
Last Updated: February 19, 2026
Introduction to Arginase Deficiency
Arginase deficiency (AD) represents one of the disorders that result from defects in the processes of the urea cycle. Arginase deficiency is a rare autosomal recessive disorder. Arginase deficiency is the result of mutations in the ARG1 gene which encodes arginase. Arginase catalyzes the release of urea from arginine while simultaneously generating ornithine.
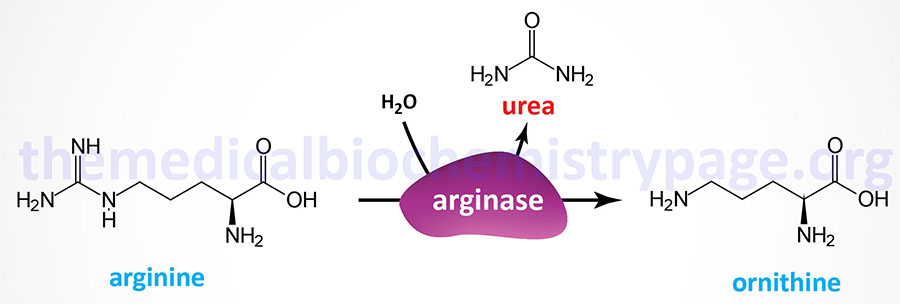
Arginase deficiency is associated with persistent hyperargininemia and debilitating cognitive, neurologic, and mobility impairments. Intracellular hepatic arginine levels in AD patients is routinely 50 times the normal level. The hepatic arginine is readily transported to the blood and subsequently accumulates in many other tissues, particularly the brain and cerebrospinal fluid (CSF). The excess arginine is metabolized to arginine-derived guanidino compounds which are thought to be the reason for the neurotoxicity of arginase deficiency.
Molecular Biology of Arginase Deficiency
Humans express two distinct arginase genes identified as ARG1 and ARG2 that encode the arginase type I and type II enzymes, respectively. The ARG1 gene encoded protein is the cytoplasmic liver enzyme contributing 98% of the urea cycle arginase activity in that organ. ARG2 encodes a mitochondrial arginase found predominately in the kidneys.
The ARG1 gene is located on chromosome 6q23.2 and is composed of 8 exons that generate three alternatively spliced mRNAs. One of these mRNAs encodes a protein of 330 amino acids (isoform 1) and this protein is often referred to as the erythroid variant. The other two ARG1-derived mRNA encodes proteins of 322 amino acids (isoform 2) and 237 amino acids (isoform 3). The functional arginase enzyme exists as a homotrimer.
Mutations in ARG1 give rise to hyperargininemia also known as arginase deficiency (AD). Mutations in ARG1 gene are rare and the frequency of arginase deficiency is approximately 1 per 363,000 live births. Mutations in the ARG1 gene resulting in argininemia include missense mutations, nonsense mutations, deletions, insertions, duplications, splicing mutations, and at least one example of mutation of the translation initiation codon. Of all the various mutations in the ARG1 gene, missense mutations are the most common type.
The ARG2 gene is located on chromosome 14q24.1 and is composed of 8 exons that encode a precursor protein of 354 amino acids. Although the precise function of the ARG2 encoded enzyme (arginase type II) remains unclear it is thought to play a role in arginine metabolism, nitric oxide production, and polyamine metabolism. Recent (2024) work in mice and human liver organoids has found that the ARG2 encoded enzyme functions in the coordination of hepatic urea cycle activity with oxidative metabolism.
Clinical Features of Arginase Deficiency
The major symptoms of AD are all progressive and include psychomotor impairment, spastic tetraplegia where the lower limbs are more affected than the upper, hyperactivity, growth failure, and seizures. Because arginase catalyzes the last reaction of the urea cycle, hyperammonemia is not as severe as in other neonatal urea cycle disorders (UCD). In arginase deficiency the ammonia levels are 3-4 times normal as opposed to >6 times normal in, for example, ornithine transcarbamylase deficiency (OTCD).
Patients with arginase deficiency do not present the typical findings associated with the other urea cycle disorders. Arginase deficiency patients usually appear healthy at birth and clinical manifestations typically begin to develop in early childhood in association with high plasma arginine levels with hyperargininemia (and not hyperammonemia) considered to be the primary
driver of disease pathology.
The most prominent laboratory findings in AD are mild to no hyperammonemia, hyperargininemia, hyperaminoaciduria (arginine, lysine, ornithine, and cystine), and orotic aciduria. Plasma arginine levels in AD patients are typically >300 μM (>300 μmol/L) whereas in normal individuals the levels range from 40 -115 μM.
Treatment of Arginase Deficiency
AD patients are treated in much the same ways as for other neonatal UCDs in that protein intake must me highly regulated and the hyperammonemia must be controlled. Hemodialysis is the only effective means to rapidly lower serum ammonia levels in these patients. Acute episodes of hyperammonemia can be treated with intravenous administration of Ammunol® and with oral Buphenyl® for chronic adjunctive therapy of hyperammonemia. The mechanism of action of these compounds is detailed in the Urea Cycle Disorders: Overview page.