
Last Updated: February 5, 2026
Introduction to Lipid Mediators of Inflammation
Lipid mediators of inflammation belong to the large family of bioactive lipids termed oxylipins. Oxylipins are derived from omega-6 and omega-3 PUFA as well as from the essential fatty acids. The primary omega-6 PUFA are dihomo-γ-linolenic acid (DGLA) and arachidonic acid. The primary omega-3 PUFA are eicosapentaenoic acid (EPA) and docosahexaenoic acid (DHA). The essential fatty acids are linoleic acid and α-linolenic acid (ALA).
The prostaglandins, thromboxanes, leukotrienes, and lipoxins are derived from arachidonic acid, a 20-carbon PUFA, and are therefore, referred to as the eicosanoids (from Ancient Greek eíkosi for 20). In addition to these four classes of oxylipins, that are derived from arachidonic acid, there are the resolvins, protectins (also called neuroprotectins in the brain), maresins, mono-, di-, and tri-hydroxy fatty acids, epoxy fatty acids, eoxins, and hepoxilins. The various oxylipins are synthesized via the actions of cyclooxygenases, lipoxygenases, and cytochrome P450 (CYP) enzymes.
Actions of Aspirin via Lipid Modulators of Inflammation
Aspirin is the acetylated form of salicylic acid. Salicylate is a common constituent of numerous medicinal plants which have been used for thousands of years to treat pain and rheumatic fever. Ancient Egyptians used the leaves of the Myrtle tree to treat rheumatic pain and Hippocrates treated eye infections with extracts from poplar trees and used extracts from willow bark in treating the pain and fever associated with childbirth. Salicylate was first chemically synthesized in 1859 and it entered into widespread use as an anti-inflammatory in 1876. Salicylate has an extremely bitter taste and causes gastric irritation so researchers set out to develop analogs that would have the same pharmacological benefits but be easier to tolerate upon ingestion. In 1897 Felix Hoffman, at the Bayer company, discovered the mechanism to acetylate salicylate giving rise to the advent of aspirin (acetylsalicylic acid).
Even though aspirin was in use for over 70 years its mode of action remained unknown. In 1960 H.O. Collier and colleagues determined that aspirin worked, in part, through modulation of the activation of the pathways involved in the synthesis of the prostaglandins (PG). For details on the synthesis of the prostaglandins go to the Eicosanoid Metabolism: Prostaglandins, Thromboxanes, Leukotrienes, and Lipoxins page.
In the 1970s it was determined that aspirin and other non-steroidal anti-inflammatory drugs (NSAID) all exerted their effects through the inhibition of PG synthesis via the inhibition of the bifunctional enzyme commonly called cyclooxygenase, COX (prostaglandin-endoperoxide synthase: PTGS). Humans express two COX isoforms, COX-1 (PTGS-1) and COX-2 (PTGS-2). However, this did not explain all of the actions that were being described for aspirin, in particular the ability of aspirin to limit leukocyte migration into sites of inflammation, thereby dampening host inflammatory responses.
Among the NSAIDs, aspirin is unique in that it not only has analgesic (pain), antipyretic (fever), and anti-inflammatory effects (exerted at the level of the PG and TX synthesis) but it also exerts beneficial effects on the cardiovascular system via anti-inflammatory pathways distinct from PG and TX inhibition.
At high doses aspirin functions to block the PG and TX synthesizing activity of COX-1 which results in inhibition of the primary pro-inflammatory, pyretic and pain-inducing action of these eicosanoids. In addition, aspirin is an important inhibitor of platelet activation by reducing the production of thromboxane A2 (TXA2). Aspirin also reduces endothelial cell production of prostacyclin (PGI2), an inhibitor of platelet aggregation and a vasodilator. Localized to the site of coagulation is a balance between the levels of platelet derived TXA2 and endothelial cell derived PGI2. This allows for platelet aggregation and clot formation but preventing excessive accumulation of the clot, thus maintaining blood flow around the site of the clot. Endothelial cells regenerate active COX faster than platelets because mature platelets cannot synthesize the enzyme, requiring new platelets to enter the circulation (platelet half-life is approximately 4 days). Therefore, PGI2 synthesis is greater than that of TXA2. The net effect of aspirin is more in favor of endothelial cell-mediated inhibition of the coagulation cascade.
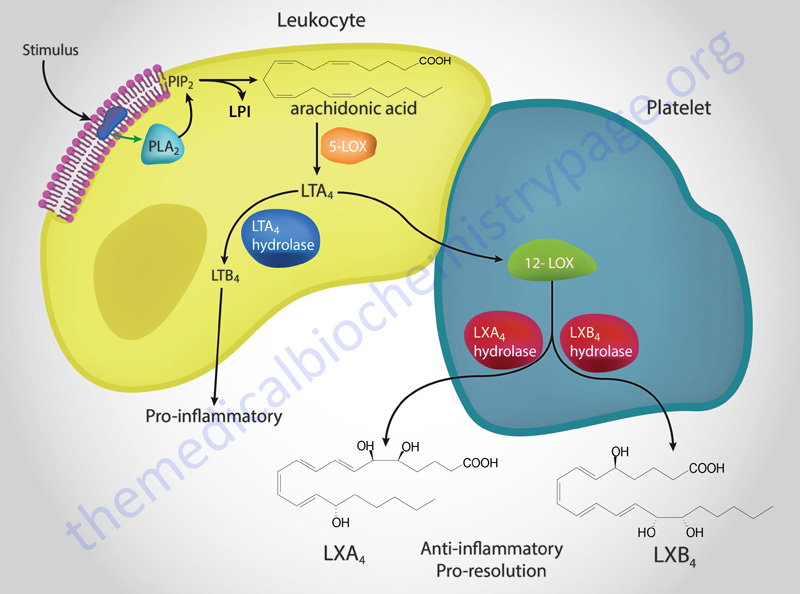
Biosynthesis of the Lipoxins
Part of the cardiovascular benefits of aspirin are related to its dose-dependent differential effects on inflammatory events. Only at low doses (e.g. 81mg) will aspirin elicit its most important anti-inflammatory benefits. The low dose anti-inflammatory effects of aspirin are due to its ability to trigger the synthesis of the lipoxins (LX: LXA4 and LXB4). Higher doses of aspirin have no significant effect on LX synthesis. The lipoxins are anti-inflammatory eicosanoids synthesized through lipoxygenase interactions (hence the derivation of the name: see Figure below). The anti-inflammatory actions of the LX are, in part, a function of their ability to inhibit the actions of the leukotrienes.
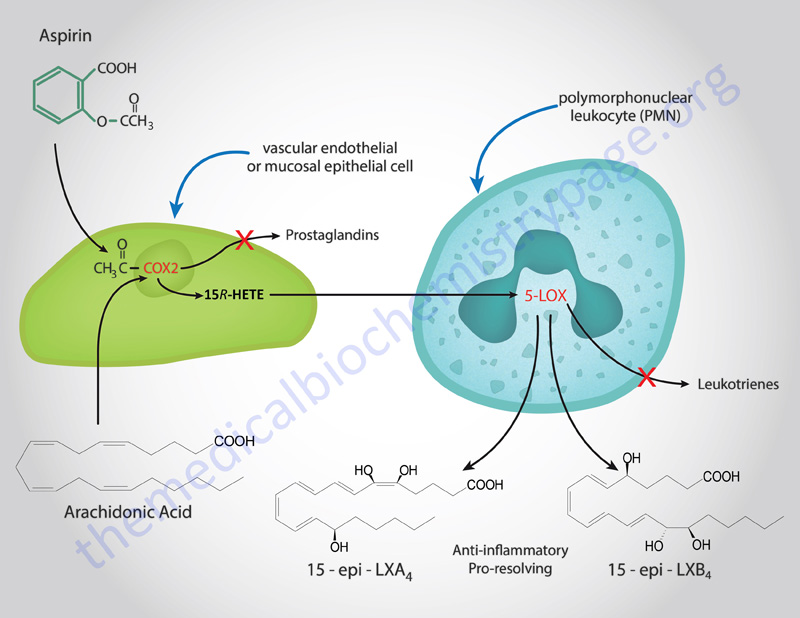
The finding that aspirin could induce the synthesis of LX was one of the most exciting discoveries in pharmocology and was deduced in 1995 by Charles Serhan and colleagues. Aspirin was shown to trigger the synthesis of stereoisomers (epimers) of LXA4 and LXB4 identified as 15 epi-LXA4 and 15 epi-LXB4 (these compounds are also referred to as aspirin-triggered lipoxins (ATLs).
It was known for many years that aspirin inhibited the action of COX-1 and COX-2 by causing the acetylation of the enzyme. However, in endothelial and epithelial cells the aspirin-induced acetylation of COX-2 alters the enzyme such that it now converts arachidonic acid to 15R hydroxyeicosatetraenoic acid (15R-HETE). This latter compound is then rapidly metabolized to the epi-LX in monocytes and leukocytes through the action of 5-lipoxygenase (5-LOX), the enzyme also responsible for initiation of leukotriene synthesis. There are two additional “classic” pathways for LX synthesis that are not triggered by aspirin as shown in the Figure above.
Biological Activities of the Lipoxins
As indicated in the Figure above, the two “classical” pathways for the synthesis of the lipoxins are the result of the concerted actions of 15-LOX (acting on arachidonic acid in epithelial cells, such as in the airway) and 5-LOX in leukocytes or through the actions of 5-LOX in leukocytes followed by 12-LOX action in platelets. This latter activity requires that platelets interact directly with adherent neutrophils as occurs only following platelet activation.
There is also an aspirin-triggered pathway leading to the synthesis of the epimeric lipoxins, 15 epi-LXA4 and 15 epi-LXB4 (also called aspirin-triggered lipoxins, ATL). As described above, this latter pathway is initiated as a consequence of the aspirin-induced acetylation of COX-2 which leads to 15R-HETE synthesis from arachidonic acid which is then converted to the epi-LX. The aspirin-triggered lipoxin synthesis pathway is initiated when activated circulating leukocytes (primarily neutrophils) adhere to the vascular endothelium. Activated leukocytes that adhere to epithelial cells as a consequence inflammation (such as gastrointestinal, airway or kidney epithelia) also result in the production of LXs. An additional stimulus that leads to LX production is epithelial cell conversion of LTA4 that is released from airway epithelia.
The lipoxins are potent anti-inflammatory eicosanoids and counteract the actions of the pro-inflammatory eicosanoids (primarily LTB4 but also PGE2 and TXA2). The lipoxins LXA4 and 15 epi-LXA4 elicit their effects by binding to a specific G protein-coupled receptor (GPCR) originally identified as ALXR. ALXR was subsequently shown to be the same as the formyl peptide receptor 2 (FPR2) protein. FPR2 is was originally assigned to the formyl peptide receptor (FPR) family of receptors that bind N-formylated peptides derived by the degradation of bacteria or host cells. Subsequently FPR2 was shown to also be a member of the leukotriene receptor family of receptors. FPR2 is also capable of interacting with several small peptide derived signaling molecules indicating that it is a multi-recognition receptor involved in immune responses.
The three original members of the FPR gene family in humans are FPR1, FPR2, and FPR3. Confusion in the literature stems from the fact that FPR1 was originally identified as FPR, FPR2 was originally identified as FPR1, and FPR3 was originally identified as FPR2. The FPR2 (ALXR) protein is encoded by the formyl peptide receptor 2 (FPR2) gene. The FPR family of receptors, including FPR2, are each coupled to the activation of Gi-type G-proteins.
Both LXA4 and LXB4 have been shown to promote the relaxation of the vasculature (both aortic and pulmonary relaxation). Lipoxins and epi-LX inhibit polymorphonuclear leukocyte (PMN) chemotaxis, PMN-mediated increases in vasopermeability, and PMN adhesion and migration through the endothelium. The LX also stimulate phagocytosis of apoptotic PMNs by monocyte-derived macrophages. PMN phagocytosis represents the resolution phase of inflammatory events, thus aspirin promotes this process and increases the rate of return to the normal tissue state. The pro-resolving activity of aspirin is exerted not only through the induced synthesis of the lipoxins, but also via the induced synthesis of an additional class of anti-inflammatory lipid mediators known as the resolvins (Rvs)and the protectins (PDs).
Additional anti-inflammatory actions of the lipoxins and aspirin-triggered lipoxins include blocking expression of the IL-8 gene (a pro-inflammatory chemokine produced by macrophages and endothelial that stimulates neutrophil migration), inhibition of the release and actions of tumor necrosis factor-α (TNF-α), and stimulation of transforming growth factor-β (TGF-β) activity. By regulating the actions of histamine the lipoxins also lead to a reduction in swelling due to edema.
In addition, the actions of LXA4 in some tissues leads to the production of prostacyclin (PGI2) and nitric oxide (NO) both of which are vasodilators and may play roles in the anti-inflammatory properties of the aspirin-triggered lipoxins (ATLs) 15 epi-LXA4 and 15 epi-LXB4. The induction of NO by aspirin is correlated, in a dose-dependent manner, with a reduction in leukocyte accumulation at sites of inflammation. No other NSAID has been shown to exert this effect on NO production making aspirin unique among this class of drug. The induced production of NO by aspirin plays a significant role in the protective effects of aspirin on the cardiovascular system.
The Resolvins, Protectins, and Maresins
As discussed in detail above, the lipoxins and the aspirin-triggered lipoxins represent a clinically significant class of bioactive lipids that are derived from the omega-6 PUFA, arachidonic acid. Numerous bioactive metabolites of the omega-3 PUFA, eicosapentaenoic acid (EPA) and docosahexaenoic acid (DHA) have also been identified. These omega-3 PUFA-derived bioactive lipids are identified as the resolvins (Rv), protectins (PD), and maresins MaR).
Complexity in the structures of the resolvins, protectins, and maresins stems from the fact that different stereoisomers (S and R) can be generated. These various bioactive lipids are derived through the concerted actions of cytochrome P450 (CYP) enzymes and different members of the lipoxygenase family (5-LOX, 12-LOX, and 15-LOX). In addition, the acetylating effects of low dose aspirin on the activity of COX-2 leads to epimeric structures being generated.
The CYP enzymes that are involved in the synthesis of the resolvins are those that possess epoxygenase activity or ω-hydroxylase activity. These epoxygenase CYP enzymes (CYP2C and CYP2J family enzymes) convert arachidonic acid, EPA, and DHA into their epoxy forms, epoxy-eicosatrienoic acids (EpETrE), epoxy-eicosatetraenoic acids (EpETE), and epoxydocosapentaenoic acids (EpDPE; sometimes designated EpDPA), respectively.
The ω-hydroxylase activities (CYP4A family enzymes) convert arachidonic acid, EPA, and DHA to hydroxyeicosatetraenoic acids (HETE), hydroxyeicosapentaenoic acids (HEPE), and hydroxydocosahexaenoic acids (HDoHE), respectively. The products of epoxygenases are rapidly metabolized via epoxide hydrolase 2 (EPHX2) to the corresponding dihydroxy (diol) derivatives. The EPHX2 encoded enzyme is commonly called soluble epoxide hydrolase (sEH).
These important EPA- and DHA-derived bioactive metabolites are often referred to as anti-inflammatory lipids. More precisely, the Rv, PD, and MaR molecules do not inhibit the onset of inflammation but stimulate the resolution of the response resulting in the shutting off of an inflammatory process. Thus far at least 16 resolvin molecules have been characterized along with 7 protectins and 5 maresin molecules. Collectively, these immune modulating EPA and DHA derivatives are referred to as specialized proresolving mediators, SPM.
Resolvins
The resolvins were initially identified by their ability to promote the resolution of the inflammatory cycle, hence the derivation of their names as resolvins. The resolvins are divided into two classes with the E-series resolvins (RvE) being synthesized from EPA and the D series resolvins (RvD) being derived from DHA.
Resolvin D Series
The RvD family currently consists of 10 characterized members that are synthesized from DHA via the actions of aspirin-mediated acetylation of COX-2, or via the activity of 15-LOX, followed by the actions of the lipoxygenases, 5-LOX and 15-LOX. The D series resolvin family also includes molecules identified as resolvin conjugates in tissue regeneration (RCTR).
DHA is first converted to 17(S)-hydroperoxydocosahexaenoic acid [17(S)-HpDHA] via the actions of 15-LOX or to 17(R)-HpDHA via the action of aspirin modified COX-2. As a result of auto oxidation 17(S)-HpDHA is converted to 17(S)-hydroxydocosahexaenoic acid [17(S)-HDHA]. Conversion of 17(S)-HDHA to the D series resolvins (RvD1-RvD6) occurs via the action of 5-LOX. At least two 17(R)-series resolvin epimers are generated from DHA.
Conversion of 17(S)-HDHA to 7(S)-hydroperoxy-17(S)-hydroxy-DHA or 4(S)-hydroperoxy-17(S)-hydroxy-DHA occurs via the actions of 5-LOX. The compound, 7(S)-hydroperoxy-17(S)-hydroxy-DHA, is ultimately converted to RvD1 [7(S),8(R),17(S)-trihydroxy-DHA], RvD2 [7(S),16(R),17(S)-trihydroxy-DHA], and RvD5 [7(S),17(S)-dihydroxy-DHA]. In the conversion of 7(S)-hydroperoxy-17(S)-hydroxy-DHA to RvD1 and RvD2 the intermediate trihydroxy compound, 7(S),8(S),17(S)-trihydroxy-DHA, is formed.
Enzymes of the glutathione-S-transferase family conjugate glutathione (GSH) to 7(S)-hydroperoxy-17(S)-hydroxy-DHA converting it to RCTR1 [7(S),17(S)-dihydroxy-DHA+GSH]. RCTR1 can, in turn, be converted to RCTR2 and then RCTR3.
The compound, 4(S)-hydroperoxy-17(S)-hydroxy-DHA, is converted RvD3 [4(S),11(R),17(S)-trihydroxy-DHA], RvD4 [4(S),5(R),17(S)-trihydroxy-DHA], and RvD6 [4(S),17(S)-dihydroxy-DHA]. In the conversion of 4(S)-hydroperoxy-17(S)-hydroxy-DHA to RvD3 and RvD4 the intermediate trihydroxy compound, 4(S),5(S),17(S)-trihydroxy-DHA, is formed.
The actions of aspirin, coupled with 5-LOX, yield the aspirin-triggered D-series resolvins (AT-RvD1 through AT-RvD6). Aspirin-modified COX-2 generates 17(R)-hydroperoxydocosahexaenoic acid [17(R)-HpDHA] which auto-oxidizes to 17(R)-hydroxy-DHA [17(R)-HDHA]. The actions of 5-LOX to convert 17(R)-HDHA to 7(S)-hydroperoxy-17(R)-HDHA or 4(S)-hydroperoxy-17(R)-HDHA. The compound 7(S)-hydroperoxy-17(R)-HDHA can be converted to AT-RvD5 [7(S),17(R)-dihydroxy-DHA] or to 7(S),8(S)-epoxy-17(R)-HDHA. The compound 7(S),8(S)-epoxy-17(R)-HDHA is then converted to AT-RvD1 [7(S),8(R),17(R)-trihydroxy-DHA] and AT-RvD2 [7(S),16(R),17(R)-trihydroxy-DHA]. The compound 4(S)-hydroperoxy-17(R)-HDHA is converted to AT-RvD6 [4(S),17(R)-dihydroxy-DHA] or to 4(S),5(S)-epoxy-17(R)-HDHA. The compound 4(S),5(S)-epoxy-17(R)-HDHA is then converted to AT-RvD3 [4(S),11(R),17(R)-trihydroxy-DHA] and AT-RvD4 [4(S),5(R),17(R)-trihydroxy-DHA].
Resolvin E Series
The RvE family currently consists of four characterized members (Rve1-RvE4) that are synthesized from EPA via the actions of CYP enzymes, aspirin modified COX-2, 5-LOX, and/or 15-LOX. Aspirin-modified COX-2, or a CYP enzyme, converts EPA to 18(R)-hydroperoxyeicosapentaenoic acid [18(R)-HpEPE] which then is converted to 18(R)-hydroxyeicosapentaenoic acid [18(R)-HEPE] via the action of a peroxidase. The 18(S) stereoisomers of RvE1 and RvE2 have also been identified and are derived from 18(S)-HEPE.
The actions of 15-LOX on 18(R)-HEPE, followed by peroxidation, result in the production of RvE3 [17(S),18(R)-dihydroxy EPA].
The actions of 5-LOX on 18(R)-HEPE result in the production of 5(S)-hydroperoxy-18(R)-HEPE. Peroxidation of 5(S)-hydroperoxy-18(R)-HEPE leads to the production of RvE2 [5(S),18(R)-dihydroxy-EPA].
Dehydration of 5(S)-hydroperoxy-18(R)-HEPE results in the production of 5(S),6(S)-epoxy-18(R)-HEPE which in then hydrolyzed generating RvE1 [5(S),12(R),18(R)-trihydroxy-EPA].
The action of 15-LOX on EPA yields 18(S)-hydroperoxyeicosapentaenoic acid [15(S)-HpEPE]. The action of 5-LOX on 15(S)-HpEPE yields RvE4 [5(S),15(S)-dihydroxy EPA].
The levels of RvE1 increase spontaneously in individuals taking aspirin or consuming EPA. RvE1 is produced in a transcellular manner involving endothelial cells and leukocytes. Within the endothelium EPA is converted to 18R-HEPE [18(R)-hydroxyeicosapentaenoic acid] by aspirin acetylated COX-2. The 18(R)-HEPE is released by the endothelial cells and taken up by adherent leukocytes where 5-LOX converts it to RvE1. Certain COX-2 inhibitors block this pathway to RvE1 synthesis but the NSAIDs, indomethacin and acetaminophen do not.
The E series resolvins reduce inflammation, regulate PMN infiltration by blocking transendothelial migration, reduce dendritic cell function (dendritic cells are potent antigen presenting cells which prime T cell mediated inflammatory responses), regulate IL-12 production and lead to resolution of the inflammatory responses. RvE1 also inhibits PMN superoxide anion generation in response to TNFα or to the bacterial peptide N-formyl-methionyl-leucyl-phenylalanine (f-MetLeuPhe). RvE1 stimulates the phagocytosis of apoptotic PMNs by macrophages.
In a rabbit model of periodontitis, administration of RvE1 led to a complete regeneration of damaged tissues that included bone. This action of RvE1 was unique in that lipoxin pre-treatment did not exert the same effect. When compared to the use of aspirin or steroids such as dexamethasone in the same periodontitis model RvE1 was vastly more potent at resolving the inflammation.
RvE1 is also able to selectively disrupt thromboxane-mediated platelet aggregation indicating further its ability to exert anti-inflammatory effects. In the presence of aspirin and DHA, COX-2 activity leads to the synthesis of the aspirin-triggered resolvins which are also members of the D series of resolvins.
Protectins
The protectin, PD1 was originally identified as NPD1 (where the N refers to neuroprotectin). PD1 is derived from DHA via the actions of 15-LOX or to the aspirin-triggered protectin 1 (AT-PD1) via the actions of aspirin modified COX-2. DHA is first converted to 17(S)-hydroperoxydocosahexaenoic acid (17(S)-HpDHA) via the actions of 15-LOX or to 17(R)-HpDHA via the actions of aspirin modified COX-2. The action of 15-LOX then converts 17(S)-HpDHA to 16(S),17(S)-epoxy-DHA which is subsequently converted to PD1 [10(R),17(S)-dihydroxy-DHA] via the actions of soluble epoxide hydrolase (sEH; encoded by the EPHX2 gene). In the aspirin-triggered pathway, 17(R)-HpDHA is converted to 16(R),17(R)-epoxy-DHA and then to AT-PD1 [10(R),17(R)-dihydroxy-DHA].
The protectins NPD1 and AT-PD1 have been shown to exert potent inflammation resolving actions by inhibiting T cell and PMN chemotaxis, by promoting T cell apoptosis, by decreasing TNFα and interferon-gamma (INF-γ) secretion, by reducing airway inflammation, and by exerting neuroprotective action during ischemia-reperfusion injury.
An additional protectin identified as protectin DX is formed through the sequential actions of two lipoxygenase reactions and this protectin has been shown to inhibit platelet aggregation, inhibit the formation of reactive oxygen species (ROS) production, and to inhibit COX-2 activity in neutrophils.
Maresins
The maresins, named maresin 1 (MaR1): [7(R),14(S)-dihydroxy-DHA] and MaR2 [13(R),14(S)-dihydroxy-DHA), are derived through the action of the lipoxygenase, 12-LOX on DHA. DHA is first converted to 14(S)-hydroperoxydocosahexaenoic acid [14(S)-HpDHA] by the actions of 12-LOX and then to the maresins via the actions of soluble epoxide hydrolase (sEH; encoded by the EPHX2 gene) or other members of the epoxide hydrolase family.
Maresin 1 is produced mainly by macrophages hence the derivation of the name maresin from macrophage mediator resolving inflammation. The maresin family of molecules also includes those identified as maresin conjugates in tissue regeneration (MCTR).
Maresin 1 exhibits potent anti-inflammatory, pro-resolving, analgesic, and wound healing activities. Major cellular targets for the actions of MaR1 are vascular smooth muscle (VSM) cells and vascular endothelial cells. In these cells MaR1 attenuates the adhesion of monocytes to the endothelium induced by TNFα.
Maresin 1 also inhibits the production of reactive oxygen species by both VSM and endothelial cells. The major mechanism through which MaR1 exerts these effects, in VSM and endothelial cells, is through down-regulation of the transcription factor, nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB).
Maresin 2 has been shown to reduce neutrophil infiltration and to enhance macrophage-mediated phagocytosis of dead and dying cells, a process termed efferocytosis.
Two related structures termed the maresin-like mediators (MaR-L1 and MaR-L2) are generated when the maresins produced by macrophages are released and acted upon by platelets.
Recent work has shown that the number of DHA-derived bioactive lipid mediators is quite extensive and includes many more metabolites than just the resolvins, protectins, and maresins. Bioactive DHA-derived lipids include the electrophilic oxo-derivatives (EFOX) which exhibit anti-proliferative and anti-inflammatory effects; the neuroprostanes (NP) which exert wound healing and cardioprotective effects; the epoxides which exhibit analgesic and antihypertensive effects; the ethanolamines which exert neuromodulatory, immune modulatory, and metabolic effects; the fatty acid esters of hydroxy fatty acids (FAHFA) which are inflammation proresolution and immune modulatory molecules; and the N-acyl amides which exert neuroprotective and metabolic regulatory effects.
Aspirin can trigger the synthesis of an epimeric protectin D1 compound as well as epimeric resolvin D series compounds. The switch from synthesis of pro-inflammatory eicosanoids, such as the prostaglandins and the thromboxanes, to the pro-resolving lipoxins, resolvins, and protectin D1, occurs via induction of 15-LOX. Disruption of COX-2 or inhibition of LOX enzymes results in a disruption in the normal process of inflammation resolution. The important role of 15-LOX in pro-resolution phenomena can be demonstrated by the fact that in animal models where 15-LOX can be over-expressed, resolution of inflammation occurs more readily. The abbreviation AT- refers to the aspirin-triggered compound.
Receptors Binding Resolvins, Protectins, and Maresins
The various lipid mediators derived from EPA and DHA exert their effects by binding to cell surface receptors, the majority of which belong to the G-protein coupled receptor (GPCR) family. The various bioactive metabolites of DHA are known to bind to the cannabinoid receptors, members of the transient receptor potential vanilloid channels (TRPV channels), N-formyl peptide receptor 2 (encoded by the FPR2 gene), GPR18, GPR32, GPR37, and adhesion G protein-coupled receptor F1 (also called GPR110).
GPR18, which binds RvD2, is also identified as D resolvin receptor 2 (DRV2).
GPR32 has been shown to bind RvD1 and the lipoxin, LXA4 (see above) and as such is also designated as D resolvin receptor 1 (DRV1). GPR32 is also activated by binding the D-series resolvins RvD3 and RvD5 and the aspirin-triggered epimer of RvD3, AT-RvD3.
Protectin D1 (PD1) interacts with the GPR37 receptor which is a Gi-type G-protein coupled receptor. Expression of GPR37 is highest in the CNS within the amygdala, basal ganglia, substantia nigra, hippocampus, frontal cortex, and hypothalamus, with exceptionally high levels of expression observed the spinal cord. GPR37 has also been identified as the parkin-associated endothelin receptor-like receptor (PaelR). Activation of GPR37 has been associated with states of chronic inflammation such as neurodegenerative disorders and cancers.
The N-formyl peptide receptor 2 (which is also identified as ALX) also binds RvD1 and LXA4.
The chemerin chemokine-like receptor 1, encoded by the CMKLR1 gene (protein formerly identified as ChemR23), is activated by the E-series resolvins RvE1 and RvE2 and as such is also identified as E resolvin receptor 1 (ERV1).
The leukotriene B4 (LTB4) receptor, BLT1, is also activated by RvE1 and RvE2.
Role of Epoxy-Oxylipins in Inflammatory Resolution
Polyunsaturated fatty acids (PUFA) are oxidized to epoxides by epoxygenases that are members of the large cytochrome P450 family of enzymes. The action of these epoxygenases generate numerous potent bioactive lipid mediators. The epoxygenase pathway primarily involves enzymes of the CYP2C and CYP2J families although the CYP1A family members, CYP1A1 and CYP1A2, also function as a lipid epoxygenases.
With respect to the metabolism of arachidonic acid, related eicosanoids, and other bioactive lipids, the CYP2C8 and CYP2C9 enzymes are the most significant. CYP2C8 functions as an expoxygenase to convert long-chain polyunsaturated fatty acids (PUFA) to their biologically active epoxide forms.
The significance of these many lipid epoxides is that they regulate vascular tone, modulate angiogenesis, have anti-inflammatory actions, have inflammation resolution properties, protect the heart against ischemia–reperfusion injury, are potent inhibitors of cardiac arrhythmias, modulate inflammatory and neuropathic pain, and reduce tumor metastasis.
Linoleic acid (9,12-octadecadienoic acid) derived epoxides are epoxyoctadecamonoenoic acids (EpOME). The EpOME are also identified as epoxyoctadecenoic acids.
Arachidonic acid derived epoxides are epoxyeicosatrienoic acids (EET; also designated EpETrE).
EPA derived epoxides are epoxyeicosatetraenoic acids (EpETE).
DHA derived epoxides are epoxydocosapentaenoic acids (EpDPE; these are sometimes designated EpDPA).
The epoxides can exist as either R,S or S,R enantiomers. For example the linoleic acid derived EpOME are 9,10-epoxyoctadecenoic acid (9,10-EpOME; coronaric acid, also known as leukotoxin), 12,13-epoxyoctadecenoic acid (12,13-EpOME; vernolic acid, also known as isoleukotoxin) and these can be 9R,10S-EpOME or 9S,10R-EpOME and 12R,13S-EpOME or 12S,13R-EpOME.
Epoxide Hydrolases
Epoxide hydrolases convert lipid epoxides to their corresponding diols which can attenuate the function of the lipid epoxides. For example soluble epoxide hydrolase (sEH) activity on the linoleic acid derived 12,13-EpOME converts it to 12,13-dihydroxyoctadecamonoenoic acid (12,13-DiHOME).
Humans express four epoxide hydrolase genes, identified as EPHX1-EPHX4, that encode enzymes of the α/β hydrolase fold (ABHD) enzyme family. The EPHX2 encoded enzyme is commonly called soluble epoxide hydrolase (sEH). Soluble epoxide hydrolase (sEH) is predominantly found in the cytosol and is highly expressed in the liver.
Inactivating mutations in the EPHX2 gene that encodes sEH, as well as pharmacologic inhibition of sEH, is associated with attenuation of the development of various diet-induced disorders, including endoplasmic reticulum (ER) stress, metabolic syndrome, insulin resistance, hepatic steatosis, hepatic fibrosis, inflammation, and endothelial dysfunction.
Effects of 12,13-EpOME Conversion to 12,13-DiHOME in Inflammation
As indicated, the epoxy-oxylipin, 12,13-EpOME, is derived from linoleic acid via the actions of CYP epoxygenases and is then converted to the diol, 12,13-DiHOME, through the actions of sEH. The levels of 12,13-EpOME have been shown to increase at sites of inflammation during the transition from the onset of inflammatory reposes to their resolution. The effects of 12,13-EpOME, at the level of inflammation, have been shown to include the inhibition NF-κB and interference with p38 MAPK phosphorylation leading to reductions in the expression of pro-inflammatory cytokines.
The conversion of 12,13-EpOME to 12,13-DiHOME via sEH action results in a reduction in the level of the inflammation resolution properties of 12,13-EpOME, as well as other epoxy-oxylipins. Inhibition of sEH activity results in a significant elevation in the levels of 12,13-EpOME and a consequent reduction in the level of intermediate monocytes during inflammation.