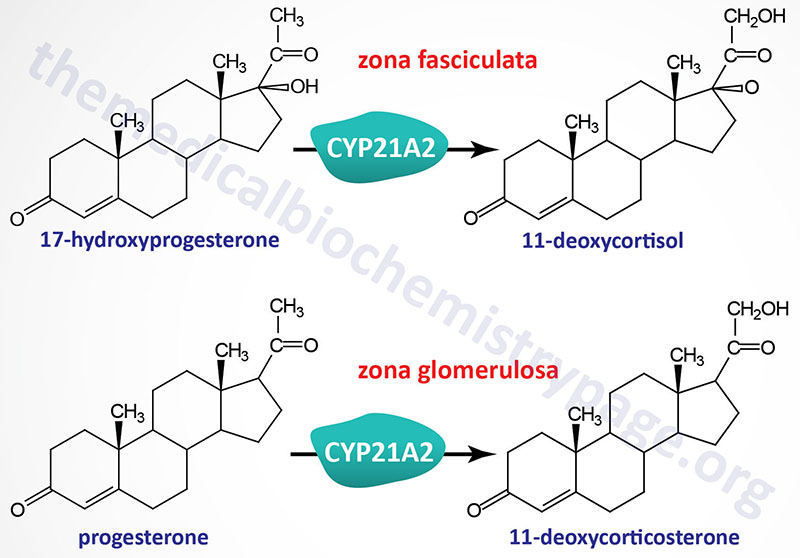
The forms of CAH that result from deficiencies in the CYP21A2 gene are inherited as autosomal recessive diseases. These forms of CAH represent the most commonly occurring forms (>95%) of the disease. The majority of the mutations in CYP21A2 that result in CAH have been identified and the severity of the disorder can be correlated to specific mutations and the consequent effect of the mutation on enzyme activity.
Molecular Biology of CYP21A2 Mutations
The 21-hydroxylase enzyme is encoded by the CYP21A2 gene (previously also identified as CYP21 or CYP21B). The CYP21A2 gene is located within the class III region of the major histocompatibility complex (MHC) on chromosome 6. There are in fact, two CYP21 genes within this MHC locus but one of the two is a pseudogene and is identified as CYP21A1P. Older nomenclature identifies the CYP21A1P gene as the 21-hydroxylase A gene (or CYP21A1) and CYP21A2 as the 21-hydroxylase B gene. All 21-hydroxylase activity is synthesized from the mRNA encoded by the CYP21A2 gene.
The CYP21A2 gene is located on chromosome 6p21.33, spans 3.35 kbp, and is composed of 10 exons that generate four alternatively spliced mRNAs that collectively encode three distinct protein isoforms.
Approximately 1300 variants in the CYP21A2 gene have been identified, with at least 460 affecting the coding region of the mRNA and 439 affecting 5′-UTR, 3′-UTR, or intronic DNA. Of these variants, at least 230 pathogenic mutations have been identified in the CYP21A2 gene. Pathogenic mutations include point mutations, small deletions, small insertions, and complex rearrangements of the gene. The pathogenic mutations in the CYP21A2 gene have been identified in all 10 exons of the gene.
The most common CAH causing mutations in the CYP21A2 gene are large deletions, large conversions, or one of several point mutations. The point mutations generate missense mutations in approximately 70% of the pathogenic variants. These point mutations result from unequal crossover between the CYP21A2 gene and a pseudogene, CYP21A1P, that is located 30 kbp from the CYP21A2 gene. The CYP21A2 and CYP21A1P genes share 98% sequence identity in their exons and 96% in their introns accounting for the likelihood of unequal crossover and gene conversion events. Indeed, 95% of pathogenic CYP21A2 mutations are the result of recombination events.
Mutations in CYP21A2 are classified according to the impact of the mutation on the activity of 21-hydroxylase. These classifications include Group Null, Group A, Group B, and Group C.
As the name of the group implies, group null mutations, which includes deletions and nonsense mutations, result in a complete loss of 21-hydroxylase function. The most common group null variants are the thirty kilobase deletion (30-kb del), the eight base pair deletion, the Q319X nonsense mutation, the L307fs mutation (fs is frame-shift), and the missense mutations that constitute what is referred to as Cluster E6 (I237N, V238E, and M240K). Homozygous or compound heterozygotes harboring mutations of the null group are often associated with the salt-wasting form of CAH. However, around 20% of cases have a simple virilizing phenotype.
Group A mutations result in 21-hydroxylase with 0%-1% of normal activity. Group A is represented by an intron variant IVS2-13A/C>G, that results in the retention of 19 nucleotides of intron 2.
Group B mutations result in 21-hydroxylase with 1%-10% of the normal activity. This level of 21-hydroxylase activity is sufficient to prevent adrenal crisis. The representative mutation in this group is the missense mutation, I173N. Group B CAH cases are associated with the simple virilizing form of CAH.
Group C mutations result in 21-hydroxylase with around 20%-60% of normal. These mutations are all associated with a mild form of CAH. The most common mutations in group C are the missense mutations, P31L, V282L, and P454S.
Deficiencies in CYP21A2 result in decreased secretion and plasma concentration of cortisol. The reduced levels of cortisol result in a reduction in the negative feedback exerted by this hormone on the hypothalamic-pituitary axis. The reduced negative feedback leads to increased secretion of corticotropin releasing hormone (CRH) and ACTH. The resultant high plasma concentrations of ACTH are responsible for the adrenocortical hyperplasia characteristic of this disorder.
CAH resulting from CYP21A2 deficiencies is divided into three distinct clinical presenting forms. The most severe enzyme impairment mutations result in the salt-wasting form. Females with this form of the disease present at birth due to ambiguity in the external genitalia. Males with the salt-losing form present with acute adrenal crisis shortly after birth or in early infancy. Females who present with no acute adrenal crisis and only mild masculinization of the external genitalia and males who present with virilism early in life are considered be suffering from the simple virilizing form of CAH. The final clinical form of CAH manifests only in females at puberty or shortly thereafter with symptoms of mild excess androgen production resulting in hirsutism, amenorrhea, and infertility. This form of the disorder is referred to as the attenuated form or the late onset or non-classic form.
Salt-wasting CYP21A2 deficiency
In the salt-wasting form of this disorder the degree of loss of enzyme activity is severe to complete as a result of mutations in the CYP21A2 gene that lead to greatly reduced or loss of both cortisol and aldosterone synthesis. Mutations in CYP21A2 that result in the salt-wasting form of CAH are of a variety of different types. Mutations have been identified that lead to loss of membrane association of the 21-hydroxylase, loss of, or disruption in, the heme and/or substrate binding sites, or impairment in structural stability of the 21-hydroxylase protein.
As a result of the level of enzyme deficiency the synthesis of cortisol is negligible. Because CYP21A2 is also needed for the synthesis of aldosterone, which is a major hormone involved in Na+ retention by the kidney, there is excessive salt loss leading to hyponatremic dehydration which can be fatal if not treated. The net result is an acute adrenal crisis. The near complete, or complete, loss in cortisol production results in loss of the normal cortisol-mediated feed-back inhibition of the hypothalamic-pituitary (CRH-ACTH) axis. The excess ACTH release from the pituitary leads to adrenal cortical hyperplasia and maximal adrenal androgen secretion with the result being masculinization of the female genitalia.
The first sign of CAH due to CYP21A2 deficiency is, in fact, the ambiguous female genitalia in neonates. The masculinization can be so extreme as to result in the fusion of the labia and the formation of a penile urethra. Diagnosis of the salt-losing form of this disorder is made in females born with masculinized genitalia, who are of normal 46,XX karyotype, with marked elevation in plasma 17α-hydroxyprogesterone and androstenedione, as well as serum chemistry showing hyponatremia, hypochloremia, and hyperkalemic acidosis.
The most commonly occurring mutations in CYP21A2 that result in the salt-wasting form of CAH are classified as the Null group mutations meaning there is little to no enzymatic activity in the resultant protein. This group of mutations includes, but is not limited to, the cluster of missense mutations in exon 6. Some of the most common missense mutations change an isoleucine for asparagine (I237N), a valine for glutamate (V238E), a methionine for lysine (M240K), and an arginine for tryptophan (R357W). In addition to these missense mutations, a glutamine for a stop codon (Q319X) and a frameshift at leucine 307 (L307fs) are also common. Homozygous or compound heterozygote variants of the Null group are often associated with the salt-wasting form, but approximately 20% of cases have the simple virilizing form.
Mutations that are associated with up to 1% residual 21-hydroxylase activity are classified as Group A mutations. The most common mutation in this group is an intron variant (IVS2-13A/C>G) which is created by an additional splice acceptor site causing retention of 19 intronic nucleotides of intron 2 of the CYP21A2 gene.
Simple virilizing CYP21A2 deficiency
In the simple virilizing form of the disorder the level of residual 21-hydroxylase activity is around 2%. The mutations in the CYP21A2 gene that result in the simple virilizing form affect the binding of 17α-hydroxyprogesterone and thus, the synthesis of cortisol, but aldosterone synthesis is retained. As a result the adrenal cortex can compensate for salt loss with increased aldosterone synthesis and release. In addition, although there is increased ACTH release there is near normal plasma cortisol levels and thus, there is no glucocorticoid deficit. However, as in the salt-losing form the CRH-ACTH axis is hyperactive leading to excessive adrenal androgen synthesis with consequent masculinization of the female genitalia.
The most common mutation in CYP21A2 causing the simple virilizing form of CAH is a missense mutation that converts isoleucine at position 172 to an asparagine (I172N).
Attenuated CYP21A2 deficiency
As the name of this form of disease implies, patients with the attenuated form exhibit only mild reductions in CYP21A2 activity, usually ranging from 20% to 70% of normal. Symptoms associated with this form of the disorder manifest in females at puberty. There are no signs of masculinization of the genitalia in females with this form of the disease. Although females have relatively normal breast development, the androgen excess results in excessive body hair (hirsutism), amenorrhea and the development of small ovarian cysts.
The most common mutations in the CYP21A2 gene that have been found to be associated with the attenuated from of CAH are missense mutations. One common mutation converts a proline for a leucine (P30L) and another converts a valine to a leucine (V281L).
CYP11B1 Deficiency
CAH due to deficiencies in 11β-hydroxylase (CYP11B1), although rare, are the second most commonly occurring forms of these disorders being found in approximately 5% of CAH patients. These forms of CAH are inherited as autosomal recessive diseases. CYP11B1 deficiency was originally identified as a hypertensive form of CAH.
Deficiency of CYP11B1 activity in the zona fasciculata results in reductions in cortisol and corticosterone synthesis. In addition, the loss of negative feedback on the hypothalamic-pituitary axis leads to increased ACTH release with the consequent increase in production of deoxycorticosterone (DOC; 11-deoxycorticosterone), 11-deoxycortisol, and 18-hydroxy DOC. The hypertension seen in this form of CAH is due to the increased secretion of DOC which is a weak mineralocorticoid activating the aldosterone receptor in the kidney (mineralocorticoid receptor) resulting in dysregulation in the renin-angiotensin-aldosterone system (RAAS).
Because there is hypersecretion of ACTH and a block in the normal pathway to corticosteroid synthesis there is an androgen excess similar to that seen in the CYP21A2 deficiency forms of CAH. As a result, females exhibit masculinized genitalia.
The CYP11B1 gene is located on chromosome 8q24.3 spanning 6.5kbp and composed of 9 exons that generate two alternatively spliced mRNAs encoding isoform 1 (503 amino acids) and isoform 2 (437 amino acids). Both isoforms encoded by the CYP11B1 gene include a 24 amino acid mitochondrial localization signal.
HSD3B2 Deficiency
CAH due to deficiencies in 3β-hydroxysteroid dehydrogenase (hydroxy-delta-5-steroid dehydrogenase, 3 beta- and steroid delta-isomerase 2) represent less than 1% of all cases. These forms of CAH are inherited as autosomal recessive diseases. When the deficiency in enzyme is severe, there is a near total absence of adrenal steroids. Thus, there are deficiencies in the glucocorticoids, mineralocorticoids, as well as adrenal androgens.
The impaired corticosteroid synthesis results in symptoms of adrenal insufficiency that can be fatal if not treated during the neonatal period. Several steroid precursors accumulate including pregnenolone and DHEA as well as their hydroxylated derivatives.
Females with this disorder will present with slight fusion of the labia and an enlarged clitoris, while males present with ambiguous genitalia. Correct diagnosis of this disorder can be made by measurement of the levels of DHEA, 17α-hydroxypregnenolone and 16α-hydroxy-DHEA in the urine and serum.
3β-hydroxysteroid dehydrogenase is encoded by the HSD3B2 gene. The HSD3B2 gene is located on chromosome 1p12 and is composed of 5 exons that generate two alternatively spliced mRNAs, both of which encode the same 372 amino acid protein.
POR Deficiency
Cytochrome P450 oxidoreductase (POR) is a flavoprotein that donates electrons to all microsomal P450 enzymes. In the context of adrenal steroidogenesis, POR functions as the electron donor for CYP17A1, CYP21A2, and CYP19A1. Deficiencies in POR are inherited as autosomal recessive diseases and result in ambiguous genitalia in both males and females.
Some of the skeletal malformations resulting from POR deficiency resemble Antley-Bixler syndrome (ABS), however ABS is not associated with disordered steroidogenesis. These skeletal malformations include midface hypoplasia, low set ears, craniosynostosis (premature fusion of the cranial sutures), choanal atresia (blockage in the back of the nasal passage), and fusion of the arm bones. CAH resulting from POR deficiencies are rare.
The POR gene is located on chromosome 7q11.23 and is composed of 19 exons that generate seven alternatively spliced mRNAs that collectively encode three distinct protein isoforms.
Placental Aromatase (CYP19A1) Deficiency
Deficiency in the CYP19A1 encoded aromatase is an extremely rare autosomal recessive disorder. The aromatase gene (also called estrogen synthetase) is expressed in ovaries, placenta, and extragonadal tissues such as adipose tissue, liver, brain, muscle, and hair follicles. The activity of the enzyme is to convert androgens to estrogens. During fetal development the fetal adrenal glands secrete DHEA-S as the substrate for placental estrogen production. The loss of CYP19A1 activity results in decreased placental estrogen production and increased androgen precursors. Female infants may have masculinized external genitalia.
The CYP19A1 gene is located on chromosome 15q21.2 spanning 70kbp and composed of 18 exons that generate eleven alternatively spliced mRNAs, all of which encode the same 503 amino acid enzyme.
CYP17A1 Deficiency
The CYP17A1 encoded enzyme catalyzes both the 17α-hydroxylase and 17,20-lyase (side-chain removal) reactions of adrenal steroidogenesis. Mutations in CYP17A1 are inherited in an autosomal recessive pattern. Similar to each of the above described CAH, the underlying clinical manifestations of CYP17A1 deficiency are due to the inability to produce normal levels of glucocorticoids with the consequences being a loss of the feedback inhibition of the hypothalamic-pituitary axis resulting in elevated ACTH secretion. However, symptoms in these patients are less severe than in other forms of CAH. Patients with 17α-hydroxylase deficiency do not make cortisol but do produce large amounts of corticosterone.
Corticosterone does bind the glucocorticoid receptor but with an affinity 1/100th that of cortisol. Deoxycorticosterone (DOC), which serves as the precursor to corticosterone, exhibits significant mineralocorticoid activity. This fact explains the hypertension exhibited in 17α-hydroxylase deficient patients. A deficiency in the 17,20-lyase activity of CYP17A1 leads to loss of C-18 and C-19 steroids from C-21 precursors leading to impaired production of androgens and estrogens. Affected males have defective genital development in utero and present with ambiguous external genitalia at birth. Development of female genitalia in utero does not require endogenous sex steroid production thus, affected females present at birth with normal external genitalia. However, female patients will present at puberty with infantile breasts, primary amenorrhea, absent or scant axillary and pubic hair, and hypogonadism.
The CYP17A1 gene is located on chromosome 10q24.32 and is composed of 8 exons encoding a 508 amino acid precursor protein. Deficiency in CYP17A1 is extremely rare with more than 90% of reported cases having deficiency in the both the 17α-hydroxylase and 17,20-lyase activities or just 17α-hydroxylase. The remainder of reported cases have deficiency in only the 17,20-lyase activity.
Lipoid CAH: StAR Deficiency
In order for cholesterol to be used for steroid hormone biosynthesis it must be transported from the outer mitochondrial membrane to the inner membrane. This transport process is mediated by steroidogenic acute regulatory protein (StAR). The transport of cholesterol to the inner mitochondrial membrane represents the rate-limiting step in steroidogenesis.
The StAR protein is encoded by the STAR gene. The STAR gene is located on chromosome 8p11.23 and is composed of 7 exons that encode a 285 amino acid protein. Expression of the STAR gene is exclusive to the adult adrenal glands, ovary, and testis with highest levels of expression within adrenal cortical cells.
Mutations in the gene (STAR) encoding steroidogenic acute regulatory protein are the primary causes of congenital lipoid adrenal hyperplasia (lipoid CAH), the most fatal form of adrenal hyperplasia. Lipoid CAH is a rare autosomal recessive disorder that severely inhibits the synthesis of all adrenal and gonadal steroids. Lipoid CAH is associated with disrupted adrenal and gonadal steroidogenesis as a result of defective conversion of cholesterol to pregnenolone.
Lipoid CAH patients exhibit significant salt loss due to impaired mineralocorticoid and glucocorticoid synthesis. Deficient fetal testicular steroidogenesis in patients with a 46,XY karyotype results in phenotypically female external genitalia. This phenotype is reflective of an absence of testosterone synthesis between 6 and 12 weeks of gestation. Most lipoid CAH patients have female external genitalia regardless of chromosomal sex and have evidence of salt loss, most often within the first 2 months of life.
Lipoid CAH infants have low but measurable levels of steroid hormones, but they will soon die from glucocorticoid and mineralocorticoid deficiency if hormone treatment is not initiated. Although StAR is essential for an acute and maximal steroidogenic response, there are also low levels of StAR-independent steroidogenesis. The demonstration of StAR-independent steroidogenesis led to the formulation of the two-hit model of lipoid CAH. The first hit results from mutations in the STAR gene which prevents StAR-dependent steroidogenesis. However, StAR-independent steroidogenesis will persist. This explains the low, but detectable, levels of steroid hormones in the blood of infants with lipoid CAH during the first months of life and which explains why these infants can survive for several months without treatment. In contrast, patients with other forms of salt wasting CAH will not survive for several months without intervention.
Despite the synthesis of low levels of steroid hormones in lipoid CAH patients, there is a failure to suppress the secretion of adrenocorticotropic hormone (ACTH), the gonadotropins, and angiotensin II. These hormones stimulate cellular uptake of LDL-cholesterol and increase production of cholesterol from acetate, resulting in the intracellular accumulation of cholesterol esters. The accumulating lipid droplets containing cholesterol esters will eventually destroy cells either via physical enlargement or by the accumulation of cholesterol oxidation products, or both. This process of cholesterol ester accumulation and cell death represents the second hit which, in turn, disrupts the low levels of StAR-independent steroidogenesis, leading to undetectable levels of steroid in older children with lipoid CAH.
Fetal ovaries do not express the genes encoding steroid hormone synthesis enzyme genes and, thus, do not make steroids. Unlike the testes and adrenal glands, the ovaries only start to make steroid hormones at the onset of puberty. Thus, the ovaries of 46,XX females affected with lipoid CAH do not receive the second hit until the onset of puberty, when luteinizing hormone stimulates low-levels of StAR-independent steroidogenesis. Each month another follicle is recruited and stimulated by gonadotropins, presenting spontaneous breast development in affected girls. However, gonadotropin stimulation quickly results in cholesterol accumulation in these cells (the second hit in lipoid CAH), so the later phase of ovarian steroidogenesis, which is the secretion of large amounts of progesterone, does not occur. Follicles that are not recruited remain unstimulated and constitute a reservoir of steroidogenic cells undamaged by the second hit of lipoid CAH, so a new undamaged follicle is recruited with each regular cycle, and estrogen is produced leading to cyclic uterine estrogen withdrawal bleeding that resembles a normal menstruation, but there is no progesterone, so these cycles are anovulatory.
Lipoid CAH has been reported in most ethnic groups but is common among the Japanese, Korean, and Palestinian Arab populations. To date, forty-eight different mutations in the STAR gene have been reported in various ethnic groups. The incidence of certain mutations is very high in specific ethnic groups. Genetic clusters consistently contain the Q258X mutation in the Japanese and Korean populations, the R182L mutation in Palestinian Arabs, the R182H mutation in eastern Saudi Arabians, and the L260P mutation in the Swiss population.
Treatment of CAH
In the treatment of CAH it is important to understand that, as discussed in detail in the Steroid Hormones and Their Receptors page, glucocorticoids not only activate the glucocorticoid receptor but can also activate the mineralocorticoid receptor. Likewise, mineralocorticoids not only activate the mineralocorticoid receptor but can also activate the glucocorticoid receptor. Indeed, as indicated below, the common synthetic mineralocorticoid used in the treatment of CAH is fludrocortisone which 10-15 times more potent as a glucocorticoid than is hydrocortisone, the short-acting glucocorticoid used in the treatment of infants with CAH.
The underlying theme for the treatment of CAH is normalization of hormone levels and prevention of adrenal overproduction of androgens. The mainstay of treatment involves glucocorticoid replacement. However, these therapies fail to replicate the normal circadian rhythms of normal cortisol synthesis and release. Oral hydrocortisone is the preferred glucocorticoid for use in children with CAH because of its short acting nature. In older children and adults with CAH intermediate- and long-acting glucocorticoids are used. The common intermediate-acting formulations include prednisone, prednisolone, and methylprednisolone. The long-acting glucocorticoid is dexamethasone
Mineralocorticoid replacement, for the correction of aldosterone deficiency is another important aspect to the treatment of CAH patients. Indeed, mineralocorticoid therapy is recommended in all patients with classic CAH. In the simple virilizing form of CAH there is minimal aldosterone production but the amount is insufficient to maintain normal vascular volume. By achieving optimum sodium balance there is reduced ACTH and vasopressin which allows for decreased dosing of glucocorticoid replacement therapy. The only currently available synthetic mineralocorticoid is fludrocortisone (9α-fludrocortisone). The correct dosing of fludrocortisone is determined by measurement of plasma renin levels as well as measurement of electrolytes and blood pressure. The kidneys of newborn babies have a physiological resistance to aldosterone which means that the dosing of mineralocorticoid therapy must be higher in these patients than in infant and older CAH patients.
During infancy, supplementation of breast milk or formula with sodium is common except in those patients receiving high doses of fludrocortisone. After the kidneys mature in the first year of life they are more sensitive to mineralocorticoids which leads to better sodium retention but also to the risk for hypertension. This means that the dosing of mineralocorticoid therapy must be altered as infants with CAH develop to prevent the potential for hypertension.