
Last Updated: April 9, 2026
Introduction to Bioactive Lipids and Their Receptors
Originally fats were generally considered mere sources of energy and as components of biological membranes. Aside from the eicosanoids (from Ancient Greek eíkosi for 20) few lipid-derived bioactive compounds had been characterized. However, research over the past 15-20 years has demonstrated a widely diverse array of biological activities associated with fatty acids and fatty acid derivatives as well as other lipid compounds.
Bioactive lipids span the gamut of structural entities from simple saturated fatty acids to complex molecules such as those derived from various omega-3 and omega-6 polyunsaturated fatty acids (PUFA) and those derived from sphingosine. Many bioactive lipids result from the activities of the various phospholipases (see below and in the Signal Transduction Pathways: Phospholipids page) and phospholipid kinases that are themselves activated by a variety of signal transducing receptors.
All bioactive lipids exert their effects through binding to specific receptors, many of which have just recently been characterized. The receptors for bioactive lipids include both the nuclear receptor class and the G-protein coupled receptor (GPCR) class. Bioactive lipids play important roles in energy homeostasis, cell proliferation, metabolic homeostasis, and regulation of inflammatory processes.
The scope of this page is not to discuss in detail all of the known activities of all of the known bioactive lipid compounds but to highlight several of the more important molecules with respect to disease and therapeutic potential. In addition, several classes of bioactive lipid, such as the eicosanoids, are discussed in more detail in separate pages.
Oxylipins
One of the largest families of bioactive lipids are the oxylipins. Oxylipins are derived from omega-6 and omega-3 PUFA as well as from the essential fatty acids. The various oxylipins are synthesized via the actions of cyclooxygenases (COX), lipoxygenases (LOX), and cytochrome P450 (CYP) enzymes.
The primary omega-6 PUFA is arachidonic acid but dihomo-γ-linolenic acid (DGLA), an intermediate in the de novo pathway of arachidonic acid synthesis, is also an important omega-6 PUFA involved in oxylipin synthesis. The omega-6 PUFA, adrenic acid, is another important precursor for bioactive oxylipin synthesis. The primary omega-3 PUFA are eicosapentaenoic acid (EPA) and docosahexaenoic acid (DHA). The essential fatty acids are linoleic acid (omega-6 PUFA) and α-linolenic acid (ALA; omega-3 PUFA).
The oxylipins include the prostaglandins, thromboxanes, leukotrienes, and lipoxins, the resolvins, the protectins (also called neuroprotectins when in the brain), the maresins, the mono-, di-, and tri-hydroxy fatty acids, the epoxy fatty acids, the eoxins (epoxyketones), and the hepoxilins (epoxyalcohols). The eoxins are 14,15-epoxy analogs of the leukotrienes.
Oxylipins Derived from Linoleic Acid
Linoleic acid is the essential fatty acid required for the synthesis of arachidonic acid with intermediates in this pathway including γ-linolenic acid (GLA) and dihomo-γ-linolenic acid (DGLA).
The oxylipins that are derived from linoleic acid include those generated via the actions of COX and those generated via the actions of CYP epoxygenases. The major products derived via the action of COX on linoleic acid include 9-hydroperoxyoctadecadienoic acid (9-HpODE) and 13-HpODE which are then converted to the corresponding hydroxyoctadecadienoic acid (HODE) via the action of glutathione peroxidase. Both 9-HODE and 13-HODE are converted to the oxo derivatives, 9-oxo-ODE and 13-oxo-ODE, respectively, via the actions of dehydrogenases. 9-HODE activates PPARγ in monocytes and induces endoplasmic reticulum (ER) stress in macrophages. 13-HpODE induces relaxation of pulmonary arteries.
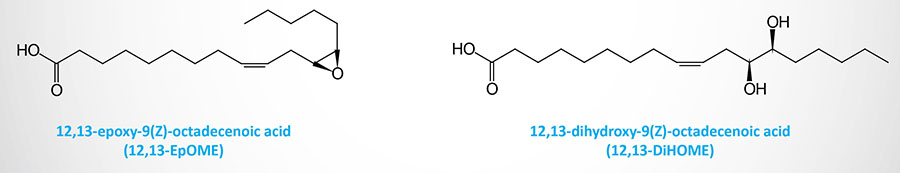
Epoxygenases of the CYP family convert linoleic acid to the epoxyoctadecenoic acid (EpOME)–derived oxylipins, 9,10-EpOME and 12,13-EpOME. Through the actions of soluble epoxide hydrolase (sEH) these molecules are converted to their dihydroxyepoxyoctadecenoic acid (DiHOME) derivatives.
The synthesis of the secreted lipokine, 12,13-dihydroxy-9Z-octadecenoic acid (12,13-diHOME), is increased in response to cold exposure and exercise. 12,13-diHOME functions as an autocrine factor for brown adipose tissue (BAT), enhances fatty acid uptake by BAT and skeletal muscle, and leads to a reduction in the level of circulating triglycerides.
Oxylipins Derived from α-Linolenic Acid (ALA)
Alpha-linolenic acid (ALA) is the essential acid that serves as the precursor for the synthesis of eicosapentaenoic acid (EPA) and docosahexaenoic acid (DHA). The LOX family enzymes and the CYP family expoxygenases convert ALA to various hydroxy fatty acids and keto fatty acids of the oxylipin superfamily.
Via the LOX pathway ALA can be converted to 9-hydroperoxyoctadecatrienoic acid (9-HpOTrE) or 13-HpOTrE. These lipids are then converted to their corresponding hydroxyoctadecatrienoic acids (HOTrE) and finally to their oxo-derivatives, 9-oxooctadecatrienoic acid (9-oxo-OTrE) and 13-oxo-OTrE.
The expogenases of the CYP family convert ALA to several epoxyoctadecadienoic acids (EpODE) that includes 9,10-EpODE, 12,13-EpODE, and 15,16-EpODE. Through the actions of soluble epoxide hydrolase (sEH) these EpODE molecules are converted to their corresponding dihydroxyoctadecatrienoic acids (DiHODE).
Oxylipins Derived from Dihomo-γ-Linolenic Acid (DGLA)
Via the action of COX enzymes DGLA is converted to the series 1 eicosanoids, PGD1, PGE1, PGF1α, PGI1, and TXA1. The significance of PGE1 and TXA1 is that they exert opposing effects to PGE2 and TXA2, respectively.
Lipoxygenase (LOX) activity on DGLA yields the 8-, 12-, and 15-hydroperoxyeicosatrienoic acid (HpETrE) which are then converted to the corresponding hydroxyeicosatrienoic acids (HETrE) via peroxidase action. The activity of 12(S)-LOX (encoded by the ALOX12 gene) generates, predominantly, 12(S)-HpETrE while 15-LOX (encoded by the ALOX15 gene) generates, predominantly, 15(S)-HpETrE. Both of these are then reduced, via peroxidase activity, to 12(S)-HETrE and 15(S)-HETrE, respectively. The formation of 8(S)-HpETrE involves the action of 5-LOX acting on γ-linolenic acid (GLA) followed by elongation in the ER and then conversion to 8(S)-HETrE.
The synthesis of 12(S)-HETrE in platelets functions in an autocrine manner by being released and activating a Gs-type GPCR whose signaling reduces platelet activation and thus the extent of thrombosis. The activity of 12(S)-HETrE is distinct from that of 12(R)-HETrE [12(R)-hydroxy-5,8,14-eicosatrienoic acid] which is produced in vascular endothelial cells from arachidonic acid.
CYP epoxygenases convert DGLA to the epoxyeicosadienoic acids (EpEDE) which are then converted, via the action of soluble epoxide hydrolase (sEH) to the dihydroxyeicosadienoic acids (DiHEDE). In humans, 8,9-EpEDE and 8,9-DiHEDE are the most well characterized functionally.
Oxylipins Derived from Arachidonic Acid
Classical Eicosanoids
The oxylipins generated from arachidonic acid include the more commonly recognized eicosanoids of the prostaglandin, thromboxane, and leukotriene families. In addition to the lipoxins, the synthesis and functions of these classical eicosanoids are described in detail in Eicosanoid Metabolism: Prostaglandins, Thromboxanes, Leukotrienes, and Lipoxins page as well as the Bioactive Lipid Mediators of Inflammation page.
Hydroxyeicosatetraenoic Acids (HETE)
The arachidonic acid derived hydroxyeicosatetraenoic acids (HETE) are a large family of molecules that includes mid-chain hydroxy and omega hydroxy derivatives. The omega HETE are those molecules where the hydroxyl group is added to carbon 19 (ω-1) or carbon 20 (ω).
The arachidonic acid-derived mid-chain HETE result from the action of lipoxygenase (LOX) family enzymes which first generate the intermediate oxylipins identified as hydroperoxyeicosatetraenoic acid (HpETE). For example, 5-LOX generates 5-HpETE. HpETE are rapidly converted to the HETE via the actions of glutathione peroxidases. The most common HETE in humans are 5-HETE, 12-HETE, and 15-HETE which are derived from the actions of 5-LOX, 12-LOX, and 15-LOX, respectively. The hydroxyl groups can exist in the R or S configuration, e.g. 12(R)-HETE or 12(S)-HETE. However, different isomers of HETE predominate in human tissues, for example 12(R)-HETE is more abundant than the 12(S)-HETE isomer.
5-HETE stimulates eosinophil and neutrophil chemotaxis, induces degranulation of neutrophils, and inhibits prostacylin (PGI2) production by coronary arterial endothelial cells.
12-HETE stimulates neutrophil chemotaxis and degranulation, enhances thrombin-induced platelet aggregation but reduces ADP-induced platelet aggregation.
15-HETE activates PPARγ, inhibits degranulation of activated polymorphonuclear (PMN) cells, and enhances thrombin-induced platelet aggregation.
Several arachidonic acid-derived HpETE are intermediates in the synthesis of the leukotrienes, the lipoxins, the hepoxilins, and the eoxins. LTA4 is derived from 5-HpETE and is then converted to LTB4 as well as the peptidoleukotrienes (LTC4, LTD4, and LTE4). The lipoxins, LXA4 and LXB4, are derived from LTA4 but can also be generated from 15-HpETE which is generated via the action of 15-LOX. The hepoxilins are derived from 12-HpETE. The eoxins are derived from 15-HpETE.
Mid-chain HETE are also generated via the actions of the CYP enzymes CYP1B1, CYP4A or CYP2B. These enzymes carry out their reactions at bis‑allylic centers and produce six regioisomeric (positional isomers) cis,trans-conjugated dienols. These dienols of arachidonic acid carry hydroxyl groups in positions 5, 8, 9, 11, 12, or 15.
Generation of the omega hydroxylated HETE is carried out primarily by CYP4A and CYP4F. The CYP1A1, CYP2C19, and CYP2E1 enzymes are involved in the (ω‑1) hydroxylation reactions. The ω-hydroxylated HETE include 16-, 17-, 18-, 19-, and 20-HETE. One of the most clinically relevant ω-hydroxylated oxylipins is 20-hydroxyeicosatetraenoic acid (20-HETE). 20-HETE exerts its effects by binding to the GPCR identified as GPR75.
Through the action of dehydrogenases the HETE can be converted to oxo-eicosatetraenoic acid (oxo-ETE). The oxylipin, 5-oxo-ETE is a potent chemoattractant for basophils and has been shown to function as a growth factor for certain types of cancer.
The LOX-derived HETE can be further modified by additional LOX activity generating the dihydroxyeicosatetraenoic acids (DiHETE). For example 5-LOX and 15-LOX, acting in any order will yield the oxylipin, 5,15-dihydroxyeicosatetraenoic acid (5,15-diHETE). The DiHETE of significance to humans are 5,15-DiHETE and 5,20-DiHETE which are derived from 5-HpETE as well as 5,12-DiHETE and 12,20-DiHETE which are derived from 12-HpETE, and 8,15-DiHETE which is derived from 15-HpETE. 5,15-DiHETE induces neutrophil and eosinophil chemotaxis and plays a central role in the activation of ferroptosis in adipose tissue. 8,15-DiHETE has chemotactic activity that is similar to that of LTB4.
Through the concerted actions of CYP family epoxygenases and soluble epoxide hydrolase (sEH), arachidonic acid is converted first to the epoxyeicosatrienoic acids (EET; also designated EpETrE) derivatives and then to the dihydroxy-eicosatrienoic acids (DiHETrE). One of the most important DiHETrE is 14,15-DiHETrE. This oxylipin has been shown to bind and activate PPARα and PPARγ.
Epoxyeicosatetraenoic Acids (EET)
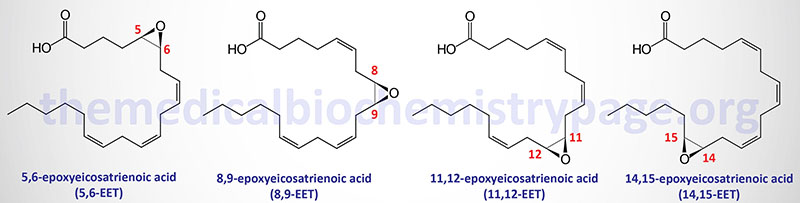
The arachidonic acid derived epoxy fatty acids are generated via the actions monooxygenases of the CYP2 family member enzymes with CYP2C and CYP2J being the most significant. These lipids are identified as epoxyeicosatrienoic acids (EET) and there are four principal cis-EET derived from arachidonic acid, 5,6-EET, 8,9-EET, 11,12-EET, and 14,15-EET. Due to the involvement of two carbon atoms to form the epoxide group in EET there are several different regioisomers and their abundance and function can be different. For example 11(R),12(S)-EET relaxes small renal arteries but 11(S),12(R)-EET does not.
Oxylipins Derived from Eicosapentaenoic Acid (EPA)
EPA is the substrate for the synthesis of the lipoxins as detailed in the Bioactive Lipid Mediators of Inflammation page.
In addition to the lipoxins, the actions of LOX family enzymes can convert EPA to several series 5 leukotrienes such as LTB5, LTC5, LTD5, and LTE5. In addition to the leukotrienes, the various LOX enzymes convert EPA to the hydroperoxyeicosapentaenoic acids (HpEPE) which are then converted to the hydroxyeicosapentaenoic acid (HEPE). 5-LOX generates 5(S)-HpEPE, 12-LOX generates 12(S)-HpEPE, and 15-LOX generates 15(S)-HpEPE, which are then converted to 5-HEPE, 12-HEPE, and 15-HEPE, respectively.
12-HEPE functions as a secreted lipokine whose synthesis in brown adipose tissue (BAT) is enhanced upon activation of β3-adrenergic receptors. When released, 12-HEPE stimulates glucose uptake by BAT and skeletal muscle. The 12-HEPE-induced glucose uptake involves the activation of an as yet unidentified Gs-type G-protein-coupled receptor (GPCR) that, when activated, stimulates the mobilization of GLUT4 transporters to the plasma membrane.
The COX1 enzyme converts EPA to the series 3 eicosanoids including PGD3, PGE3, PGF3α, PGI3, and TXA3. The COX2 enzyme that has been acetylated by low dose aspirin converts EPA to 18(R)-HpEPE which is then converted to 18(R)-HEPE. The actions of soluble epoxide hydrolase (sEH) convert 18-HEPE to three of the E series resovins, RvE1, RvE2, and RvE3. In addition to the resolvins, 18-HEPE can be converted to various dihydroxyeicosatetraenoic acids (DiHEPE) that includes 8,18-DiHEPE, 11,18-DiHEPE, and 12,18-DiHEPE.
Through the actions of the CYP epoxygenases, EPA is converted to various epoxyeicosatetraenoic acids (EpETE; some sources use the acronym: EETeTr) which are then converted to the corresponding dihydroxyeicosatetraenoic acids (DiHETE) via the actions of sEH. These DiHETE include 5,6-, 8,9-, 11,12-, 14,15-, and 17,18-DiHETE.
Oxylipins Derived from Docosahexaenoic Acid (DHA)
Bioactive lipid derivatives of DHA are extensive and many are described in greater detail in the Omega-3 & Omega-6 Fatty Acid Synthesis, Metabolism, Functions page. Bioactive DHA-derived oxylipins include the resolvins, protectins, and maresins, the fatty acid esters of hydroxy fatty acids (FAHFA), the acylglycerols, the neuroprostanes (NP), the neuroketals, the ethanolamines, the electrophilic oxo-derivatives (EFOX), the epoxides, and the N-acyl amides.
In addition to the FAHFA being discussed in the Omega-3 and Omega-6 Fatty Acid Synthesis, Metabolism, Functions page, with respect to the DHA derived FAHFA, this class of bioactive lipids is also discussed in the Fatty Acid Esters of Hydroxy Fatty Acids, FAHFA section below.
Additional DHA-derived oxylipins include the hydroxydocosahexaenoic acids (HDoHE) generated via the LOX pathway and the CYP ω-hydroxylase pathways, and the dihydroxydocosapentaenoic acids (DiHDPE) generated via the CYP epoxygenase pathways.
Oxylipins Derived from Omega-3 Docosapentaenoic Acid (DPAn-3)
Docosapentaenoic acid (DPA; designated 22:5). DPA can exist in both an omega-3 fatty acid form and an omega-6 fatty acid form. The omega-3 form (designated DPAn-3) is all-cis-7,10,13,16,19-docosapentaenoic acid (trivial name clupanodonic acid). DPAn-3 is formed from EPA. The omega-6 form (designated DPAn-6) is all-cis-4,7,10,13,16-docosapentaenoic acid (trivial name: osbond acid). DPAn-6 is formed from arachidonic acid.
The major oxylipins derived from DPAn-3 are D-series resolvins that are distinct from those derived from DHA. These D-series resolvins are designated RvDn-3DPA. The RvDn-3DPA molecules, in particular RvD5n-3DPA, repress the production of pro-inflammatory mediators and also modify leukocyte-derived activities which serve to limit tissue damage. In addition to the RvDn-3DPA molecules, DPAn-3 serves as the precursor for the synthesis of several members of the protectin family, PD1n-3DPA and PD2n-3DPA. These DPAn-3-derived protectins play a role in the coordination of the monocyte to macrophage differentiation process.
Oxylipins Derived from Adrenic Acid
Elongation of arachidonic acid, via the actions of either ELOVL2 or ELOVL5, generates the 22 carbon (22:4) omega-6 PUFA identified as all-cis-7,10,13,16-docosatetraenoic acid (DTA; also known as adrenic acid, AdA). Docosatetraenoic acid is called adrenic acid due to its abundance in the adrenal glands.
Adrenic acid is also found at high levels in the brain, kidney, and vasculature. Similar to the metabolism of arachidonic acid, the COX, LOX, and CYP enzymes convert adrenic acid into numerous bioactive lipid molecules of the oxylipin superfamily.
Cyclooxygenase converts adrenic acid into dihomo-PGH2 which is the precursor for the synthesis of numerous dihomo-eicosanoids. These dihomo-eicosanoids include dihomo-PGE2, dihomo-PGI2, dihomo-PGF2α, dihomo-TXA2 and dihomo-TXB2, and dihomo-PGI2.
Lipoxygenases metabolize adrenic acid into the dihomo-hydroperoxydocosatetraenoic acids (dihomo-HpETE). 5-LOX synthesizes dihomo-7-HpETE, 12-LOX synthesizes dihomo-14-HpETE, and 15-LOX synthesizes dihomo-17-HpETE. The dihomo-HpETE are rapidly converted to dihomo-hydrodocosatetraenoic acids (dihomo-HETE). There are numerous dihomo-HETE with dihomo-13-HETE and dihomo-17-HETE being the most well studied. The dihomo-HETE can be further metabolized to the corresponding dihydroxy compounds such as dihomo-10,17-DiHETE derived from dihomo-17-HETE.
Metabolism of adrenic acid via the CYP pathway yields the dihomo-epoxyeicosatrienoic acids (dihomo-EET). Dihomo-EET are also identified as dihomo-epoxyeicosatrienoic acids (dihomo-EpETrE). The dihomo-EET can be further converted to their respective dihomo-dihydroxyeicosatrienoic acids (dihomo-DiHETrE) such as dihomo-16,17-DiHETrE. The dihomo-DiHETrE are also identified as dihomo-DHET.
Synthesis of the dihomo derivatives of adrenic acid occurs in renal medullary zona glomerulosa cells (dihomo-prostaglandins and dihomo-thromboxanes), platelets (dihomo-thromboxanes and dihomo-HETE), and vascular endothelial cells (dihomo-prostacyclin: dihomo-PGI2).
Adrenic acid metabolites have been shown to induce concentration-dependent and endothelium-dependent relaxation of coronary arteries. This effect can be blocked by inhibition of potassium channels as well as by inhibition of the synthesis of adrenic acid metabolites by the COX and CYP pathways. The principal metabolites of adrenic acid that induce these effects and that are synthesized by coronary arteries include dihomo-prostaglandins and dihomo-epoxyeicosatrienoic acids (dihomo-EET). With respect to the dihomo-EET, the greatest effect is exerted by dihomo-16,17-EET which is the dihomo analog of 14,15-EET. This compound relaxes the vasculature by activation of smooth muscle calcium-activated potassium channels (BKCa channels) thereby inducing hyperpolarization.
Adrenic acid can be converted to the omega-6 version of docosapentaenoic acid (22:5n-6; DPAn-6; trivial name: osbond acid) via the Δ4 desaturase activity of the FADS2 encoded desaturase. The formation of DPAn-6 can also be accomplished through a series of reactions involving elongation, desaturation and β-oxidation. These three reactions convert adrenic acid to tetracosatetraenoic acid (24:4n-6) via the ELOVL2 enzyme, then the Δ6 desaturase activity of the FADS2 encoded desaturase generates tetracosapentaenoic acid (24:5n-6), and finally β-oxidation generates DPAn-6.
Elovanoids
The elovanoids are a novel class of bioactive lipids that were originally characterized and named in 2017. The elovanoids are derived from very long-chain fatty acids (VLCFA), predominantly from omega-3 VLC-PUFA. The VLCFA that have more than 30 carbon atoms are often referred to as ultra long-chain fatty acids, ULCFA. The synthesis of VLCFA is the function of the enzyme encoded by the ELOVL4 gene.
ELOVL4 converts the omega-3 PUFA DHA (22:6n-3) or EPA (20:5n-3) into VLC-PUFAn-3 in the brain, retina, testes, and skin. In the skin, the predominant VLCFA are saturated fatty acids (VLCSFA). The VLC-PUFA are predominantly incorporated at the sn-1 position of phosphatidylcholines (PC). Within the skin the VLCSFA are incorporated into sphingolipids which form a critical part of the permeability barrier of the skin.
Specifically, the elovanoids are the dihydroxylated derivatives of 32:6n-3 and 34:6n-3. These elovanoids are designated ELV-N32 [20(R),27(S)-dihydroxy 32:6n-3] and ELV-N34 [22(R),29(S)-dihydroxy 34:6n-3]. Both ELV-N32 and ELV-N34 exert neuroprotective effects. The mechanisms by which the elovanoids exert their identified effects is as yet incompletely resolved as no distinct and specific receptor(s) for the elovanoids have been identified.
Lipid Epoxides
Polyunsaturated fatty acids (PUFA) are oxidized to epoxides by epoxygenases that are members of the large cytochrome P450 family of enzymes. The action of these epoxygenases generate numerous potent bioactive lipid mediators. The epoxygenase pathway primarily involves enzymes of the CYP2C and CYP2J families although the CYP1A family members, CYP1A1 and CYP1A2, also function as a lipid epoxygenases.
The significance of these many lipid epoxides is that they regulate vascular tone, modulate angiogenesis, have anti-inflammatory actions, and protect the heart against ischemia–reperfusion injury, are potent inhibitors of cardiac arrhythmias, modulate inflammatory and neuropathic pain, and reduce tumor metastasis.
With respect to the metabolism of arachidonic acid, related eicosanoids, and other bioactive lipids, the CYP2C8 and CYP2C9 enzymes are the most significant. CYP2C8 functions as an expoxygenase to convert long-chain polyunsaturated fatty acids (PUFA) such as linoleic acid (18:2), arachidonic acid (20:4), and the physiologically significant omega-3 PUFA, eicosapentaenoic acid (EPA; 20:5) and docosahexaenoic acid (DHA; 22:6) to their biologically active epoxide forms.
CYP epoxygenases also convert the ethanolamide (EA) derivatives of EPA and DHA to epoxides. The CYP epoxygenases are also involved in the formation of anandamide (an endocannabinoid) containing either an 11,12-epoxide or a 14,15-epoxide.
Linoleic acid (9,12-octadecadienoic acid) derived epoxides are epoxyoctadecamonoenoic acids (EpOME).
Arachidonic acid derived epoxides are epoxyeicosatrienoic acids (EET; also designated EpETrE).
EPA derived epoxides are epoxyeicosatetraenoic acids (EpETE).
DHA derived epoxides are epoxydocosapentaenoic acids (EpDPE; these are sometimes designated EpDPA).
The epoxides can exist as either R,S or S,R enantiomers. For example the linoleic acid derived EpOME are 9,10-epoxyoctadecenoic acid (9,10-EpOME; coronaric acid, also known as leukotoxin), 12,13-epoxyoctadecenoic acid (12,13-EpOME; vernolic acid, also known as isoleukotoxin) and these can be 9R,10S-EpOME or 9S,10R-EpOME and 12R,13S-EpOME or 12S,13R-EpOME.
The principal EETs formed from arachidonic acid are 5,6-EET, 8,9-EET, 11,12-EET, and 14,15-EET. These R,S or S,R enantiomers result in a total of eight regioisomers (positional isomers) of EETs. These EETs are primarily produced in endothelial cells but expression of the CYP2C9 and CYP2J2 genes is found in astrocytes, cardiac myocytes, and monocytes. CYP2C9 is also an important epoxygenase generating EET from arachidonic acid. The EETs exert effects on the processes of angiogenesis, cellular proliferation, inflammation, nociception (pain), and myocardial preconditioning. For more details on the formation and actions of the EET go to the Eicosanoid Metabolism: Prostaglandins, Thromboxanes, Leukotrienes, and Lipoxins page.
The CYP epoxygenases preferentially oxidize the ω-3 carbon-carbon double bond in both EPA and DHA. As a result of this preference the primary EPA derived EpETEs are 17,18-EpETEs (predominantly 17S,18R-EpETE) while the primary DHA derived EpDPEs are 19,20-EpDPEs with the 13,14-EpDPEs also being prevalent. The major CYP2J2 derived EpETE from EPA is 17,18-EpETE and the major EpDPE derived from DHA is 19,20-EpDPE.
The EPA and DHA derived epoxides exhibit potent anti-arrhythmic actions and they protect the heart from ischemia–reperfusion injury. These cardiac effects of the EpETE and EpDPE are more potent than those exerted by EET. The EpETE and EpDPE also exert an analgesic effect against neuropathic pain and inflammation-induced pain. The DHA derived epoxides inhibit angiogenesis and reduce tumor metastasis.
Epoxide Hydrolases
Humans express four epoxide hydrolase genes, identified as EPHX1-EPHX4, that encode enzymes of the α/β hydrolase fold (ABHD) enzyme family. The EPHX2 encoded enzyme is commonly called soluble epoxide hydrolase (sEH). Soluble epoxide hydrolase (sEH) is predominantly found in the cytosol and is highly expressed in the liver.
The EPHX2 gene is located on chromosome 8p21.2–p21.1 and is composed of 19 exons that generate 12 alternatively spliced mRNAs encoding 11 distinct protein isoforms. The EPHX2 encoded enzyme possesses two independently functioning domains. The N-terminal domain is a phosphatase and the C-terminal domain harbors the epoxide hydrolase activity. The functional sEH enzyme is a homodimer.
Epoxide hydrolases convert lipid epoxides to their corresponding diols which can attenuate the function of the lipid epoxides. For example sEH activity on the linoleic acid derived 9,10-EpOME converts them to 9,10-dihydroxyoctadecamonoenoic acid (9,10-DiHOME). The arachidonic acid derived EET are converted to dihydroxyeicosatrienoic acids (DHET) by sEH. The EPA-derived EpETE are converted to dihydroxyeicosatetraenoic acids (DiHETE) by sEH. The DHA-derived EpDPE are converted to diyhdroxydocosapentaenoic acid (DiHDPE) by sEH.
Inactivating mutations in the EPHX2 gene that encodes sEH, as well as pharmacologic inhibition of sEH, is associated with attenuation of the development of various diet-induced disorders, including endoplasmic reticulum (ER) stress, metabolic syndrome, insulin resistance, hepatic steatosis, hepatic fibrosis, inflammation, and endothelial dysfunction.
The EPHX1 encoded enzyme (commonly called microsomal epoxide hydrolase, mEH) and EPHX3 encoded enzyme (also identified as ABHD9) also carry out the metabolism of PUFA-derived epoxides.
Fatty Acids and Bioactive Lipid-Sensing GPCR
Numerous receptors of the GPCR family have been shown to be activated by fatty acids and fatty acid derivatives. As discussed in detail in the Eicosanoid Metabolism: Prostaglandin, Thromboxane, Leukotriene, and Lipoxin page, the receptors for these bioactive eicosanoids belong to the GPCR family.
During the course of the human genome project numerous genes were characterized whose encoded proteins had structures similar to the well characterized G-protein coupled receptor (GPCR) family of proteins. However, when initially identified, the ligands for these novel GPCR were unknown. At the time these receptors were referred to as orphan GPCR and given the designation GPR with a number such as GPR40. Following their initial isolation and characterization several orphan GPCR were shown to bind and be activated by free fatty acids and/or lipid molecules.
In a search for novel galanin receptor subtypes a group of three tandemly encoded intronless genes was identified on chromosome 19q13.1 downstream of the CD22 gene. These three orphan GPCR were identified as GPR40, GPR41, and GPR43. Subsequent to their isolation and characterization GPR40 was shown to bind and be activated by medium- and long-chain free fatty acids, whereas, GPR41 and GPR43 were shown to be activated by short-chain free fatty acids.
These three lipid-sensing receptors are encoded by the free fatty acid receptor 1 (FFAR1: GPR40), FFAR2 (GPR43), and FFAR3 (GPR41) genes. The FFAR3 gene is located immediately downstream of the FFAR1 gene and recent evidence has shown that bicistronic mRNA is generated by transcription through both genes. The translation of FFAR3 protein from this bicistronic mRNA is accomplished via the internal ribosome entry site (IRES) mediated process. Another orphan GPCR that was subsequently shown to be activated by free fatty acids is GPR120 (described in greater detail below) which is encoded by the FFAR4 gene.
Metabolite- and Lipid-Sensing Receptors
GPR18 was originally discovered as an orphan receptor (i.e. no known ligand) abundantly expressed in the spleen and testis. GPR18 is a member of the Class A orphan receptor family of GPCR. GPR18 is now known to be activated by several different naturally occurring molecules including bioactive lipids, specifically resolvin D2 (RvD2). As such it has been proposed that GPR18 be renamed DVR2 for D resolvin receptor 2. In addition to RvD2, GPR18 is activated by binding N-arachidonylglycine (NAGly), a metabolite of the endocannabinoid, anandamide.
GPR31, also known as the 12(S)-HETE receptor 1 (12-HETER1), has been shown to be a high affinity receptor for the arachidonic acid derivative, 12(S)-hydroxyeicosatetraenoic [12(S)-HETE]. The synthesis of 12-HETE is catalyzed via the action of 12-lipoxygenase (12-LOX) on arachidonic acid. Ligand binding to GPR31 results in the activation of a MAP kinase signaling pathway involving MEK and ERK1/2. In addition to 12(S)-HETE, evidence suggests that GPR31 is also activated by TCA cycle intermediates. GPR31 has been shown to be involved in immune system regulation and in the progression of several different cancers.
GPR32 was originally discovered as an orphan receptor expressed in macrophage lineage cells. GPR32 is a member of the Class A orphan receptor family of GPCR. GPR32 has also been shown to be activated by molecules of the D resolvin family, specifically RvD1, RvD3, and RvD5. As such it has been proposed that GPR32 be renamed DVR1 for D resolvin receptor 1. GPR32 is also activated by aspirin-triggered D resolvins.
GPR37 has also been identified as the parkin-associated endothelin receptor-like receptor (PaelR). GPR37 is a member of the Class A orphan receptor family of GPCR. Expression of GPR37 is highest in the CNS within the amygdala, basal ganglia, substantia nigra, hippocampus, frontal cortex, and hypothalamus, with exceptionally high levels of expression observed in the spinal cord. Activation of GPR37 has been associated with states of chronic inflammation such as in neurodegenerative disorders and with most cancers. GPR37 has been shown to signal via a Gi-type G-protein. Although its high level expression within the CNS suggests that the ligand(s) for the receptor may be a neuropeptide, this has yet to be determined. However, recent experiments indicate that a natural ligand for GPR37 is the bioactive lipid, protectin D1.
GPR44 was originally identified as an orphan GPCR but was subsequently shown to bind and be activated by prostaglandin D2 (PGD2). The gene encoding the originally identified GPR44 protein is now termed PTGDR2 and represents one of two genes encoding receptors for prostaglandin D family lipids. The other gene in this family of prostaglandin D receptors is PTGDR1. The protein encoded by the PTGDR1 gene is commonly identified as DP1 and the protein encoded by the PTGDR2 gene is commonly identified as DP2. In addition to PGD2, the DP2 receptor binds the non-enzymatic metabolite of PGD2 identified as 15-deoxy-Δ(12,14)-prostaglandin J2 (15-dPGJ2).
GPR65 is one of four identified proton-sensing GPCR that were originally classified as orphan GPCR. GPR65 is a member of the Class A orphan receptor family of GPCR. GPR65 was originally identified in apoptotic thymocytes and subsequent studies have shown that the highest levels of GPR65 expression are in immune cells. GPR65 is also referred to as T-cell death-associated gene 8 (TDAG8). GPR65 appears to signal via a Gi-type G-protein.
GPR68 is one of four identified proton-sensing GPCR that were originally classified as orphan GPCR. GPR68 is a member of the Class A orphan receptor family of GPCR. GPR68 is also referred to as ovarian cancer G protein-coupled receptor 1 (OGR1). GPR68 appears to signal via a Gq-type G-protein.
GPR81 (now identified as HCA1; encoded by the HCAR1 gene) is a metabolite responsive GPCR that is activated by L-lactate but poorly, if at all, by D-lactate.
GPR84 was identified as an orphan GPCR in a screen of differentially expressed genes in granulocytes. GPR84 is a member of the Class A orphan receptor family of GPCR. GPR84 appears to be a Gi-type G-protein coupled receptor. Natural ligands for GPR84 are the medium-chain fatty acids (MCFA) fatty acids decanoic (capric) acid (C10:0) and dodecanoic (lauric) acid (C12:0). Expression of the GPR84 gene is upregulated in numerous cancers of hematopoietic origin such as leukemias.
GPR91 (SUCNR1) is activated by succinate and as a result is now identified as the succinate receptor 1 and is encoded by the SUCNR1 gene. The SUCNR1 encoded protein was at one time thought to be a member of the P2Y receptor family. SUCNR1 is coupled to both Gi– and Gq-type G-proteins. SUCNR1 has been shown to be essential for tumor metastasis in patients that harbor mutations in one or more genes encoding the subunits of succinate dehydrogenase.
GPR92 has been shown to be activated by farnesyl pyrophosphate (FPP), N-arachidonoylglycine (NAGly), and lysophosphatidic acid (LPA). The binding of LPA to GPR92 prompted the receptor to be designated as LPAR5 and the gene encoding GPR92 is the LPAR5 gene. The activation of LPAR5 in macrophages present in pancreatic islets is involved in the control of β-cell function, particularly in a setting of high fat diet-induced obesity.
GPR99 is activated by 2-oxoglutarate (α-ketoglutarate) and as a result is now identified as 2-oxoglutarate receptor 1 and is encoded by the OXGR1 gene. The OXGR1 encoded protein was also identified as GPR80 and at one time was also thought to be a member of the P2Y receptor family (P2RY15). GPR99 has also been shown to be activated by cysteinyl leukotriene and as such is also identified as CysLTR3.
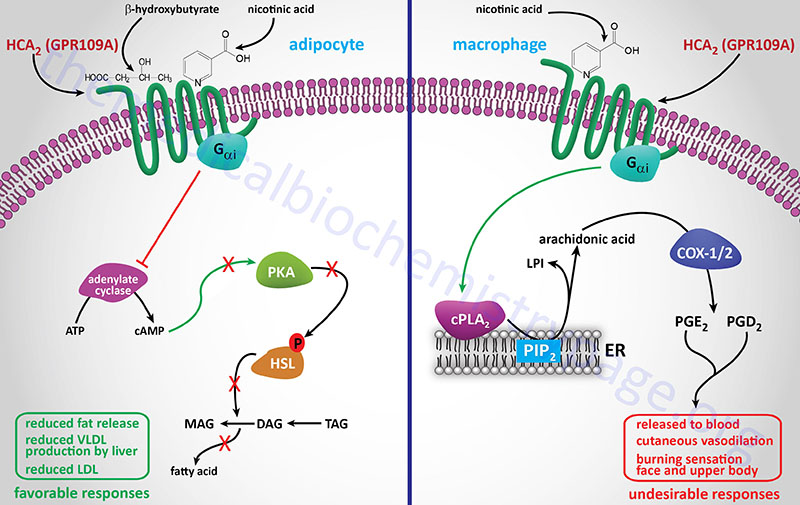
GPR109A (now identified as HCA2; encoded by the HCAR2 gene) is activated by the ketone, β-hydroxybutyrate (BHB). HCA2 within the gastrointestinal system binds gut microbiota-derived butyrate. As indicated below GPR109A (HCA2) was originally identified as being a receptor for nicotinic acid.
GPR119 and GPR120 (FFAR4) were identified as a result of the human genome sequencing project and were shown to be members of the class A (rhodopsin-like) family of GPCR.
GPR131, which was also previously identified as TGR5, is now identified as G-protein coupled bile acid receptor 1 (encoded by the GPBAR1 gene). The GPBAR1 encoded protein represents one of the several receptors activated by bile acids. GPBAR1 appears to be coupled to a Gs-type G-protein.
GPR132 is another receptor for lactate that, like HCA1, is involved in lactate-mediated modulation of immune responses. GPR132, which is also identified as G2A (G2 accumulation), was originally identified as a proton-sensing receptor. GPR132 is a member of the Class A orphan receptor family of GPCR. GPR132 is coupled to a Gq-type G-protein that leads to the activation of PLCβ which when active hydrolyzes membrane localized phosphatidylinositol-4,5-bisphosphate (PIP2; PtdIns-4,5-P2) generating inositol-1,4,5-trisphosphate (IP3; Ins-1,4,5-P3). The resulting IP3 binds to receptors in the endoplasmic reticulum (ER) resulting in release of stored Ca2+. GPR132 has also been shown to be associated with a Gs-type G-protein that activates adenylate cyclase leading to increased production of cAMP with subsequent activation of PKA. Additional ligands for GPR132 include oxidized fatty acids such as the glycine amide, N-palmitoylglycine (PalGly), and 9-hydroxyoctadecadienoic acid (9-HODE).
GPR170 is now identified as the oxoeicosanoid receptor 1 which is encoded by the OXER1 gene. OXER1 is the high affinity receptor for the arachidonic acid derived metabolites generated via the actions of 5-lipoxygenase (5-LOX). The most potent ligand is 5-oxo-eicosatetraenoic acid (5-oxo-ETE) but the receptor also binds, and is activated by other structurally related lipids such as 5-hydroxyeicosatetraenoic acid (5-HETE) and 5-hydroperoxyeicosatetraenoic acid (5-HpETE). The OXER1 receptor is coupled to the activation of a Gi-type G-protein where release of the Gβγ subunits is primarily responsible for the signaling pathways activated by the receptor.
GPR34 (LPS1)
GPR34 is a member of the class A GPCR family (rhodopsin-like) and belongs to the P2Y subfamily (specifically the P2Y12-like subgroup) of GPCR to which other emerging newly identified lysophospholipid receptors, such as LPA4 (P2Y9 or GPR23), LPA5 (GPR92) and LPA6 (P2Y5) belong. GPR34 is a Gi-type G-protein coupled receptor.
The natural ligand for GPR34 has been shown to be lysophosphatidylserine (lysoPS) which is the product of the action of phosphatidylserine (PS)-specific PLA1 (PS-PLA1). The PLA1 family of lipases is discussed in the Signal Transduction Pathways: Phospholipids page. In addition to PS-PLA1, an enzyme of the secreted PLA2 (sPLA2) family can also generate lysoPS by removal of the fatty acid at the sn-2 position of PS. Cells of the immune system, specifically dendritic cells, macrophages, and microglia, express GPR34 and control of its expression correlates with immune function. Activation of GPR34 by lysoPS is greatest when there is a fatty acid on the sn-2 position, a 1-acyl-lysoPS. Activation of GPR34 has been shown to play an important role in the activation of a subset of innate lymphoid cells (ILC) that reside in the epithelial tissues of the lamina propria of the gut.
In addition to GPR34, lysoPS has been shown to activate two additional GPCR, P2Y10 and GPR174. These three lysoPS receptors have been proposed to be identified as LPS1 (GPR34), LPS2 (P2Y10), and LPS3 (GPR174),
The GPR34 gene is located on the X chromosome (Xp11.4) and is composed of 3 exons that generate two alternatively spliced mRNAs both of which encode the same 381 amino acid protein.
GPR35
GPR35 is a member of the class A (rhodopsin-like) GPCR family . GPR35 appears to be a Gi-type G-protein coupled receptor. GPR35 was first described to be activated by kynurenic acid which is an intermediate in tryptophan catabolism that has neurotransmitter activity as an anti-excitotoxic and anticonvulsant. Additional studies indicated that the endogenous ligand for GPR35 was the phospholipid, 2-arachidonyl lysophosphatidic acid. Recently GPR35 has been shown to be the receptor for the chemokine (C-X-C motif) identified as CXCL17 and as such it has been suggested that GPR35 be identified as CXCR8 (CXC motif chemokine receptor 8).
The emerging function of GPR35 demonstrates that it may be an important target involved in pain, heart disease, inflammatory bowel disease (IBD), cancer, and asthma. One significant pathway activated in response to GPR35 activation is the hypoxia inducible factor (HIF) pathway.
The GPR35 gene is located on chromosome 2q37.3 and is composed of 7 exons that generate four alternatively spliced mRNAs that encode two distinct protein isoforms. The major GPR35 protein (identified as GPR35 isoform a) is 309 amino acids in length. The splice variant (identified as GPR35 isoform b) was originally found in gastric cancer cells and contains an additional 31 amino acids on the N-terminus.
Expression of GPR35 is seen at highest levels in the stomach, small intestine, and colon. Expression, albeit at lower levels than in the GI, is seen in lung, uterus, spinal cord, and several types of white cells including basophils, eosinophils, mast cells, peripheral monocytes, and macrophages.
Free Fatty Acid Receptor 1: FFAR1 (GPR40)
Free fatty acid receptor 1 (FFAR1) was originally identified as an orphan GPCR called GPR40. FFAR1 is coupled to a Gq-type G-protein that activates PLCβ upon ligand binding to the receptor. FFAR1 is abundantly expressed in pancreatic β-cells and is also found in the gut in enteroendocrine cells.
The preferred ligands for FFAR1 are medium to long-chain saturated fatty acids (C12–C16) as well as unsaturated fatty acids (C18–C22) such as the important omega-3 fatty acid docosahexaenoic acid, DHA. The thiazolidinedione class of drugs, used to treat type 2 diabetes, have also been shown to bind and activate FFAR1.
The activation of FFAR1 in pancreatic β-cells results in increased cytosolic Ca2+ via IP3-mediated release from the ER. The increased cytosolic Ca2+ can depolarize the β-cell leading to an influx of additional Ca2+ leading to increased secretion of insulin. This is an important mechanism by which fatty acids enhance glucose-stimulated insulin secretion (GSIS).
The FFAR1 gene is located on 19q13.12 and is composed of 2 exons encoding a protein of 300 amino acids. Each of the GPR40 family member genes (including the FFAR2 and FFAR3 genes) is clustered in this region of chromosome 19.
A synthetic agonist for FFAR1 called TAK-875 (fasiglifam; developed by Takeda Pharmaceuticals, Ltd.) was in Phase III clinical trials as a potentially useful orally active antidiabetic drug demonstrating little or no risk of hypoglycemia. Patients in the Phase II trials of fasiglifam showed significant reductions in HbA1c compared to placebo while hypoglycemia was similar to the placebo treated control group. However, during the phase III trials, it was determined that the benefits of fasiglifam did not outweigh the risks for the development of liver toxicity. Thus, trials of this drug were terminated by Takeda.
Overexpression of FFAR1 is associated with breast and hepatic cancers. Excess oleic acid has been found to contribute to the invasiveness of the hepatocellular carcinoma (HCC) through its ability to activate FFAR1.
FFAR2 (GPR43) and FFAR3 (GPR41)
FFAR2 is coupled to the activation of both Gq– and Gi-type G-proteins. FFAR3 is coupled to the activation of a Gi-type G-protein. FFAR2 and FFAR3 are activated by short-chain fatty acids (SCFA) such as acetate, propionic acid, butyric acid, and pentanoic acid. Both of these receptors are expressed at highest levels in adipose tissue and immune cells but are also found expressed in enteroendocrine cells of the gut. The activation of FFAR2 and FFAR3 is involved in adipogenesis and the production of leptin by adipose tissue.
In the gut, FFAR2 and FFAR3 are involved in responses to SCFA, particularly propionate, derived from gut microbiota metabolism of complex carbohydrates. Intestinal FFAR2 also binds intestinal microbiota-derived acetate.
The FFAR2 and FFAR3 genes are clustered on chromosome 19q13.12 along with the FFAR1 gene. The FFAR2 gene is composed of 5 exons that generate two alternatively spliced mRNAs, both of which encode a 330 amino acid protein. The FFAR3 gene is composed of 2 exons that encode a 346 amino acid protein.
Intestinal FFAR3 plays a critical role in energy homeostasis and as well as control of feeding behaviors through the activated release of gut hormones such as GLP-1 and PYY. Experiments with mice have shown that animals colonized in a sterile environment (i.e. free of gut microbiota) are resistant to high-fat diet (HFD)-induced obesity. However, when the guts of these sterile mice are colonized with saccharolytic bacteria from non-sterile mice they will become obese even on a diet of standard lab chow. However, in sterile FFAR3 knock-out mice this effect of saccharolytic bacteria colonization is ablated. In addition, the normal increase in PYY secretion upon bacterial colonization is also significantly reduced in FFAR3 knock-out mice.
GPR55
GPR55 was initially cloned as an orphan GPCR from human striatum. GPR55 is a member of the class A (rhodopsin-like) GPCR family and is coupled to the activation of both Gq and G12/13-type G-proteins. Expression of GPR55 is highest in certain regions of the brain, followed by the GI system, adrenal glands, testis, and endothelial cells, as well as being associated with numerous cancers. GPR55 was initially suggested to be a cannabinoid receptor for cannabinoid and endocannabinoid responses that are not mediated by the classical cannabinoid receptors; CB1 and CB2.
Despite the fact that certain endocannabinoids, phytocannabinoids, and synthetic cannabinoids can act as GPR55 agonists or antagonists, the most potent GPR55 agonist characterized to date is 2-arachidonoyl lysophosphatidylinositol (2-ALPI). More detailed information on the biological activities of various lysophospholipids (LPL) can be found below in the Lysophospholipids section.
Another potential ligand for GPR55 is the fatty acid amine, palmitoylethanolamide, PEA (described below). Interestingly, 2-arachidonoylglycerol (2-AG), a major endocannabinoid, can be metabolized to 2-arachidonoyl lysophosphatidic acid (2-ALPA) through the action of a monoacylglyceride kinase. Also, the endogenous GPR55 ligand, 2-ALPI, can be degraded either to 2-AG by a phospholipase C (specifically PLCβ1) or to 2-ALPA (2-arachidonyl lysophosphatidic acid) by phospholipase D (PLD). Therefore, it appears that mutual interconversion is possible between 2-ALPA and 2-AG within a cell.
The GPR55 gene is located on chromosome 2q37.1 and is composed of 4 exons that encode a protein of 319 amino acids.
GPR75
GPR75 is the receptor for the ω-hydroxylated form of arachidonic acid, 20-hydroxyeicosatetraenoic acid (20-HETE). The formation of ω-hydroxylated arachidonic acid is catalyzed by several members of the CYP4 family including CYP4A11. 20-HETE has been shown to be involved in the activation of pro-inflammatory pathways, and the signaling pathways leading to hypertension, both of which contribute to the development of obesity, type 2 diabetes, and cardiovascular disease. In addition to 20-HETE, GPR75 has been shown to bind, and be activated by, the chemokine, CCL5 [chemokine (C-C motif) ligand 5]. CCL5 is also known as RANTES (Regulated on Activation, Normal T-cell Expressed and Secreted).
GPR75 is coupled to a Gq-type G-protein and, thus its activation results in increased IP3 (Ins-1,4,5-P3) and DAG production. The increased production of IP3 results in release of ER stored Ca2+ and activation of various Ca2+-mediated signaling cascades. With respect to the vasculature, the activation of GPR75 by 20-HETE in vascular smooth muscle cells leads to inhibition of the large conductance calcium- and voltage-activated potassium channel (designated KCa1.1 and encoded by the KCNMA1 gene). The KCNMA1 encoded potassium channel is also known as MaxiK or BK (big potassium). The inhibition of the KCNMA1 channel underlies the hypertension-inducing effects of 20-HETE.
The signaling cascade initiated by GPR75 activation in vascular endothelial cells includes the Ca2+-mediated release of the SRC tyrosine kinase from the receptor complex. The released SRC then interacts with the vascular endothelial growth factor receptor (VEGFR; encoded by the FLT1 gene) resulting in activation of a signaling cascade that includes the inhibition of endothelial nitric oxide synthase (eNOS). Endothelial cell NOS generates nitric oxide (NO) that normally passes through to the underlying vascular smooth muscle cells resulting in vasodilation. Thus, the activity of 20-HETE in the endothelium results in inhibition of vascular relaxation contributing to a hypertensive state.
In addition to its effects in the vascular, GPR75 is also involved in the regulation of whole body lipid homeostasis. Experiments in mice have demonstrated that loss of GPR75 function results in increased activity and reduced levels of body fat. The reduction in body fat induced by GPR75 knockout is associated with an amelioration of the effects of metabolic dysfunction-associated fatty liver disease (MAFLD), also known as metabolic dysfunction-associated steatotic liver disease (MASLD). In humans, polymorphisms in the GPR75 gene have been identified that are associated with a reduced incidence of hepatic steatosis. MAFLD was formerly referred to as non-alcoholic fatty liver disease, NAFLD.
The GPR75 gene is located on chromosome 2p16.2 and is composed of 2 exons that encode a 540 amino acid protein. Although expression of the GPR75 gene is observed in numerous tissues, the highest levels of expression are found in the brain and are nearly 4-times higher there than in any other tissue.
GPR84
GPR84 is a member of the class A (rhodopsin-like) GPCR family. GPR84 is coupled to a pertussis toxin-sensitive Gi/o-type G-protein. GPR84 was originally shown to be activated by lipopolysaccharide (LPS) suggesting that medium-chain free fatty acids could be regulating inflammatory responses via interaction with GPR84. Subsequently it was demonstrated that GPR84 is a receptor for medium-chain saturated fatty acids such as capric acid (C10:0; decanoic acid), undecanoic acid (C11:0), and lauric acid (C12:0).
GPR84 is highly expressed in leukocytes and when the receptor is activated in the monocyte lineage there is an amplification of the LPS-stimulated IL-12 production. In macrophages that are influenced by local inflammatory conditions there is an increased level of expression of GPR84 in these cells.
The GPR84 gene is located on chromosome 12q13.13 and is composed of 3 exons that encode a 396 amino acid protein.
Hydroxycarboxylic Acid Receptors: HCA1 (GPR81), HCA2 (GPR109A), HCA3 (GPR109B)
These three receptors each share significant sequence homology and have also been shown to bind hydroxycarboxylic acid (HCA) metabolites. For this reason these three GPCR have been proposed to constitute a subfamily called the HCA receptors.
GPR81 is now designated HCA1 (encoded by the HCAR1 gene) and as indicated above binds L-lactic acid (2-hydroxy-propanoic acid). GPR81 is coupled to the activation of a transmembrane form of adenylate cyclase (tmAC).
GPR109A was originally identified as a GPCR whose expression was induced in macrophages by interferon-γ (IFN-γ) treatment and identified as PUMA-G (Protein Upregulated in MAcrophages by IFN-Gamma). GPR109A is now designated HCA2 (encoded by the HCAR2 gene) and as indicated above binds ketones, specifically β-hydroxybutyrate (3-hydroxybutyric acid). In separate experiments HCA2 (GPR109A) was also shown to be a receptor for nicotinic acid and identified as HM74A. Another previous designation for HCA2 is NIACR1 because of its niacin binding activity. In addition to adipose tissue, HCA2 is expressed in macrophages, neutrophils, keratinocytes, and intestinal epithelial cells. Within the gut the HCA2 protein serves as a receptor for gut bacteria produced butyrate.
GPR109B is now designated HCA3 (encoded by the HCAR3 gene) and is activated by 3-hydroxyoctanoic acid, an intermediate in mitochondrial fatty acid β-oxidation. HCA3 has also been designated as HM74 and NIACR2. All three of these receptors are coupled to Gi-type G-proteins and all three are expressed at highest levels in adipocytes. HCA3 is also expressed in macrophages, neutrophils, and intestinal epithelial cells.
HCA1, HCA2, and HCA3 are each involved in the inhibition of lipolysis, while both HCA2 and HCA3 also activate immune cells.
All three of the hydroxycarboxylic acid receptors are derived from single exon intronless genes clustered on chromosome 12q24.31. The HCA1 protein is a 346 amino acid protein encoded by the HCAR1 gene. The HCA2 protein is a 363 amino acid protein encoded by the HCAR2 gene. The HCA3 protein is a 387 amino acid protein encoded by the HCAR3 gene.
HCA1
The HCAR1 gene is expressed at the highest levels in adipocytes of both white and brown adipose tissue (WAT and BAT, respectively). Expression of HCAR1 also is found, but at levels much lower than in adipose tissue, breast tissue, skeletal muscle, liver, kidney, neurons and astrocytes in the brain, and in dendritic cells of the immune system. In addition, expression of HCAR1 is found to be elevated in numerous cancers.
HCA1 function in adipose tissues allows for lactate-induced activation of the receptor and the resultant decrease in cAMP leading to inhibition of lipolysis, a response that might at first seem counterintuitive. This is because an obvious cause of elevated blood lactate levels is intensive exercise and it would be expected that this type of exercise would require increased fatty acid release from adipocytes for skeletal muscle energy generation. However, adipocytes are another source of lactate and their reduction of glycolytic pyruvate to lactate increases as a result of insulin-stimulated glucose uptake into these cells. Therefore, the likely normal function of lactate-induced stimulation of HCA1 is to contribute to the insulin-induced inhibition of adipocyte lipolysis. Indeed, in HCA1 knock-out mice there is an associated decrease in insulin-mediated inhibition of lipolysis. This indicates that lactate functions in an autocrine and paracrine manner to mediate insulin-induced regulation of adipocyte lipolysis.
As indicated, HCA1 expression is also found on cancer cells and dendritic cells of the immune system. These sites of expression contribute to the role of lactate in cancer metabolism and immune system evasion. Due to the altered metabolism of glucose in cancer cells they produce large amounts of lactate. The lactate is transported out of the cells via transporters of the monocarboxylate transporter (MCT) family (predominantly MCT4) which are encoded by genes of the SLC16 family. The lactate then binds to HCA1 receptors on these same cells resulting in the activation of a transcription program that contributes to immune evasion.
Signaling processes related to immune modulation via lactate-mediated activation of HCA1 include inhibition of the NLRP3 inflammasome. NLRP3 refers to Nucleotide-binding oligomerization domain and Leucine-rich repeat-containing Receptor Protein 3. The NLRP3 inflammasome is a complex consists of a sensor (NLRP3 protein), an adaptor (apoptosis-associated speck-like protein, ASC), and an effector (caspase-1). Lactate activated HCA1 also inhibits expression of several pro-inflammatory proteins, such as NF-κB, via an AMPK-mediated pathway. Activated AMPK then phosphorylates and activates several tumor suppressor kinases which inhibit the mechanism of inhibition of NF-κB and other pro-inflammatory proteins.
One significant gene whose transcription is enhanced in lactate-stimulated cancer cells is CD274. The protein encoded by the CD274 gene is called programmed death ligand 1 (PD-L1). The role of PD-L1 is suppression of the adaptive immune response, thereby contributing to reduced immune system activation of T-cells. When HCA1 is activated on dendritic cells of the immune system they express reduced levels of MHC molecules as well as the interleukins, IL-6 and IL-12. IL-12 is involved in the maturation of immature T-cells and thus, reduced release of IL-12 by dendritic cells further contributes to reduced T-cell-mediated adaptive immunity.
HCA2
Although HCA2 was originally identified as a receptor for nicotinic acid, and remains an important target for the antidyslipidemic effects of nicotinate, its primary naturally occurring ligand is the ketone, β-hydroxybutyrate. The activation of HCA2 by this ketone indicates that within the adipocyte the receptor serves as a sensor for the level of ketones produced in the liver. The ketones, such as β-hydroxybutyrate, are produced in the liver, primarily during periods of fasting, from fatty acids released from adipocytes. Given that β-hydroxybutyrate activates adipocyte HCA2 and results in inhibition of lipolysis, this effect represents a classic negative feedback mechanism that controls the rate of fatty acid release from adipose tissue during starvation in order to prevent excessive triglyceride degradation.
GPR119
GPR119 is a member of the class A family (rhodopsin-type) of GPCRs and is coupled to the activation of a Gs-type G-protein. The GPR119 protein had previously been referred to as glucose-dependent insulinotropic peptide receptor (GIPR), however, the true GIP receptor has subsequently been identified.
GPR119 binds numerous lipophilic ligands including oleoylethanolamide (OEA), 2-oleoylglycerol (2-OG), palmitoylethanolamide (PEA) and linoleoylethanolamide (LEA). The ligand with one of the highest affinities for GPR119 is OEA, with an EC50 of as low as 0.2 μM. Additional high affinity ligands for GPR119 are 5-hydroxy-eicosapentaenoic acid (5-HEPE), linoleoylethanolamide (LEA), palmitoylethanolamide (PEA), oleoyl-lysophosphatidylcholine (18:1-lysoPC), and palmitoyl-lysophosphatidylcholine (16:0-lysoPC). In addition, the lipid modified amino acid, N-oleoyl-tyrosine, has been shown to have high affinity for GPR119.
The role of GPR119 in OEA-mediated effects is described in more detail below in the section on fatty acid derivatives which focuses on OEA.
The GPR119 gene is located on the X chromosome (Xq26.1) and is composed of 2 exons, only one of which encodes a protein of 335 amino acids. GPR119 is expressed at the highest levels in the pancreas and fetal liver with expression also seen in the gastrointestinal tract, specifically the ileum, cecum, and colon. The intestinal expression pattern of GPR119 is very similar to that of the glucagon gene (GCG) and the peptide tyrosine tyrosine (PYY) gene.
Within the ileum and colon the expression of GPR119 is highest in enteroendocrine L cells. The enteroendocrine L cells secrete GLP-1, GLP-2, PYY, oxyntomodulin (OXM), and serotonin. Secretion of GLP-1 is induced by fat ingestion and this response is mediated by GPR119. When intestinal GPR119 is activated, gastric emptying is slowed by both gut hormone-dependent and gut hormone-independent mechanisms.
FFAR4 (GPR120): Obesity and Diabetes
FFAR4 is one of four members of the free fatty acid sensing GPCR family of receptors that was originally identified as an orphan GPCR and designated GPR120. Ligand binding to FFAR4 is associated with the activation of a Gq-type G-protein.
FFAR4 is specifically activated by long-chain non-esterified fatty acids (NEFAs) in particular in the intestines by α-linolenic acid (ALA), an omega-3 polyunsaturated fatty acid (PUFA). Activation of FFAR4 in the intestines results in increased GLP-1 secretion from enteroendocrine L cells. This results due to receptor-mediated activation of the intracellular signaling kinases ERK and PI3K.
The FFAR4 gene is located on chromosome 10q23.33 and is composed of 4 exons that generate two alternatively spliced mRNAs. These two mRNAs encode a short form (originally identified as GPR120-S) and a long form (originally identified as GPR120-L) of the FFAR4 protein. The GPR120-S protein is 361 amino acids and the GPR120-L protein is 377 amino acids.
FFAR4 is highly expressed in adipose tissue, and proinflammatory macrophages. The high expression level of FFAR4 in mature adipocytes and macrophages is indicative of the fact that FFAR4 is likely to play an important role in biologic functions of these cell types. In contrast, negligible expression of FFAR4 is seen in muscle, pancreatic β-cells, and hepatocytes. Although not expressed at appreciable levels in hepatocytes expression of FFAR4 is highly inducible in liver resident macrophage-like cells known as Kupffer cells. FFAR4 can be activated with a synthetic agonist (GW9508) as well as omega-3 PUFA. FFAR4 is also expressed in enteroendocrine L cells of the gut. These are the cell types that express the incretin peptide hormone GLP-1. Previous work on FFAR4 focused on the potential ability of this receptor to stimulate L cell GLP-1 secretion.
Short-chain fatty acids are known to be proinflammatory and unsaturated fatty acids are generally neutral. In contrast the omega-3 PUFA, DHA and EPA, exert potent anti-inflammatory effects through FFAR4. It has been found that FFAR4 functions as an omega-3 fatty acid receptor/sensor in proinflammatory macrophages and mature adipocytes. By signaling through FFAR4, DHA and EPA, mediate potent anti-inflammatory effects to inhibit both the Toll-like receptor (TLR) and tumor necrosis factor-α (TNF-α) inflammatory signaling pathways. The TLRs are a class of non-catalytically active transmembrane receptors that are involved in mediating responses of the innate immune system. Their name is derived from the fact that they have sequence similarities to the Toll gene found in Drosophila.
It is known that chronic tissue inflammation is an important mechanism resulting in the development of insulin resistance. Therefore, the anti-inflammatory actions of omega-3 PUFA can exert potent insulin sensitizing effects. It has recently been demonstrated in obese mouse models that the in vivo anti-inflammatory and insulin sensitizing effects of omega-3 PUFA are dependent on expression of FFAR4. Given that FFAR4 is highly expressed in proinflammatory macrophages and functions as an omega-3 PUFA receptor, this receptor is critical in mediating the anti-inflammatory effects of the omega-3 PUFA class of lipids.
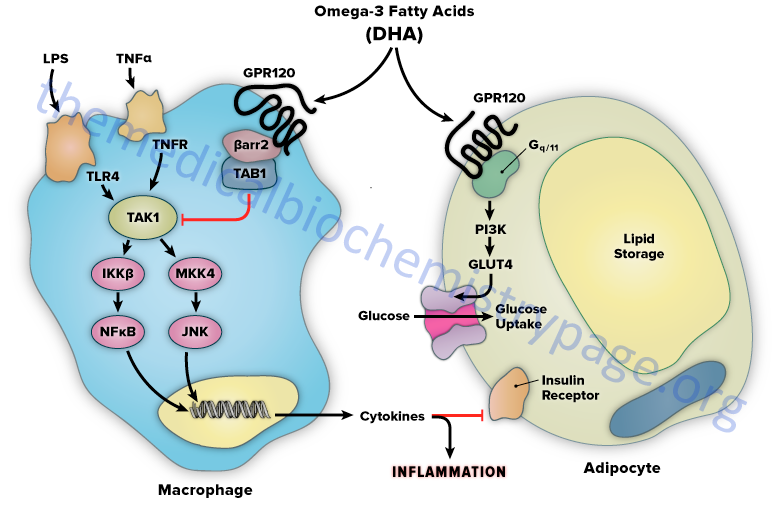
The mechanism of FFAR4-mediated anti-inflammation involves inhibition of transforming growth factor β–activated kinase 1 (TAK1) through a β-arrestin-2 dependent effect. β-arrestins are a class of protein that serve as scaffold or adaptor proteins for a wide range of GPCRs, as well as several other groups of receptor subtypes. After ligand binding, β-arrestins can associate with the cytoplasmic domains of GPCRs and couple the receptor to specific downstream signaling pathways, as well as mediate receptor endocytosis.
Stimulation of FFAR4 by DHA has been shown to inhibit both the TLR2/3/4 and TNF-α proinflammatory cascades. Activation of the kinases, inhibitor of nuclear factor-kappa B kinase subunit beta (IKKβ) and c-JUN N-terminal kinase (JNK), is common to TLR and TNF-α signaling. Nuclear factor-kappa B (NF κB) is one of the most important transcription factors regulating the expression of proinflammatory genes. Given that activation of FFAR4 by DHA results in inhibition of both the TLR and TNF-α cascades it indicates that the locus of FFAR4 inhibition is at or proximal to the IKKβ and JNK kinases. TAK1 activation stimulates both the IKKβ/NFκB and JNK/AP1 pathways, and the TLR and TNF-α signaling pathways converge at this step.
Stimulation of FFAR4 has been shown to specifically inhibit TAK1 phosphorylation and activation, providing a common mechanism for the inhibition of both TLR and TNF-α signaling. Activation of FFAR4 by DHA results in the association of the receptor with β-arrestin2. DHA stimulation results in the recruitment of β-arrestin2 to the plasma membrane where it co-localizes with FFAR4. Following association between FFAR4 and β-arrestin2 the complex is internalized and the complex is colocalized in the cytoplasmic compartment. TAB1 is the activating protein for TAK1 and following DHA-stimulated internalization of the FFAR4/β-arrestin2 complex, β-arrestin2 associates with TAB1 (TAK1 binding protein).
The interaction of β-arrestin2 blocks the ability of TAB1 to associate with TAK1, thereby inhibiting TAK1 activation and downstream signaling to the IKKβ/NFκB and JNK/AP1 system. The anti-inflammatory effects mediated by FFAR4 are entirely dependent on β-arrestin2. However, not all of the biological effects of DHA exerted via activation of FFAR4 rely on β-arrestin2 association with the receptor.
FFAR4 is expressed in mature adipocytes, but not preadipocytes. DHA stimulation of FFAR4 in adipocyte precursor cells in culture results in increased GLUT4 translocation to the cell surface with a subsequent increase in glucose transport into the cells. The DHA-mediated effects on glucose uptake through FFAR4 stimulation in adipocytes turns out to be independent of β-arrestin2. The effects of DHA on glucose uptake in adipocytes in culture are additive to those of a submaximally stimulating concentration of insulin.
Although it is possible to propose that the insulin sensitizing effects of omega-3 PUFA in adipocytes contributes to the overall insulin sensitizing actions of these fatty acids, muscle glucose uptake accounts for the great majority of insulin stimulated glucose disposal but FFAR4 is not expressed in muscle. In addition, experiments with muscle cells in culture demonstrate that DHA does not stimulate glucose uptake. However, since chronic, low grade tissue inflammation is an important cause of obesity-related insulin resistance, the anti-inflammatory effects of FFAR4 stimulation are most likely coupled to insulin sensitizing actions in vivo.
Comparing effects of omega-3 PUFA in wild-type and FFAR4 knock-out (KO) mice demonstrates the link between inflammation and insulin sensitivity. When fed a normal diet, lean FFAR4 KO mice are glucose intolerant, hyperinsulinemic and they have decreased skeletal muscle and liver insulin sensitivity. These FFAR4 KO mice also have a 2- to 5-fold higher level of expression of several proinflammatory genes. Feeding a high fat diet to both the FFAR4 KO and the wild-type mice will result in obesity and insulin resistance. Of significance is the fact that when these mice are supplemented with omega-3 PUFA (such as DHA) the wild-type, but not the FFAR4 KO mice, show a dramatic increase in insulin sensitivity. In addition, omega-3 PUFA supplementation results in a decrease in adipose tissue markers of inflammation as well as anti-inflammatory effects in macrophages only in the wild-type mice. The in vivo anti-inflammatory actions of omega-3 PUFA are consistent with the insulin sensitizing effects of these lipids and are completely dependent on the presence of FFAR4.
Lipid profile effects of omega-3 PUFA are also directly related to activation of FFAR4. In wild-type and FFAR4 KO mice fed a high fat diet there is an increase in total triglyceride, diglyceride, short-chain fatty acids, monounsaturated fatty acids and omega-6 fatty acids in the blood. All of these lipid changes are ameliorated with omega-3 PUFA treatment in wild-type but not FFAR4 KO mice. Results such as these, obtained in experimental animals, are consistent with the view that the reversal of metabolic dysfunction-associated fatty liver disease (MAFLD) by omega-3 PUFA treatment is mediated, in part, by activation of FFAR4. MAFLD is the preferred designation for the liver disease originally identified as non-alcoholic fatty liver disease, NAFLD.
The results of animal studies on the functions of omega-3 PUFA in inflammation, insulin sensitization, and lipid profiles mediated through activation of FFAR4 indicates that this GPCR is a critically important control point in the integration of anti-inflammatory and insulin sensitizing responses, which may prove useful in the future development of new therapeutic approaches for the treatment of diabetes. However, there is some controversy regarding the direct anti-inflammatory and insulin sensitizing effects of the omega-3 PUFA, EPA and DHA being mediated through GPR120 activation since studies in mice in which the GPR120 (FFAR4) gene was knocked out still exhibited EPA- and DHA-mediated increases in insulin sensitivity and decreases in inflammatory responses.
Recent genomic screening in obese children and adults identified two polymorphisms in the FFAR4 gene. One polymorphism results in the substitution of His(H) for Arg(R) at amino acid 270 (identified as the R270H variant), while the other results in substitution of Cys(C) for Arg(R) at amino acid 67 (identified as the R67C variant). The R270H variant is strongly associated with obesity and insulin resistance. This discovery, in human subjects, fits well with previously characterized results in FFAR4 KO mice as described above. Biochemically, the R270H mutation in FFAR4 is significant because it functions as a dominant-negative mutation. This means that even in the presence of a normal copy of the FFAR4 gene (heterozygous individuals) the R270H mutation will manifest near full inhibition of normal FFAR4 signaling.
GPR119 Ligand Oleoylethanolamine (OEA)
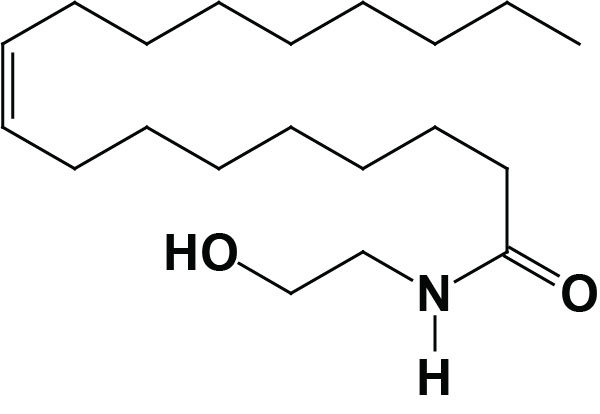
Oleoylethanolamide (OEA; also identified as N-oleoylethanolamine) is a member of the fatty-acid ethanolamide (N-acylethanolamines, NAE) family that includes linoleoylethanolamide (LEA), palmitoylethanolamide (PEA), N-arachidonoylethanolamine (anandamide), N-docosahexaenoyl ethanolamine (synaptamide), and N-stearoylethanolamine (SEA).
Anandamide was identified as an endogenous ligand (endocannabinoid) for the cannabinoid receptors. Details of the endocannabinoids and the cannabinoid receptors can be found in the Endocannabinoids in Feeding Behavior and Energy Homeostasis page. Synaptamide is an endocannabinoid-like compound produced from the omega-3 PUFA, docosahexaenoic acid, DHA.
Excellent vegetarian and vegan sources of oleic acid are olive oil in which up to 85% of the fatty acid in the triglycerides in olive oil is oleic acid. Other vegetable and nut oils also contain high levels of oleic acid in their triglycerides with canola oil (60%–65%) having the second highest amount compared to olive oil. Pecan oil has 60%–75% oleic acid in its’ triglycerides. Another excellent source of oleic acid (43%) is argan oil (from the argan tree which is abundant in Morocco). In addition to the presence of oleic acid, argan oil has added benefits in that it is high in numerous antioxidant plant phenolic compounds as well as vitamin A and vitamin E. Peanut oil (35%–70%), sunflower oil (20%–80%), and grape seed oil (15%–20%) are also excellent plant sources of oleic acid. Animal fats are also high in oleic acid with lard and tallow containing 44%–47% in the triglyceride component.
Synthesis of OEA
Much of the OEA that is absorbed from the intestines is generated by the actions of gut microbiota. However, OEA is also produced by mucosal cells in the proximal small intestine from dietary oleic acid. The pathway of OEA synthesis, like the synthesis of the endocannabinoid anandamide, involves the enzyme N-acyl phosphatidylethanolamine-specific phospholipase D (NAPE-PLD).
Synthesis of OEA occurs on demand within the membrane of the cell by two concerted reactions. The first reaction involves the transfer of a fatty acid residue from the sn-1 position of a phosphatidylcholine (PC) to the free amine of phosphatidylethanolamine (PE). This transfer is catalyzed by a member of the N-acetyltransferase (NAT) family of enzymes. The products of this reaction are known as N-oleoylphosphatidylethanolamines (NOPE) which represent a subclass of the family of lipid molecules called N-acyl-phosphatidylethanolamines (NAPE). At least two activities are termed N-acetyltransferase that are involved in NAPE synthesis (including the synthesis of NOPE), one that is Ca2+-dependent and one that is Ca2+-independent. Although these NAPE synthesis enzymes have been identified and characterized the genes encoding them have not yet been characterized. The Ca2+-dependent NAT is localized to the membrane, whereas the Ca2+-independent NAT is localized to the cytosol indicating that the synthesis of OEA is most likely to be catalyzed by the Ca2+-dependent enzyme.
The second step in synthesis involves cleavage of the NAPE to produce the corresponding fatty acid ethanolamide. This latter reaction is catalyzed by NAPE-specific phospholipase D (NAPE-PLD) which is encoded by the NAPEPLD gene. The NAPEPLD gene is located on chromosome 7q22.1 and is composed of 8 exons that generate two alternatively spliced mRNAs, both of which encode the same 393 amino acid protein. The NAPEPLD encoded enzyme is unrelated to other members of the PLD family of lipid hydrolases. Intestinal bile acids play a role in the synthesis of OEA due to their ability to stabilize the activity of NAPE-PLD.
Once synthesized OEA is eliminated via hydrolysis to oleic acid and ethanolamine. Two enzymes are known to be responsible for OEA hydrolysis. One is known as fatty acid amide hydrolase (FAAH) and the other is N-acylethanolamine (NAE)-hydrolyzing acid amidase (NAAA; also known as PEA-preferring acid amidase, PAA). FAAH is present in the membranes of most mammalian tissues with highest levels observed in brain and liver.
Biological Activities of OEA
OEA has been shown to activate the fatty acid-sensing GPCR identified as GPR119 as well as the non-selective gated cation channel TRPV1 (transient receptor potential vanilloid 1), and to interact with intestinal FAT/CD36 for uptake from the gut. TRPV1 is also known as the capsaicin receptor and is the receptor responsible for the sensation of heat produced by spicy peppers. Evidence suggests that OEA may be the endogenous ligand for GPR119, however, its’ interaction with FAT/CD36 is required for the satiety response elicited by this bioactive lipid.
OEA is one of the most potent ligands, and likely represents the endogenous ligand, for GPR119. The demonstration that OEA is the most active endogenous ligand for GPR119 is of particular interest since previous work has demonstrated that OEA, when administered to laboratory animals, causes a significant reduction in food intake and body weight gain. When intestinal GPR119 is activated by OEA there is clear induction of satiety. However, outside of the intestine the satiety-inducing effects of OEA do not require non-intestinal GPR119 since the effects of systemic OEA administration are still observed in mice in which the gene encoding GPR119 has been knocked out.
GPR119 is, nonetheless, involved in the control of feeding behaviors as demonstrated by studies in GPR119 knock out mice. In response to food deprivation these mice have lower blood glucose and a higher level of body weight reduction when compared to wild-type mice. The response of GPR119 knockout mice to food, following food deprivation, is hyperphagia and the time between eating is reduced.
The satiety-inducing effects of OEA involve the activation of the nuclear receptor PPARα resulting in increased expression of fatty acid translocase and modification of feeding behavior and motor activity. The effects are specific to activation of PPARα since data has shown that OEA does not bind PPARγ nor the obligate heterodimer partner of the PPARs, the retinoid X receptors (RXR).
With respect to anorexic effects of fatty acid ethanolamides, OEA is quite specific. This is demonstrated by the use of close structural analogs of OEA which have no effects on feeding behaviors. PEA and anandamide also do not elicit inhibition of feeding. In fact anandamide increase feeding behavior due to activation of the cannabinoid receptor pathway. Reduced feeding in response to OEA is due solely to an enhanced state of satiety and not to enhanced stress, malaise, anxiety, or taste aversion. When OEA is administered to rodents prior to the dark period when they prefer to eat, there is a marked increase in feeding latency (delay in onset of feeding) and a decrease in meal frequency. However, there is no effect of OEA on the size of the meal the animals consume. Of significance is the fact that PPARα agonists exert very similar effects on feeding behaviors.
Since OEA is produced in the gut its means of effecting changes in feeding behavior involve engagement of vagal sensory afferent (to the brain) fibers that converge on the nucleus of the solitary tract (NTS) in the brain stem. For more information on the neural circuits functioning between the gut and the brain see the Gut-Brain Interrelationships and Control of Feeding Behaviors page. The activation of PPARα by OEA within intestinal enterocytes leads to the activation of the vagal afferent neurons. In animals in which the vagal nerve has been cut OEA fails to reduce feeding behaviors. Further evidence for the role of the vagus nerve in OEA activity is demonstrated by the fact that peripheral administration of OEA results in reduced feeding whereas, intracerebroventricular (ICV) injection does not have an effect. In addition to reducing feeding behavior, OEA administration results in reduced body-weight gain in lean and obese animals. The effects of OEA on weight gain are likely due to increases in energy expenditure since the compound rapidly increases the rate of lipolysis.
There is an self-sustaining activation loop involved in the production of OEA and its effects elicited within the NTS. Activation of neurons within the NTS leads to activation of preganglionic sympathetic neurons within the rostral ventrolateral medulla (RVLM). These preganglionic neurons then activate postganglionic sympathetic neurons that impinge on cells of the submucosal layer and the myenteric plexus. The norepinephrine that is released from the postganglionic sympathetic neurons activates β-adrenergic receptors in these cells leading to increased OEA production. OEA itself exerts effects on the cells of the submucosal layer and the myenteric plexus, likely through binding to GPR119, that result in decreased gastric motility slowing down digestion and activating satiety sensations.
In addition, activation of GPR119 in the pancreas is correlated with enhanced glucose-stimulated insulin secretion (GSIS). Equally important is that activation of GPR119 in the gut results in increased secretion of the incretin hormones GLP-1 and GIP. These observations indicate that GPR119 activation is associated with a dual mechanism of reducing blood glucose: acting directly through pancreatic β-cells to promote GSIS and in the gut via stimulated secretion of the incretins, GLP-1 and GIP, both of which increase insulin release from the pancreas in response to food intake. Currently there are several small molecule agonists of GPR119 in clinical trials being tested for their efficacy in treating the hyperglycemia of type 2 diabetes as well as for their efficacy in treating obesity.
Fatty Acid Amides and Cellular Signaling
Many fatty acid amides have been shown to be highly significant endogenous signaling molecules. The fatty acid amides are grouped into two classes, the fatty acid ethanolamides (acylethanolamides), which includes anandamide and oleoylethanolamide, OEA (see above), and the fatty acid primary amides, of which oleamide is the most well characterized molecule. The acylethanolamides exert prominent effects within the central nervous system and their synthesis occurs on demand resulting in their accumulation in response to neuronal injury. Although anandamide is more well known as a CNS-acting molecule due to its known effects as a natural cannabinoid receptor ligand, other acylethanolamides, such as oleoyl-, palmitoyl-, and stearoylethanolamide, are found at much higher levels in the brain.
Anandamide
Anandamide (N-arachidonylethanolamine, AEA) is an endocannabinoid that functions by binding to the cannabinoid receptors (CB1 primarily, and CB2). This lipid-derived signaling molecule is known to be involved in the modulation of diverse physiological processes such as the regulation of appetite, learning and memory, nociception (pain sensation), anxiety, and inflammation. The details of anandamide synthesis and function are covered in the Endocannabinoids in Feeding Behavior and Energy Homeostasis page.
Oleoylethanolamide (OEA)
Oleoylethanolamide is discussed in above.
Palmitoylethanolamide (PEA)
Palmitoylethanolamide, [N-(2-hydroxyethyl)hexadecanamide: PEA] is synthesized from different precursors than anandamide but the pathway involves a similar intermediate, N-acylated phosphatidylethanolamine (NAPE). Although several enzymatic pathways have been shown to be involved in the synthesis of PEA, the major pathway utilizes the same enzyme involved in the anandamide synthesis pathway, membrane-associated NAPE-phospholipase D (NAPE-PLD).
Like the metabolism of anandamide, PEA metabolism occurs primarily through the action of the ER-associated enzyme fatty acid amide hydrolase (FAAH). An additional metabolic pathway involved in the hydrolysis of PEA involves the lysosomal enzyme, N-acylethanolamine (NAE)-hydrolyzing acid amidase (NAAA). NAAA is also referred to as PEA-preferring acid amidase (PAA)
Palmitoylethanolamide exerts numerous effects within the immune system including reductions in allergic reactions and inflammation, primarily exerted via inhibition of mast cell degranulation. The anti-inflammatory actions of PEA were originally identified in 1957 when the lipid was extracted from soy beans and peanuts. PEA action also modulates the production of nitric oxide (NO) which contributes to the anti-inflammatory as well as the anti-nociception properties of this lipid. PEA has also been shown to exert anti-inflammatory effects on adipocytes suggesting that drugs that mimic the action of PEA could be useful in the treatment of obesity-induced insulin resistance. Although the structure and synthesis of PEA is similar to anandamide, this lipid is not active through the cannabinoid receptors, CB1 and CB2.
PEA has been shown to bind to and activate the orphan GPCR, GPR55, at nanomolar concentrations. The receptor binding characteristics of PEA coupled with its observed biological functions suggests that this particular N-acylethanolamide may represent a novel endocannabinoid-like system. PEA also binds to GPR119 but in the micromolar range.
The nuclear receptor peroxisome proliferator-activated receptor-α (PPARα) has been identified as the molecular target responsible for the anti-inflammatory properties of PEA. PEA is also known to down-regulate and/or inhibit the activity of FAAH. Combined, the effects of PEA on PPARα and FAAH activities are referred to in the context of “entourage effect”. The entourage effect was a termed coined in 1998 by researchers in the cannabinoid field to reflect the observations that many of the compounds in marijuana work together to produce a synergy of effects. With respect to PEA, the entourage effect is primarily due to the resultant inhibition of FAAH which in turn leads to reduced metabolism of anandamide. Prolonged anandamide levels in turn lead to enhanced action through the cannabinoid receptors.
PEA can also exert effects via the transient receptor potential cation channel subfamily V, member 1 (TRPV1). In addition to receptor activation, PEA has been shown to inhibit the functions of certain voltage-gated potassium channels such as Kv1.5 and Kv4.3. The Kv1.5 protein is encoded by the KCNA5 gene (potassium voltage-gated channel, subfamily A, member 5). The KCNA5 encoded protein plays a role the restoration of the resting membrane potential of pancreatic b-cells and as such contributes to the regulation of insulin secretion. Kv4.3 is an α-subunit of the Shal-related family of A-type voltage-gated ion channels and it is encoded by the potassium voltage-gated channel subfamily D, member 3 (KCND3) gene. The KCND3 encoded protein is important in the repolarization phase of action potentials in cardiac myocytes.
Stearoylethanolamide (SEA)
Stearoylethanolamide (SEA) is an N-acylethanolamide (NAE) derived from the fully saturated C18 fatty acid, stearic acid. The pathway for SEA synthesis has not been as well characterized as those for anandamide and PEA but it is presumed that it is produced by an analogous process. As indicated earlier, SEA and PEA represent the major NAEs found in the body. Also like PEA, SEA exerts many of its most pronounced effects within the CNS and is also a potent anti-inflammatory lipid. Additional similarities between SEA and PEA activities are evident by the fact that SEA does not bind to the cannabinoid receptors, has been shown to down-regulate (competitive inhibition) the activity of the metabolizing enzyme, FAAH, activates PPARα, activates the cation channel, TRPV1, and inhibits the potassium channels Kv1.5 and Kv4.3.
Linoleoylethanolamide (LEA)
Linoleoylethanolamide (LEA) is an NAE derived from the polyunsaturated fatty acid, linoleic acid. Like SEA, the biosynthesis pathway to LEA is less well characterized than anandamide and PEA but is believed to occur via a similar pathway. Fewer studies have been conducted on the functions of LEA but one important function for this lipid is similar to that of OEA. LEA is produced in the small intestine where it activates PPARα and subsequently plays a role in the regulation of food intake by selective prolongation of feeding latency and interval period between meals. LEA has also been shown to activate the GPR119 receptor.
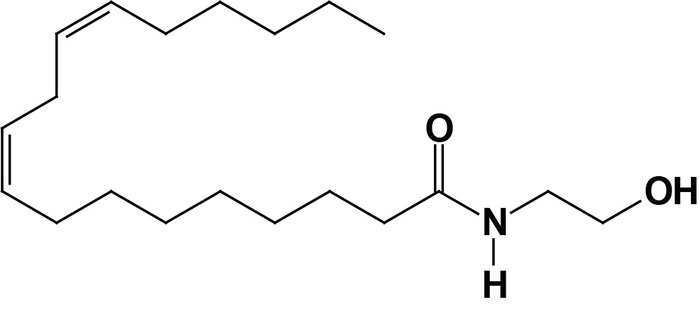
Oleamide
Fatty acid primary amides represent a distinct class of bioactive lipids. This class includes the amides of oleic acid (oleamide; the most well characterized member), palmitic acid (C16:0), palmitoleic acid (C16:1), linoleic acid (C18:2), and elaidic acid (C18:1). Elaidic acid is a C18 monounsaturated fatty acid, like oleic acid, but where the site of unsaturation exists in the trans configuration. The biosynthesis of oleamide may occur via two distinct pathways. Evidence indicates that glutamine can serve as the amino donor in the amidation of oleic acid and additional evidence has shown that oleamide can be derived from the glycine adduct of oleic acid. The synthesis of oleamide from the glycine adduct involves a well characterized enzyme called peptidylglycine α-amidating monooxygenase (PAM). ) PAM is a multifunctional protein complex composed of two distinct enzymatic activities: peptidylglycine α-hydroxylating monooxygenase (PHM) and peptidyl-α-hydroxyglycine α-amidating lyase (PAL). The function of PAM is critical in the generation of numerous C-terminally amidated neuroendocrine peptides.
The bioactivity of oleamide was initially characterized as a signaling molecule involved in the regulation of sleep. Subsequent research demonstrated that oleamide exerted distinct cannabinoid-like activities. These latter activities are the result of oleamide acting as a direct agonist on CB1 cannabinoid receptors. Oleamide has also been shown to interact with several other receptors systems and ion channels. Oleamide acts indirectly at an allosteric site on the GABAA receptor similar to the binding of benzodiazepines to GABAA receptors. Oleamide has also been shown to modulate the activity of several subtypes of the serotonin receptors that includes 5-HT1A, 5-HT2A, and 5-HT7. In addition, oleamide interacts with voltage-gated sodium channels.
Although oleamide has been shown to exert multiple effects, its most prominent is its ability to induce natural physiological sleep, i.e. induction of sleep that is indistinguishable that which occurs naturally. This activity of oleamide is stereo- and structurally specific. The trans-isomeric amide produced from elaidic acid does not induce sleep, nor is sleep induced from other C18 monounsaturated fatty acid amides nor from the fully saturated C18 amide of stearic acid.
The Glycine Amides
Another potentially important class of bioactive lipid amides are the N-acylglycinamides harboring fatty acid acyl groups, although much less is known about their actual bioactivity. It is not yet clear whether this class of lipid serves as independent signaling molecules or whether they are simply biosynthetic precursors to the more well characterized fatty acid primary amides described above. Identified fatty acyl glycinamides include N-arachidonoylglycine (NAGly), N-palmitoylglycine (PalGly), N-oleoylglycine, (OlGly), N-stearoylglycine (StrGly), N-linoleoylglycine (LinGly), and N-docosahexaenoylglycine (DocGly).
Preliminary research suggests that NAGly functions as an endogenous ligand for GPR92 which is encoded by the LPAR5 (lysophosphatidic acid receptor 5) gene. N-arachidonoylglycine has been shown to bind to GPR18.
Platelet Activating Factor, PAF
Platelet activating factor (PAF) is one of the most potent bioactive lipids produced by the human body. PAF represents a group of structurally related choline plasmalogens (1-O-1′-enyl-2-acetyl-sn-glycero-3-phosphocholine). The most common form of PAF contains a 16-carbon fatty alkylation at the sn-1 position. All PAF molecules are unique alkyl phospholipids because they all contain an acetyl group at the sn-2 position whereas, all other plasmalogens and phospholipids possess fatty acyl groups at that position.
PAF functions as a mediator of hypersensitivity, acute inflammatory reactions and anaphylactic shock. PAF is synthesized in response to the formation of antigen-IgE complexes on the surfaces of basophils, neutrophils, eosinophils, macrophages and monocytes. The synthesis and release of PAF from cells leads to platelet aggregation and the release of serotonin from platelets. PAF also produces responses in liver, heart, smooth muscle, and uterine and lung tissues.
PAF is produced by stimulated basophils, monocytes, polymorphonuclear neutrophils, platelets, and endothelial cells primarily through lipid remodeling. A variety of stimuli can initiate the synthesis of PAF. These stimuli include macrophages going through phagocytosis and endothelial cells that have taken up thrombin. There are two different pathways in which PAF can be synthesized: the de novo pathway and the remodeling pathway. The remodeling pathway is activated by inflammatory agents and it is thought to be the primary source of PAF under pathological conditions. The de novo pathway is used to maintain PAF levels during normal cellular function.
The precursor to the remodeling pathway is a membrane plasmalogen, which is typically a choline plasmalogen. The fatty acid is removed from the sn-2 position of the three-carbon backbone of the phospholipid by phospholipase A2 (PLA2) to produce a lyso-plasmalogen. An acetyl group is then added by lysophosphatidylcholine acetyltransferase 1 (LPCAT1) to produce PAF. LPCAT1 has also been referred to as lyso-PAF acetyltransferase.
Using the de novo pathway, PAF is produced from precursor lipids of the acyldihydroxyacetone phosphate (acyl-DHAP) family. The enzyme alkylglycerone phosphate synthase (AGPS) converts an acyl-DHAP and a long-chain alcohol to an alkyl-DHAP. The alkyl-DHAP is then reduced to an alkyllysoglycerol phosphate by an NADPH-dependent oxidoreductase. An acetyltransferase then converts the alkyllysoglycerol phosphate to an alkylacetylglycerol phosphate. A phosphohydrolase removes the phosphate group yielding an alkylacetylglycerol. Choline is then added from CDP-choline by a specific PAF choline phosphotransferase.
PAF exerts its effect through interactions with a cell surface receptor whose expression is restricted to hematopoietic cells and cells of the immune system. The PAF receptor is a member of the GPCR family and is coupled to the activation of both Gq– and Gi-type G-proteins. The Gq-mediated signaling pathway is the mediator of pro-inflammatory processes as well as the activation of the synthesis of additional PAF. The Gi-mediated signaling pathway prevents the activation of PKA and thus the activation of anti-inflammatory processes initiated by PKA.
The PAF receptor is encoded by the PTAFR gene. The PTAFR gene is located on chromosome 1p35.3 and is composed of 4 exons that generate four alternatively spliced mRNAs, each of which encode the same 342 amino acid protein.
Fatty Acid Esters of Hydroxy Fatty Acids, FAHFA
The FAHFA family of lipids produced by mammalian cells can be divided into two major classes. These classes are the branched-chain FAHFA and the omega-FAHFA (ω-FAHFA). The branched-chain FAHFA are bioactive lipids whereas the ω-FAHFA function as biosurfactants.
Branched fatty acid esters of hydroxy fatty acids (FAHFA) are recently identified endogenous lipids, the synthesis of which is regulated by fasting and high-fat feeding and associated with insulin sensitivity. FAHFA belong to the lipid class referred to as estolides which are intermolecular esters of fatty acids. The FAHFA were initially identified in mice in which expression of the GLUT4 gene was overexpressed specifically within adipose tissue. Their synthesis was shown to be regulated by both fatty acid content of the diet (e.g. high-fat diets) and by fasting. Strikingly, the level of synthesis of FAHFA isomers is highly correlated to insulin sensitivity with their synthesis being dramatically reduced in adipose tissue of humans exhibiting insulin resistance.
In humans the FAHFA isomers are found in numerous tissues as well as in the blood with highest concentrations being found in white and brown adipocytes. When FAHFA isomers were administered to mice it was observed that circulating levels of glucose declined while the levels of the gut and pancreatic hormone GLP-1 increased as did the level of insulin secretion. These results indicate that certain FAHFA exhibit anti-diabetic actions. The insulin sensitizing effects of several branched-chain FAHFA are likely to be the result of binding to and activating the free fatty acid receptor, FFAR1.
Structurally, the initially characterized FAHFA isomers were found to be comprised of a C-16 or C-18 fatty acid either saturated [palmitic (C16:0) or stearic (C18:0)] or monounsaturated [palmitoleic (16:1) or oleic (18:1)] linked to a hydroxylated C-16 or C-18 lipid, again either saturated or monounsaturated.
During the studies in which FAHFA were originally identified, palmitic-acid-9-hydroxy-stearic acid (9-PAHSA) was shown to be elevated the most in serum and adipose tissue of the GLUT4 overexpressing mice. This FAHFA is formed from palmitic acid esterification to 9-hydroxy stearic acid. When examining the presence of FAHFA in humans, 9-PAHSA was found to be the most predominant isomer. The levels of 9-PAHSA are reduced in the serum and adipose tissues in the insulin-resistant state of type 2 diabetes in humans.
Since their initial discovery (in 2014) several other FAHFA isomers have been identified such as those composed of the omega-3 polyunsaturated fatty acid (PUFA), docosahexaenoic acid (DHA) linked via an ester linkage to carbon 9 or carbon 13 of the hydroxylated form of the essential fatty acid, linoleic acid. These two FAHFA are identified as 9-DHAHLA and 13-DHAHLA, respectively. 13-DHAHLA is shown in the Figure.
Another DHA containing FAHFA contains DHA esterified to 14-hydroxy DHA and is identified as 14-DHAHDHA. The level of synthesis of DHA containing FAHFA in white adipose tissue (WAT) is similar to the level of protectins and resolvins derived from DHA in WAT. The activity of 13-DHAHLA has been shown to be both anti-inflammatory and pro-resolving.
The most well studied branched-chain FAHFA families are those that are composed of palmitic acid and hydroxystearic acid (PAHSA) and those composed of palmitic acid and hydroxypalmitic acid (PAHPA). These two families of FAHFA include the 5-, 7-, 8-, 9-, 10-, 11-, 12-, and 13-hydroxy regioisomers. Regioisomers are chemical isomers that possess the same functional groups attached at different positions of the molecular backbone.
The enzymes involved in the synthesis of the various FAHFA are not yet fully characterized. The human candidate enzymes that have been suggested to carry out the requisite acylation reactions are involved in bile acid metabolism. One of these enzymes is bile acid CoA: amino acid N-acyltransferase (encoded by the BAAT gene). The other enzyme that has been suggested to be involved in FAHFA synthesis is acyl-CoA:amino acid N-acyltransferase, however, in humans the gene that encodes this protein is a pseudogene and thus, not likely to be involved in FAHFA synthesis.
Two enzymes that are FAHFA-specific hydrolases, involved in the metabolism of FAHFA, have recently been identified. Although not previously characterized as hydrolytic enzymes these two proteins have now been shown to represent a novel class of threonine hydrolases that are involved in the metabolism of bioactive lipids. These two enzymes are both multipass transmembrane proteins and they were originally were originally identified as androgen induced 1 (encoded by the AIG1 gene) and its sequence-related homologous protein called androgen dependent tissue factor pathway inhibitor (TFIP) regulating protein (encoded by the ADTRP gene).
Fat Transport and Scavenger Receptors
When fatty acids are released from adipose tissue stores they enter the circulation as free fatty acids (FFAs) and are bound to albumin for transport to peripheral tissues. When the fatty acid–albumin complexes interact with cell surfaces the dissociation of the fatty acid from albumin represents the first step of the cellular uptake process. Uptake of fatty acids by cells involves membrane proteins with high affinity for fatty acids.
There are several members of the fatty acid receptor family including fatty acid translocase (FAT/CD36) and at least six fatty acid transport proteins (FATPs). FAT is also known as CD36 which is a member of the scavenger receptor class (class B scavenger receptors) of receptors that bind lipids and lipoproteins of the LDL family. The FATPs are a family of at least six fatty acid transport proteins (FATP1–FATP6) that are also members of the solute carrier family of transporters thus, FATP1 is SLC27A1, FATP2 is SLC27A2, FATP3 is SLC27A3, FATP4 is SCL27A4, FATP5 is SLC27A5, and FATP6 is SCL27A6.
The FATPs facilitate the uptake of very long-chain and long-chain fatty acids (VLCFA and LCFA). FATP2 is also known as very long-chain acyl-CoA synthetase (VLCS). FATP4 is the major intestinal long-chain fatty acid transporter. FATP5 is also known as very long-chain acyl-CoA synthetase-related protein (VLACSR) or very long-chain acyl-CoA synthetase homolog 2 (VLCSH2) and is capable of activating 24- and 26-carbon VLCFAs. FATP6 is also known as very long-chain acyl-CoA synthetase homolog 1 (VLCSH1) and exhibits a preference for the transport of palmitic acid and linoleic acid but does not transport fatty acids less than 10 carbons long.
Table of Mammalian Fatty Acid Transporters
| Fat Transporter | Comments |
| FAT/CD36 | fatty acid translocase; FAT is also known as CD36 which is a member of the scavenger receptor class (class B scavenger receptors) of receptors that bind lipids and lipoproteins of the LDL family; located on chromosome 7q11.2 and com[posed of 18 exons that generate eight alternatively spliced mRNAs that encode four distinct isoforms of the protein |
| FABPpm | plasma membrane-associated fatty acid-binding protein; originally characterized as a plasma membrane-associated fatty acid transporter this protein was later demonstrated to be the mitochondrial isoform of glutamate-oxalate transaminase (gene symbol: GOT2) |
| FATP1 | FATP1 is SLC27A1; FATP1 is also known as acyl-CoA synthetase very long-chain family, member 5 (ACSVL5); highest levels of expression in adipose tissue, skeletal and heart muscle; located on chromosome 19p13.11 spanning 13 kb, composed of 15 exons encoding a 646 amino acid protein |
| FATP2 | FATP2 is SLC27A2; FATP2 is also known as acyl-CoA synthetase very long-chain family, member 1 (ACSVL1) as well as very long-chain acyl-CoA synthetase (VLCS); highest levels of expression in liver and kidney; present in peroxisome and microsomal membranes; located on chromosome 15q21.2 composed of 10 exons that generate two alternatively spliced mRNAs |
| FATP3 | FATP3 is SLC27A3; FATP3 is also known as acyl-CoA synthetase very long-chain family, member 3 (ACSVL3); located on chromosome 1q21.3 composed of 10 exons encoding a 730 amino acid protein |
| FATP4 | FATP4 is SLC27A4; FATP4 is also known as acyl-CoA synthetase very long-chain family, member 4 (ACSVL4); is the major intestinal long-chain fatty acid transporter; located on chromosome 9q34.11 spanning 17 kb, composed of 13 exons encoding a 643 amino acid protein |
| FATP5 | FATP5 is SLC27A5; FATP5 is also known as acyl-CoA synthetase very long-chain family, member 6 (ACSVL6), very long-chain acyl-CoA synthetase-related protein (VLACSR), or very long-chain acyl-CoA synthetase homolog 2 (VLCSH2); ER-associated enzyme; highest levels of expression in the liver; capable of activating 24- and 26-carbon VLCFAs; located on chromosome 19q13.43 composed of 10 exons encoding a 690 amino acid protein |
| FATP6 | FATP6 is SLC27A6; FATP6 is also known as acyl-CoA synthetase very long-chain family, member 2 (ACSVL2), very long-chain acyl-CoA synthetase homolog 1 (VLCSH1); expressed at highest levels in the heart; protein only detected in heart and testis; exhibits a preference for the transport of palmitic acid and linoleic acid, does not transport fatty acids less than 10 carbons long; located on chromosome 5q23.3 spanning 67 kb and composed of 12 exons that generate two alternatively spliced mRNAs encoding the same 619 amino acid protein |
The result of the interaction of fatty acids with plasma membrane receptors/binding proteins is a transmembrane concentration gradient. At the plasma membrane the apparent pKa of the fatty acid shifts from about 4.5 in aqueous solutions to about 7.6. This pKa change is independent of fatty acid type. As a consequence, about half of the fatty acids are present in the un-ionized form. This local environment effect promotes a transfer (flip-flop) of uncharged fatty acids from the outer leaflet across the phospholipid bilayer. At the cytosolic surface of the plasma membrane, fatty acids can associate with the cytosolic fatty acid binding protein (FABPc) or with caveolin-1. Caveolin-1 is a constituent of caveolae (Latin for little caves) which are specialized “lipid rafts” present in flask-shaped indentations in the plasma membranes of many cells types that perform a number of signaling functions by serving as lipid delivery vehicles for subcellular organelles.
In order that the fatty acids that are thus taken up to be directed to the various metabolic pathways (e.g. oxidation or triglyceride synthesis) they must be activated to acyl-CoA. Members of the fatty acid transport protein (FATP) family have been shown to possess acyl-CoA synthetase (ACS) activity. Activation of fatty acids by FATPs occurs at the highly conserved cytosolic AMP-binding site of these proteins. The overall process of cellular fatty acid uptake and subsequent intracellular utilization represents a continuum of dissociation from albumin by interaction with the membrane-associated transport proteins, binding to FABPc and caveolin-1 at the cytosolic plasma membrane, activation to acyl-CoA (in many cases via FATP action) followed by intracellular trafficking via FABPc and/or caveolae to sites of metabolic disposition.
Fatty acid translocase (FAT, also called CD36). CD36 is also recognized as a member of the class B scavenger receptor family of lipid binding receptors. The lipid mediator oleoylethanolamide (OEA, see above for details) has been shown to require interaction with FAT/CD36 in the intestine to elicit an activation of PPARα and subsequent hypothalamic sensations of satiety. Although GPR119 may be the receptor for OEA in the body, evidence clearly indicates that intestinal FAT/CD36 is required for OEA-induced satiety.
Lysophospholipids
Lysophospholipids (LPL or LysoPL) are minor lipid components compared to the major membrane phospholipids such as phosphatidylcholine (PC), phosphatidylethanolamine (PE), and sphingomyelin. The lysoPL are generated via the actions of phospholipase A (PLA) family enzymes on glycerophospholipids or sphingolipids. The lysoPL were originally presumed to be simple metabolic intermediates in the de novo biosynthesis of phospholipids. However, subsequent studies demonstrated that the lysoPL exhibited biological properties resembling those of extracellular growth factors or signaling molecules.
The most biologically significant lysoPL are lysophosphatidic acid (LPA or LysoPA), lysophosphatidylinositol (LPI or LysoPI), lysophosphatidylcholine (LPC or LysoPC), lysophosphatidylserine (LPS or LysoPS), sphingosine 1-phosphate (S1P; more information below), and sphingosylphosphorylcholine (SPC).
Each of these lysoPL functions via interaction with specific G-protein coupled receptors (GPCR) leading to autocrine or paracrine effects. The first lysoPL receptor identified was called LPA1 because it bound lysoPA (LPA). The first GPCR shown to bind S1P was called S1P1.
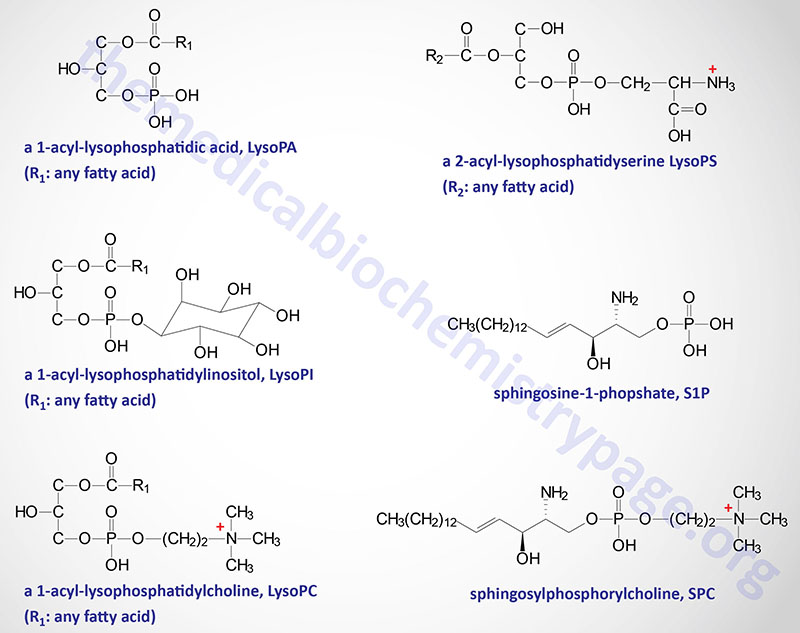
Currently there are fifteen characterized LPL receptors. Because several of the LPL receptors were independently identified in unrelated assays, there are several different names for some members of this receptor family. In particular, there is a group of genes that were originally identified as GPCRs and called endothelial differentiation genes (EDGs) that were later found to be the same as several of the LP receptors. Thus LPA1 is also known as EDG-2, LPA2 as EDG-4, and LPA3 as EDG-7. S1P1 is also known as EDG-1, S1P2 as EDG-5, S1P3 as EDG-3, S1P4 as EDG-6, and S1P5 as EDG-8.
Lysophosphatidic acid, LysoPA (LPA)
LysoPA (also abbreviated LPA), although being simple in structure, exerts a wide variety of cellular responses in many different cell types. LysoPA is known to enhance platelet aggregation, smooth muscle contraction, cell proliferation and migration, neurite retraction, and the secretion of chemokines and cytokines. These effects of LysoPA are the result of binding to specific GPCR. There are at least six characterized LysoPA receptors that, in addition to LPA1–LPA3 indicated above, includes LPA4, LPA5, and LPA6. As indicated above LPA1–LPA3 are members of the EDG family, while LPA4–LPA6 are members of the P2Y family of GPCRs that are referred to as the purinergic receptors. Purinergic receptors were first characterized as a family of GPCRs that were activated by binding adenine and uridine nucleotides. LysoPA has also been shown to interact intracellularly with PPARγ.
Table of Lysophosphatidic Acid (LPA) Receptors
| LPA Receptor | Alternative Name | Gene Symbol | G-Proteins | Comments |
| LPA1 | EDG2 | LPAR1 | Gi/o, Gq, G12/13 | expressed in a wide variety of tissues; highest expression seen in brain, heart, stomach, intestines, and kidney; also expressed in uterus, testis, lung, spleen, thymus, placenta, and skeletal muscle; activation by LPA promotes cell proliferation and survival, cell–cell contact, cell migration, cytoskeletal changes, Ca2+ mobilization, and inhibition of adenylate cyclase; inhibited when LPA4 is activated |
| LPA2 | EDG4 | LPAR2 | Gi/o, Gq, G12/13 | highest levels of expression in the testes and in leukocytes, also in prostate, spleen, thymus, and pancreas; is involved in cell survival and migration, immune function, myelination, and nervous system functions and development |
| LPA3 | EDG7 | LPAR3 | Gi/o, Gq | highest expression in the testis, pancreas, prostate, and heart; lower levels of expression in lung, brain, stomach, spleen, and thymus; activation results in Ca2+ mobilization, MAPK activation, and modulation of adenylate cyclase |
| LPA4 | P2Y9, GPR23 | LPAR4 | Gs, Gi/o, Gq, G12/13 | highest expression only in ovary; low levels of expression in thymus, pancreas, colon, brain, heart, small intestine, testis, prostate, and spleen; is related to the P2Y family of receptors; when activated there is inhibition of LPA1 |
| LPA5 | GPR92 | LPAR5 | Gq, G12/13 | more restricted pattern of expression that other LPARs, highest level seen in spleen; lower expression levels in heart, small intestine, placenta, colon, and liver |
| LPA6 | P2Y5 | LPAR6 | Gs, Gi/o, G12/13 | expressed in a wide variety of tissues; mutations associated with autosomal recessive hypotrichosis; is related to the P2Y family of receptors; |
| GPR87 | GPR87 | Gq, G12/13 Gi | is related to the P2Y family of receptors | |
| P2Y10 | P2RY10 | Gq | highest levels of expression in spleen and lymph; low levels of expression in stomach, intestines, liver, and lung |
LysoPA Synthesis and Degradation
LysoPA is produced by activated platelets, activated adipocytes, neuronal cells, as well as several other cell types. The modes of lysoPA synthesis intracellularly involves at least four different pathways that include the monoacyglycerol kinase (MAGK) pathway, the phosphatidic acid-phospholipase A1 (PA-PLA1) or A2 (PA-PLA2) pathway, the glycerophosphate acyltransferase
(GPAT) synthesis pathway, and the oxidative modification of low-density lipoprotein (LDL) pathway.
In the MAGK pathway various monoacylglycerols (MAG) are phosphorylated by MAGK to yield various lysoPA.
In the PA-PLA1 and PA-PLA2 pathway diacylglycerols are first phosphorylated to phosphatidic acids via the action of various diacylglycerol kinases (DGK). Phosphatidic acids can also be generated from membrane phospholipids via the actions of phospholipase D family member enzymes. The phosphatidic acids are then converted to various lysoPA via the actions of PA-PLA1 or PA-PLA2. The products of PA-PLA1 are 2-acyl-lysoPA and the products of PA-PLA2 are 1-acyl-lysoPA.
In the GPAT synthesis pathway, glycerol-3-phosphate is esterified on the sn-1 hydroxyl by an enzyme of the GPAT family (GPAT2, GPAT3, GPAT4, or GPAM) to yield a lysoPA.
LysoPA is also produced in the serum through the action of several different enzymes including monoacylglycerol kinase (MAGK), secreted phospholipase A1 (sPLA1), secretory phospholipase A2 (sPLA2), and lysophospholipase D (lysoPLD). LysoPLD is also called autotaxin (ATX) which was the name given to a tumor autocrine motility factor. ATX was also shown to be an ecto-nucleotide phosphodiesterase.
Degradation of the various lysoPA occurs via lysophospholipase, lipid phosphate phosphatases (phospholipid phosphatases: PLPP), and 1-acylglycerol-3-phosphate O-acyltransferases (AGPAT). The AGPAT enzyme family consists of 11 members. Some of the AGPAT enzymes are also called lysophosphatidic acid acyltransferases (LPAAT). Lysophospholipase converts lysoPA to glycerol-3-phosphate. Lipid phosphate phosphatases convert lysoPA to monoacylglycerides. Members of the AGPAT family convert lysoPA to phosphatidic acids.
Lysophosphatidylinositol, LysoPI
Studies on the biological activities of lysophosphatidylinositol, LysoPI, are not as numerous or as extensive as those on LysoPA and sphinosine-1-phosphate (S1P, details below). However, it is known that numerous cell types produce LysoPI and it has been shown to exert a range of effects. The earliest associations to LysoPI production were in studies on cellular transformation which were associated with accumulation of LysoPI. Studies on potential functions of LPI were limited until very recently when it was shown that the receptor for LysoPI is the orphan GPCR identified as GPR55. It should be noted that other studies have shown that LysoPI can bind and activate another GPCR identified as GPR119 which is discussed above in the context of the biological activities of OEA.
LysoPI activation of GPR55 results in increased intracellular calcium concentrations. This effect of LysoPI is associated with increased insulin release from the pancreas, arterial contraction, and the proliferation and migration of several cell type. In addition, LPI induces calcium flux in hepatic mitochondria. The effects of LysoPI on intracellular calcium mobilization are blocked by GPR55 antagonists as well as in cells where GPR55 levels have been downregulated demonstrating the specificity of the role of GPR55 in LysoPI action.
As indicated, early increased production of LysoPI is associated with cellular transformation. The increased synthesis and release of LysoPI is also associated with induction of proliferation in these same cells. Recent evidence has demonstrated a strong link between GPR55 and LysoPI in an autocrine loop regulating the proliferation of prostate and ovarian cancer cells. In cancer cells cPLA2 generates a pool of LysoPI that is transported from the cell via the action of the ABCC1 transporter. Once released, the LysoPI binds to GPR55 and activates the signaling cascades resulting in increased proliferation. Of significance is the fact that when GPR55 is downregulated in ovarian and pancreatic cancer cell lines their proliferation is inhibited.
Lysophosphatidylserine, LysoPS
LysoPS has been shown to exert important effects in the central nervous system (CNS), immune system, liver, and colon. LysoPS exerts its effects by binding to at least three distinct receptors of the GPCR family, GPR34, P2Y10, and GPR174. It has been proposed that these three receptors be identified as LPS1, LPS2, and LPS3, respectively.
Whereas the various forms of LysoPA are generated through biosynthetic pathways, the production of LysoPS is the result of the catabolism of phosphatidylserine (PS). The enzymes that have been identified forming lysoPS outside the cell include a phosphatidylserine-specific PLA1 (PS-PLA1) and a secreted PLA2 (sPLA2). Another enzyme, α/β-hydrolase domain-containing protein 16 A (ABHD16A) has been shown to generate lysoPS intracellularly. The degradation of lysoPS most likely occurs through the action of other members of the α/β-hydrolase domain-containing protein family, ABHD12 and ABHD6.
Although lysoPS molecules can have a fatty acid at either the sn-1 or sn-2 position, similar to the other LPL, the most active forms of lysoPS possess a fatty acid at the sn-2 position. Several LysoPS species have been characterized with the fatty acid varying in carbon number and degree of unsaturation with C16:0 (palmitic acid), C18:1 (oleic acid), and C22:6 (docosahexaenoic acid) being the most prevalent.
LysoPS has been shown to enhance IgE-triggered histamine degranulation, enhance nerve growth factor (NGF)-induced neurite outgrowth, inhibit mitogen-induced T cell proliferation, induce the chemotactic migration of fibroblasts and glioma cells, and activate Toll-like receptor 2 (TLR2) on dendritic cells (DCs).
Sphingosine-1-phosphate, S1P
Synthesis of S1P occurs exclusively from sphingosine via the action of sphingosine kinases. For details on the synthesis of sphingosine go to the Sphingolipid Metabolism and the Ceramides page.
Sphingosine is phosphorylated in humans through the action of two related sphingosine kinases encode by the SPHK1 and SPHK2 genes. The intermediate in sphingosine synthesis, dihydrosphingosine, is also a substrate for sphingosine kinases. The importance of the action of the sphingosine kinases can be shown by the fact that when both are knocked out in transgenic mice, embryos are not viable due to the incomplete development of the brain and the vascular system.
In vertebrates, S1P is secreted into the extracellular space by specific transporters, one of which is called spinster 2 homolog-2 encoded by the SPNS2 gene. Plasma levels of S1P are high, whereas interstitial fluids contain very low levels. This results in an S1P gradient in different compartments.
Hematopoietic cells and vascular endothelial cells are the major sources of the high plasma S1P concentrations. Lymphatic endothelial cells are also thought to secrete S1P into the lymphatic circulation. The majority of plasma S1P is bound to HDL (65%) with another 30% bound by albumin. Recent work has demonstrated that the ability of HDL to induce vasodilation and migration of endothelial cells, as well as to serve a cardioprotective role in the vasculature is dependent on S1P. These studies suggest that the beneficial property of HDL to reduce the risk of cardiovascular disease may be due, in part, on its role as an S1P chaperone.
Degradation of S1P occurs through dephosphorylation and cleavage. The dephosphorylation occurs via the action of enzymes of the lipid phosphatase family, specifically the two enzymes in the sphingosine-1-phosphate phosphatase subfamily (S1P phosphatase-1 and -2) as well as lipid phosphate phosphatase 3 (LPP3). LPP3 is a member of the phospholipid phosphatase (PLPP) family and is encoded by the PLPP3 gene. Cleavage of S1P is catalyzed by sphingosine-1-phosphate lyase 1 (S1P lyase: encoded by the SGPL1 gene).
The different S1P phosphatases remove the phosphate, thus, regenerating sphingosine which can re-enter the sphingolipid metabolic pathway. When used as a substrate for phospholipid synthesis, S1P is degraded by S1P lyase to yield hexadecenal and phosphoethanolamine. Phosphoethanolamine is the direct precursor for the synthesis of the phospholipid phosphatidylethanolamine (PE). The hexadecenal is converted into hexadecenoic acid by hexadecenal dehydrogenase and then into palmitoyl-CoA. The degradation of S1P by the S1P lyase pathway serves as an important pathway for the conversion of sphingolipids into glycerolipids.
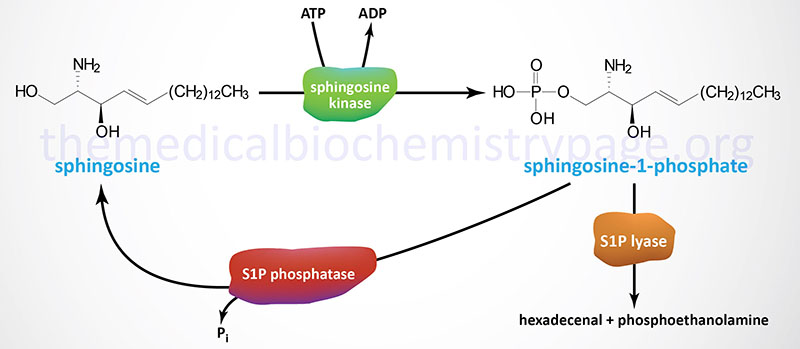
Each of the LPL functions via interaction with specific G-protein coupled receptors (GPCR) leading to autocrine or paracrine effects. The first GPCR shown to bind S1P was called S1P1. Currently there are five characterized S1P receptors. Because several of the LPL receptors were independently identified in unrelated assays, there are several different names for some members of this receptor family. In particular, there is a group of genes that were originally identified as GPCRs and called endothelial differentiation genes (EDGs) that were later found to be the same as several of the LP receptors. Thus S1P1 is also known as EDG1, S1P2 as EDG5, S1P3 as EDG3, S1P4 as EDG6, and S1P5 as EDG8.
The biological activities attributed to S1P interaction with any of the five identified receptors are broad. These activities include involvement in vascular system and central nervous system development, viability and reproduction, immune cell trafficking, cell adhesion, cell survival and mitogenesis, stress responses, tissue homeostasis, angiogenesis, and metabolic regulation.
Table of Sphingosine-1-phosphate (S1P) Receptors
| S1P Receptor | Alternative Name | Gene Symbol | G-Proteins | Comments |
| S1P1 | EDG1 | S1PR1 | Gi/o | expressed in brain, heart, spleen, liver, kidney, skeletal muscle, thymus, pancreatic β-cells, and numerous white blood cells; within the immune system, activation of S1P1 has been shown to block B cell and T cell chemotaxis and infiltration into tissues, in addition S1P1 activation results in inhibition of late-stage maturation processes associated with T cells; within the central nervous system S1P1 is involved in astrocyte migration and increased migration of neural stem cells; within the vasculature S1P1 is involved in early vascular system development and endothelial cell functions such as adherens junction assembly and vascular smooth muscle cell development; within the pancreas S1P1 functions in islet cell survival and insulin secretion. |
| S1P2 | EDG5 | S1PR2 | Gi/o, Gq, G12/13 | expressed in the brain, heart, spleen, liver, lung, kidney, skeletal muscle, and thymus. S1P2 is involved in the development of epithelial cells, enhancing the survival of cardiac myocytes to ischemic-reperfusion injury, and hepatocyte proliferation and matrix remodeling; within the vasculature S1P2 promotes mast cell degranulation and decreases vascular smooth muscle cell responses to PDGF-induced migration. In the eye S1P2 activation can result in pathologic angiogenesis and disruption in adherens junction formation. |
| S1P3 | EDG3 | S1PR3 | Gi/o, Gq, G12/13 | expressed in the brain, heart, spleen, liver, lung, kidney, skeletal muscle, testis, and thymus. S1P3 activation is associated with a worsening of sepsis, increased inflammation and coagulation; however, with respect to cardiac tissues S1P3 promotes survival in response to ischemic-reperfusion injury |
| S1P4 | EDG6 | S1PR4 | Gi/o, G12/13 | expressed in lymphoid tissues (leukocytes) and within a restricted subset of cells in the lung that includes airway smooth muscle cells; the primary responses to S1P4 activation are increased T cell migration and secretion of cytokines. |
| S1P5 | EDG8 | S1PR5 | Gq, G12/13 | expressed in the brain, spleen, and the skin; within the brain S1P5 activation is associated with inhibition of migration of oligodendrocyte progenitors while increasing the survival of oligodendrocytes; S1P5 also stimulates natural killer (NK) cell trafficking. |
Recent studies have shown that SPHK2 (which contains a nuclear localization signal) and its product S1P are found in the nucleus associated with transcriptional co-repressor complexes that contain histone deacetylase 1 (HDAC1) and HDAC2. The S1P-containing HDAC complexes are prevented from deacetylating lysine residues in the histone tails, thereby altering gene expression patterns. One gene whose expression is upregulated in cells with S1P-HDAC complexes is the cell cycle regulating protein p21CIP which is an inhibitor of cyclin-dependent kinases (CDK) and is involved in p53-mediated apoptosis. This observation suggests that enhancing SPHK2-S1P-associated complexes in the nucleus could be of clinical benefit in p53-deficient types of cancer. Given that mutations in p53 are some of the most commonly occurring genetic abnormalities in cancer, this could prove highly significant.