
Last Updated: February 16, 2026
Introduction to Argininosuccinate Synthetase Deficiency
Arginosuccinate synthetase (also identified as argininosuccinate synthase) deficiency (ASD) is an autosomal recessive disorder of the urea cycle that also affects the synthesis of arginine. This disorder is more commonly referred to as citrullinemia type I (CTLN1) or classic citrullinemia due to the accumulation of citrulline in addition to the associated hyperammonemia. The frequency of ASD is approximately 1 per 57,000 live births.
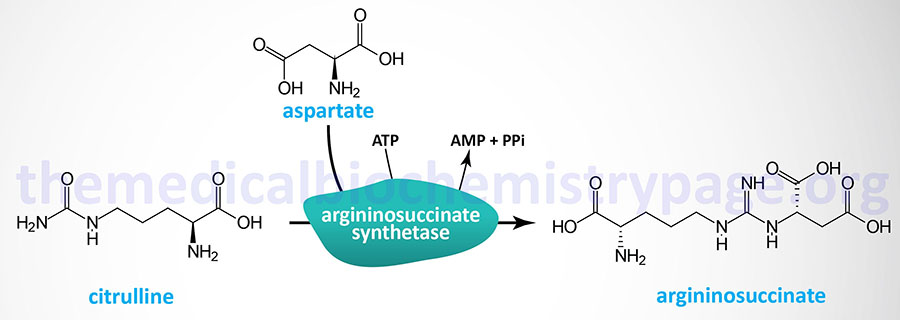
Argininosuccinate synthetase (also identified as argininosuccinate synthase) catalyzes the urea cycle reaction that condenses aspartic acid with citrulline forming argininosuccinate. This reaction requires the equivalent of two mole of ATP to drive it in the forward direction.
Molecular Biology of Argininosuccinate Synthetase Deficiency
Argininosuccinate synthetase deficiency results from mutations in the arginosuccinate synthetase gene (ASS1). The ASS1 gene is located on chromosome 9q34.11 spanning 63 kb and composed of 16 exons that generate two alternatively spliced mRNAs, both of which encode the same 412 amino acid protein. Surprisingly, there are at least 14 AS pseudogenes found on various chromosomes, including two pseudogenes on chromosome 9 but distant from the location of the active ASS1 gene.
Functional argininosuccinate synthetase exists as a homotetramer.
There are at least 22 known mutations in the AS gene that result in argininosuccinate synthetase deficiency (ASD). Mutations include missense, nonsense, and exon deletions.
Clinical Features of Argininosuccinate Synthetase Deficiency
ASD is, like the other neonatal onset forms of urea cycle disorders (UCD), most severe when presenting in newborn infants. As with each of the four neonatal onset UCD, ASD is characterized by the accumulation of ammonia and glutamine with clinical manifestations appearing in full-term infants with no prior obstetric risk factors. The classic symptoms appear between 24hrs and 48hrs after birth (but not prior to 24hrs) and include convulsions, hyperventilation, ataxia, hypothermia, lethargy, vomiting, and poor feeding. If left untreated the hyperammonemia with result in coma and death. The severe effects of hyperammonemia are described in the Nitrogen Metabolism and the Urea Cycle page.
Even though sepsis is a rare event in a normal term infant with no prior obstetric complications, this disorder is misdiagnosed in almost half of neonatal UCD cases. Initial laboratory findings will include respiratory alkalosis which is the earliest objective indication of encephalopathy. The encephalopathy will progress to the point where mechanical ventilation is required. Another routine laboratory finding is reduced serum (blood) urea nitrogen (BUN) which may be as low as 1mg/dl (normal for newborns is 3–12mg/dl). If plasma ammonia levels are not measured the infants’ death will be attributed to sepsis, intracranial hemorrhage, or some other disorder that would normally be associated with a pre-term delivery.
Treatment of Argininosuccinate Synthetase Deficiency
ASD patients are treated in much the same ways as for other neonatal UCDs in that protein intake must me highly regulated and the hyperammonemia must be controlled. Hemodialysis is the only effective means to rapidly lower serum ammonia levels in these patients. Acute episodes of hyperammonemia can be treated with intravenous administration of Ammunol® and with oral Buphenyl® for chronic adjunctive therapy of hyperammonemia. The mechanism of action of these compounds is detailed in the Urea Cycle Disorders: Overview page.
Additionally, ASD is treated with oral arginine. The utility of arginine therapy stems from the conversion, ultimately, to citrulline by other enzymes of the urea cycle. The arginine is cleaved to urea and ornithine by the action of arginase. Ornithine and carbamoyl phosphate are condensed to citrulline by the action of ornithine transcarbamylase (OTC). The citrulline is then excreted in the urine. Unlike the utility of oral arginine therapy in the treatment of argininosuccinate lyase deficiency (ALD), which leads the excretion of 2 moles of waste nitrogen as argininosuccinate, citrulline only contains 1 mole of waste nitrogen and excretion of citrulline in the urine is not very efficient. Therefore, it is necessary to include sodium phenylbutyrate (or Buphenyl®) in the treatment regimen.