
Last Updated: April 3, 2026
Introduction to Amino Acid Catabolism
All tissues have some capability for synthesis of the non-essential amino acids, amino acid catabolism and remodeling, and conversion of non-amino acid carbon skeletons into amino acids and other derivatives that contain nitrogen. However, the liver is the major site of nitrogen metabolism, which includes amino acid catabolism. Another major tissue for amino acid catabolism is skeletal muscle.
In times of dietary surplus, the potentially toxic nitrogen of amino acids is eliminated via transaminations, deamination, and urea formation. During amino acid catabolism the carbon skeletons are generally conserved as carbohydrate, via gluconeogenesis, or as fatty acid via fatty acid synthesis pathways. In this respect amino acids fall into three categories: glucogenic, ketogenic, or glucogenic and ketogenic. Glucogenic amino acids are those that give rise to a net production of pyruvate or TCA cycle intermediates, such as 2-oxoglutarate (α-ketoglutarate) or oxaloacetate, all of which are precursors to glucose via gluconeogenesis. All amino acids except lysine and leucine are at least partly glucogenic. Lysine and leucine are the only amino acids that are solely ketogenic, giving rise only to acetyl-CoA or acetoacetyl-CoA, neither of which can bring about net glucose production.
Glucogenic amino acids are those that give rise to a net production of pyruvate or TCA cycle intermediates, such as 2-oxoglutarate (α-ketoglutarate) or oxaloacetate, all of which are precursors to glucose via gluconeogenesis. All amino acids except lysine and leucine are at least partly glucogenic. Lysine and leucine are the only amino acids that are solely ketogenic, giving rise only to acetyl-CoA or acetoacetyl-CoA, neither of which can bring about net glucose production.
A small group of amino acids comprised of isoleucine, phenylalanine, threonine, tryptophan, and tyrosine give rise to both glucose and fatty acid precursors and are thus, characterized as being glucogenic and ketogenic.
Finally, it should be recognized that amino acids have a third possible fate. During times of starvation the reduced carbon skeleton is used for energy production, with the result that it is oxidized to CO2 and H2O with production of reduced electron carriers, predominantly NADH, used to drive ATP synthesis.
Alanine Catabolism
Alanine serves an important function in nitrogen transport, particularly from skeletal muscle, as part of the glucose-alanine cycle. This pathway delivers waste nitrogen from skeletal muscle to the liver where it can be incorporated into urea. The alanine catabolic pathway involves a simple aminotransferase reaction that directly produces pyruvate. The transamination is carried out by alanine transaminase, ALT (also called alanine aminotransferase).
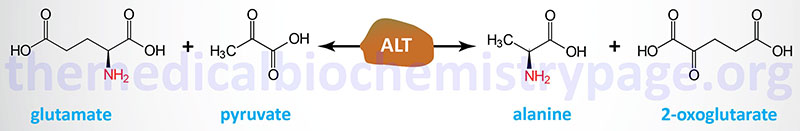
ALT was originally referred to as glutamate pyruvate transaminase, GPT. Humans express two genes encoding alanine transaminase. These genes are identified as GPT (also designated GPT1) and GPT2. The enzyme encoded by the GPT gene is localized to the cytosol, whereas the GPT2 encoded enzyme is localized to the mitochondria.
The GPT gene is located on chromosome 8q24.3 and is composed of 12 exons that generate three alternatively spliced mRNAs, all of which encode the same 496 amino acid protein. The tissues with the highest levels of GPT expression are the liver, the intestines, the kidneys, and adipose tissue.
The GPT2 gene is located on chromosome 16q11.2 and is composed of 13 exons that generate two alternatively spliced mRNAs that encode proteins of 523 amino acids (isoform 1) and 423 amino acids (isoform 2). The tissues with the highest levels of GPT2 expression are the liver, the intestines, the kidneys, adipose tissue, brain, and pancreas.
Generally, the pyruvate produced from alanine has two distinct fates that are controlled by the energy demands of the liver and the metabolic needs of the organism as a whole. When energy needs are high in hepatocytes the pyruvate is oxidized by the PDH complex (PDHc) to acetyl-CoA and diverted into the TCA cycle. Alternatively, during the fasted state when blood glucose levels are low, the pyruvate is diverted into the gluconeogenic pathway so that the liver can release glucose to the blood. This makes alanine a glucogenic amino acid.
Interrelationships of Arginine, Ornithine and Proline Catabolism
The major pathway for the catabolism of arginine begins within the context of the urea cycle. It is ultimately hydrolyzed to urea and ornithine by arginase.
Ornithine, in excess of urea cycle needs, is transaminated to form glutamate γ-semialdehyde which is in equilibrium with its tautomeric compound, Δ1-pyrroline-5-carboxylate. Catabolism of Δ1-pyrroline-5-carboxylate back to glutamate is catalyzed by aldehyde dehydrogenase 4 family, member A1 (also called delta-1-pyrroline-5-carboxylate dehydrogenase, P5CDH).
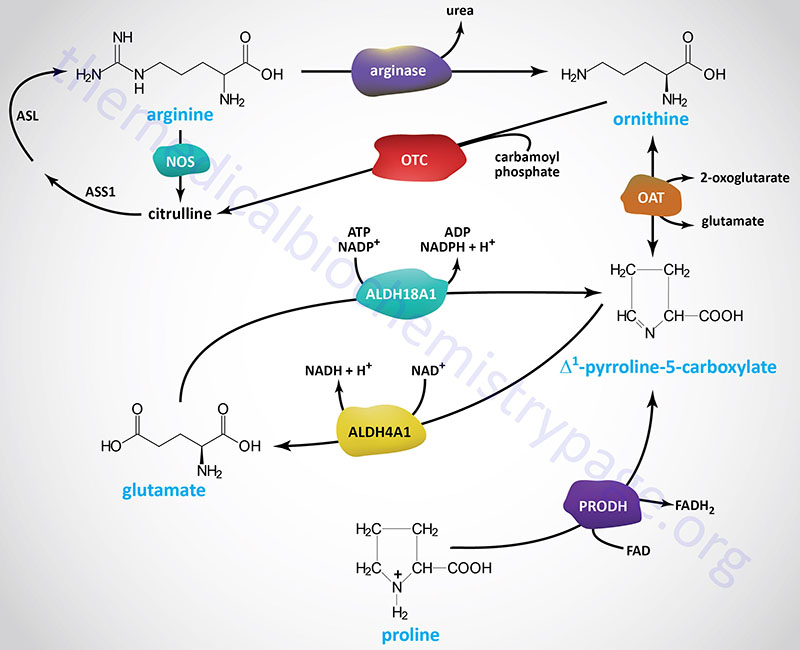
Aldehyde dehydrogenase 4 family, member A1 is encoded by the ALDH4A1 gene. The ALDH4A1 gene is located on chromosome 1p36.13 and is composed of 16 exons that generate four alternatively spliced mRNAs, two of which encode the same protein.
Proline catabolism involves a two-step process that is essentially a reversal of its synthesis process outlined above. Proline is first converted back to Δ1-pyrroline-5-carboxylate by the enzyme proline dehydrogenase 1.
Proline dehydrogenase 1 is encoded by the PRODH gene. The PRODH gene is located on chromosome 22q11.21 and is composed of 15 exons that generate two alternatively spliced mRNAs encoding precursor proteins of 600 amino acids (isoform 1) and 492 amino acids (isoform 2). As for the catabolism of ornithine, the resulting Δ1-pyrroline-5-carboxylate is converted to glutamate via the action of ALDH4A1.
The glutamate that results from ornithine and proline catabolism can then be converted to 2-oxoglutarate (α-ketoglutarate) in a transamination reaction. Therefore, ornithine and proline are both glucogenic. Since arginine is metabolized to urea and ornithine, and the resulting ornithine is a glucogenic precursor, arginine is also a glucogenic amino acid.
Asparagine and Aspartate Catabolism
As outlined in the Amino Acid Biosynthesis page, aspartate and asparagine are related via the transamination reaction catalyzed by asparagine synthetase and the deamination reaction catalyzed by asparaginase. Ultimately the carbon atoms of aspartate and asparagine are the TCA cycle intermediate, oxaloacetate.
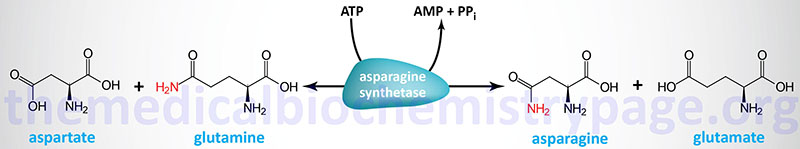
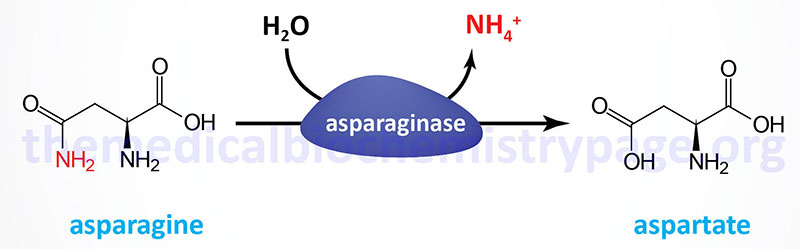
Asparaginase is widely distributed within the body, where it converts asparagine into ammonia and aspartate. Aspartate can serve as an amino donor in transamination reactions yielding oxaloacetate as outlined in the Glutamate and Glutamine Catabolism section. The resulting oxaloacetate can be used to fill up (anaplerosis) the TCA cycle or diverted into the gluconeogenic pathway to glucose.
N-Acetylaspartate Synthesis and Functions
Neurons in the brain possess unique pathways, relative to other tissues of the body, for the synthesis of lipids as well as for energy generation. The acetyl-CoA necessary for cytosolic fatty acid synthesis in cells in the brain, particularly oligodendrocytes (myelin producing cells), is derived from N-acetylaspartate (NAA), not from citrate as is the case for other tissues. Indeed, NAA is considered the second most abundant metabolite in the brain. NAA is synthesized in neurons from aspartate and acetyl-CoA within the matrix of nerve cell mitochondria via the action of N-acetylaspartate synthetase which is encoded by the NAT8L (N-acetyltransferase 8 like) gene.
The NAT8L gene is located on chromosome 4p16.3 and is composed of 3 exons that encode a 302 amino acid protein. The highest levels of expression of the NAT8L gene are in the brain and adipose tissue.
The NAA produced in neurons is transported to the cytosol of oligodendrocytes (as well as astrocytes) via the action of the sodium-dicarboxylate cotransporter encoded by the SLC13A3 gene. The SLC13A3 encoded transporter is also identified as NaDC3. Within the cytosol of oligodendrocytes the enzyme, aspartoacylase, hydrolyzes N-acetylaspartate into acetate and aspartate. The acetate can then be converted to acetyl-CoA by ATP-dependent acetyl-CoA synthetases (AceCS: encoded by acyl-CoA synthetase short-chain family member genes: ACSS). Humans express cytosolic/nuclear and mitochondrial acetyl-CoA synthetases, encoded by the ACSS2 and ACSS1 genes, respectively.
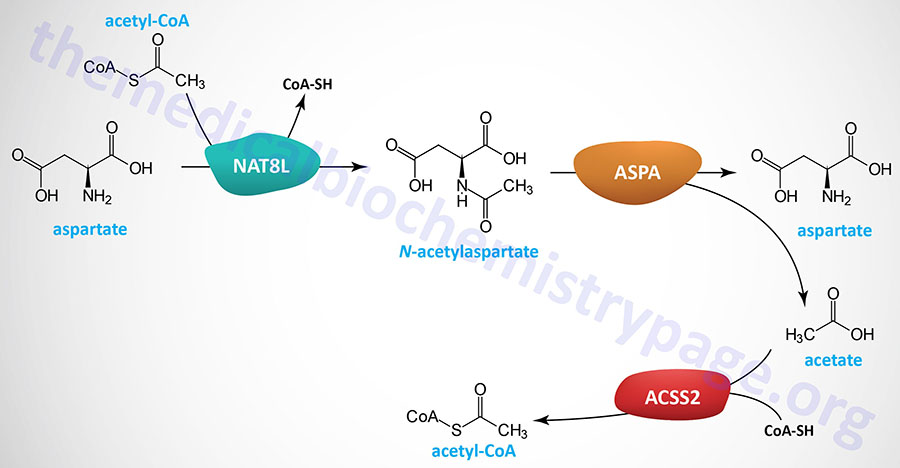
The clinical significance of brain N-acetylaspartate was ascertained from studies of the rare but fatal neonatal leukodystrophy known as Canavan disease. Canavan disease was found to be associated with toxic levels of NAA in the brain due to mutations in the gene (ASPA) encoding aspartoacylase. Aspartoacylase has also been called acylase II and N-acetyl-L-aspartate amidohydrolase.
The ASPA gene is located on chromosome 17p13.2 and is composed of 8 exons that generate two alternatively spliced mRNAs, both of which encode the same 313 amino acid protein.
Aside from the role of NAA in the mobilization of neuronal acetate from the mitochondria of neurons to the cytosol of oligodendrocytes where it serves in fatty acid biosynthesis, NAA also has osmotic effects in the brain. The role of NAA in osmotic regulation accounts for some of the pathology of Canavan disease which is associated with osmotic dysregulation in the brain which then results in disruption in the normal process of myelination as well as subcortical vacuolation.
The synthesis of NAA, although significant in the brain, is not exclusive to this tissue. As indicated, expression of the NAT8L gene is high in adipose tissue as well as the brain. Recent studies have demonstrated that adipose tissue contributes to the level of NAA in the blood and that this contribution is controlled by the activity of aspartoacylase. When NAA accumulates in adipocytes there is a concomitant increase in glucose carbon diversion into pyrimidine nucleotides which is evidenced by increased levels of orotidine 5′-monophosphate (OMP). NAA interacts with the rate-limiting enzyme of pyrimidine nucleotide biosynthesis, which is a trifunctional enzyme encoded by the CAD gene. This interaction results in enhanced pyrimidine synthesis accounting for the increase in glucose diversion into pyrimidines. As adipocyte NAA levels increase there is an associated increase in whole-body glucose disposal which occurs exclusively in white adipose tissue (WAT) in part due to the increase in pyrimidine synthesis. Increasing adipocyte pyrimidines results in increased plasma pyrimidine levels which has been associated with overall energy homeostasis and body temperature homeostasis.
In addition to providing oligodendrocytes and astrocytes with a molecule from which acetyl-CoA can be derived, the function of NAA is also to act as a source of nitrogen for the regeneration of glutamate. Within oligodendrocytes, aspartate serves as an amino donor for aspartate transaminase (AST) which transfers the nitrogen to 2-oxoglutarate (α-ketoglutarate) generating glutamate and oxaloacetate (OAA). The glutamate can be transferred to glutamatergic neurons or to GABAergic neurons where, in the latter cells, the glutamate serves as a substrate for GABA synthesis. Within oligodendrocytes the OAA and acetyl-CoA serve to supply intermediates to the TCA cycle thereby, supporting energy production from the reduced electron carriers (NAHD and FADH2) generated by the TCA cycle.
Cysteine Catabolism
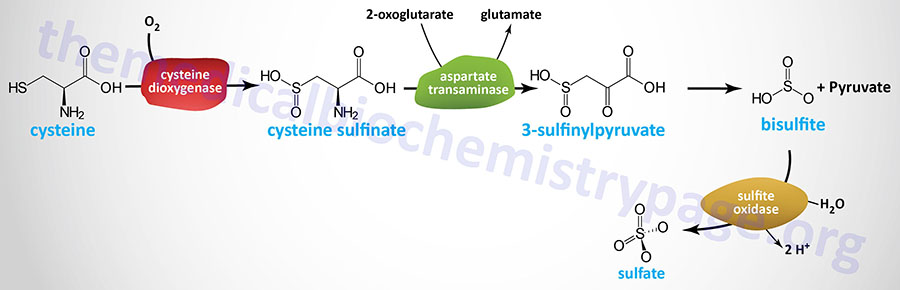
There are several pathways for non-protein disposition of cysteine that include both metabolism and catabolism. The major cysteine catabolic pathway in humans occurs via the action of cysteine dioxygenase type 1 which oxidizes the sulfhydryl group of cysteine to sulfinate, producing the intermediate cysteine sulfinate.
Cysteine dioxygenase type 1 is encoded by the CDO1 gene. The CDO1 gene is located on chromosome 5q22.3 and is composed of 8 exons that generate four alternatively spliced mRNAs, each of which encode a distinct protein isoform.
Catabolism of cysteine sulfinate proceeds through transamination (catalyzed by cytosolic AST) to β-sulfinylpyruvate (3-sulfinylpyruvate) which then undergoes spontaneous desulfuration yielding pyruvate and bisulfite, (HSO3–) which is in equilibrium with sulfite (SO32–) at pH 7.2.
The enzyme, sulfite oxidase, then catalyzes the conversion of sulfite to sulfate. Sulfite oxidase is somewhat unique in that it uses the oxygen atom from H2O to convert sulfite to sulfate, (SO42–) and releases two protons (H+).
Sulfite oxidase is encoded by the SUOX gene. The SUOX gene is located on chromosome 12q13.2 and is composed of 8 exons that generate three alternatively spliced mRNAs, each of which encode the same 545 amino acid protein.
The enzyme cysteine desulfurase is another important enzyme associated with cysteine catabolism. Cysteine desulfurase removes the sulfur from cysteine yielding alanine. The sulfur remains associated with cysteine desulfurase and is subsequently transferred to numerous enzymes that possess iron-sulfur clusters for their activity. Cysteine desulfurase is a member of the pyridoxal phosphate (B6)-dependent aminotransferase family.
Cysteine desulfurase is encoded by the NFS1 gene. The NFS1 gene is located on chromosome 20q11.22 and is composed of 14 exons. In addition to alternative splicing (generating two mRNAs), there are alternative in-frame translational start sites in the NFS1 derived mRNAs. The use of these alternative translational start sites generates mitochondrial, cytoplasmic, and nuclear forms of the enzyme.
The enzyme cystathionase (cystathionine γ-lyase) can also transfer the sulfur from one cysteine to another generating thiocysteine and pyruvate. Transamination of cysteine yields β-mercaptopyruvate (3-mercaptopyruvate, 3-MP) which then reacts with sulfite, (SO32–), to produce thiosulfate, (S2O32–) and pyruvate. Both thiocysteine and thiosulfate can be used by thiosulfate sulfurtransferase (TST; also known as rhodanese) to incorporate sulfur into cyanide ion, (CN–), thereby detoxifying the cyanide to thiocyanate.
Another rhodanese domain-containing sulfurtransferase is the enzyme mercaptopyruvate sulfurtransferase (MPST). The MPST gene generates alternatively spliced mRNAs that encode a cytosolic isoform and another isoform that localizes to the mitochondria and the cytosol.
MPST catalyzes the metabolism of 3-mercaptopyruvate (3-MP) to pyruvate and hydrogen sulfide, H2S. As indicated in the section titled Cysthathione β-Synthase and Cystathionine γ-lyase Activity and Longevity in the Amino Acid Biosynthesis page, H2S functions as a gasotransmitter that relaxes vascular smooth muscle cells resulting in vasodilation.
Within the mitochondria, the production of H2S by MPST contributes to enhanced electron flow and mitochondria bioenergetics, the consequences of which are enhanced cellular longevity.
Taurine Biosynthesis and Functions
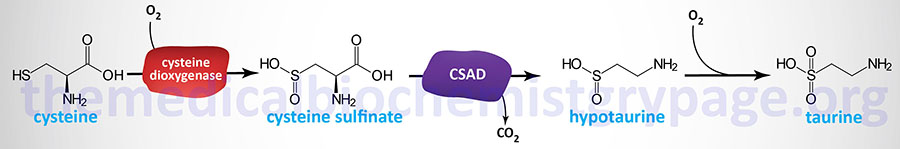
Taurine can be acquired in the diet or synthesized from cysteine as outlined in the Figure below. Taurine is utilized in the synthesis of the bile acid conjugates taurocholate and taurochenodeoxycholate. Taurine also serves numerous other important functions in the body and is commonly referred to as a conditionally essential amino acid.
Taurine is one of the most abundant amino acids in skeletal muscle, brain, and retina. In the retina taurine functions in photoreceptor development. In the brain taurine serves as a cytoprotectant against stress-related neuronal damage and other pathological conditions. Taurine is also an organic osmolyte involved in the regulation of cell volume. Taurine also plays important roles in the modulation of intracellular free calcium concentration and in the protection from mitochondrial stress.
Taurine synthesis occurs primarily in the liver since this is the only bile acid synthesizing tissue in the body. The other major tissues carrying out de novo taurine biosynthesis are the brain (astrocytes and neurons) and the retina. Taurine is derived from the cysteine catabolism intermediate, cysteine sulfinate. Cysteine sulfinate is converted to hypotaurine by the rate-limiting enzyme in taurine synthesis, cysteine sulfinic acid decarboxylase (also called sulfinoalanine decarboxylase).
Cysteine sulfinic acid decarboxylase is encoded by the CSAD gene. The CSAD gene is located on chromosome 12q13.3 and is composed of 23 exons that generate three alternatively spliced mRNAs each of which encode a unique isoform of the enzyme.
Oxidation of hypotaurine to taurine is thought to occur spontaneously (non-enzymatically) although it may involve an as yet unidentified hypotaurine dehydrogenase. Evidence suggests that the catalytic conversion of hypotaurine to taurine is the function of the enzyme encoded by the FMO1 (flavin containing dimethylaniline monoxygenase 1) gene.
Following its synthesis in the liver, taurine is transported to the blood where it is disseminated to other tissues of the body. Although neurons in the brain and cells of the retina can synthesize taurine, the majority of taurine in these tissues is derived from hepatic synthesis. Uptake of taurine into cells is mediated by the SLC family transporter, referred to as TauT, which is encoded by the SLC6A6 gene. Within the brain the SLC6A6 encoded transporter is primarily expressed in astrocytes but is also found at lower levels in neurons. Uptake of taurine into specific cells in the brain also occurs through the action of the GABA transporter encoded by the SLC6A13 gene.
Taurine exists in its zwitterionic form within the range of physiological pH. This biochemical characteristic of taurine allows for it to serve in the regulation of osmolarity. Indeed, taurine synthesis has been shown to be stimulated in cultured neurons exposed to hypertonic conditions. In addition, hyperosmolarity is associated increased expression of the SLC6A6 gene allowing for increased taurine uptake. Conversely, when cultured cells are exposed to hypo-osmotic conditions they transport taurine out to the surrounding medium.
Taurine plays a role in neurotransmitter by serving as a modulator of inhibitory neurotransmission. Taurine has been shown to interact with GABA receptors and glycine receptors. Indeed, evidence indicates that taurine is a potent ligand of the glycine receptor. Taurine is involved in the regulation of both cytoplasmic and intra-mitochondrial Ca2+ homeostasis. These effects of taurine allow it to function in the regulation of glutamate-mediated neurotransmission by reducing glutamate-induced Ca2+ transients in neurons.
Taurine is involved in mitochondrial homeostasis via its ability to act as a mitochondrial matrix pH buffer, by regulating mitochondrial potential, by modulating Ca2+-induced mitochondrial swelling, and by regulating the activity of mitochondrial dehydrogenases and ATP concentration. By exerting all of these effects, taurine helps to preserve normal mitochondrial physiology. These function of taurine are extremely important to overall brain function given that mitochondrial activity in neurons and astrocytes is crucial for brain activity.
Taurine function in overall mitochondrial homeostasis is also exerted at the level of apoptosis. With respect to brain function, the ability of taurine to reduce apoptosis in response to numerous noxious stimuli leads to improved brain outcomes following ischemic trauma. With respect to stress induced pathologies, taurine has also been shown to prevent endoplasmic reticulum (ER) stress. Taurine has been shown to attenuate mitochondrial apoptosis through inhibition of reductions in the level of the anti-apoptotic protein, Bcl-xL, and to prevent increases in the pro-apoptotic protein, Bax. In addition, taurine, prevents cytochrome c release from the mitochondria and inhibits the activation of the executioner caspase, caspase-3.
Synthesis of 3′-Phosphoadenosine-5′-Phosphsulfate: PAPS
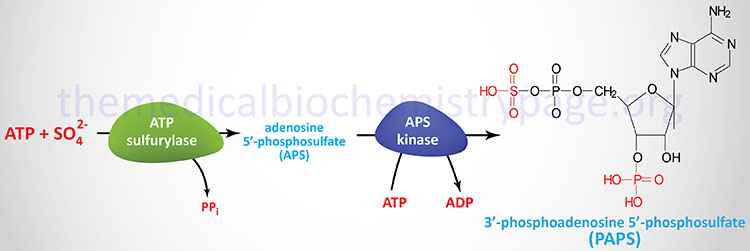
One of the important uses of the sulfate that is derived from the catabolism of cysteine is as a precursor for the formation of 3′-phosphoadenosine-5′-phosphosulfate, (PAPS). PAPS is used for the transfer of sulfate to biological molecules such as the sugars of the glycosphingolipids.
Glutamate and Glutamine Catabolism
As detailed in the Amino Acid Biosynthesis page, as well as the Nitrogen Metabolism and the Urea Cycle page, glutamate and glutamine are interrelated via reactions catalyzed by glutamine synthetase, glutaminase, and glutamate dehydrogenase. Therefore, the pathways of their catabolism are also interrelated.
The carbons of glutamate are oxidized in the TCA cycle following aminotransferase-mediated transfer of the amino group to a suitable α-keto acid. The product of the transamination reactions is 2-oxoglutarate (α-ketoglutarate) which can enter the TCA cycle for oxidation. The most common α-keto acid acceptor is oxaloacetate and the enzyme is aspartate transaminase, AST.

Glutamate, as well as aspartate, are important in collecting and eliminating amino nitrogen via glutamine synthetase and the urea cycle, respectively. The catabolic path of the carbon skeletons involves simple 1-step aminotransferase reactions that directly produce net quantities of a TCA cycle intermediate. The glutamate dehydrogenase reaction operating in the direction of 2-oxoglutarate (α-ketoglutarate) production provides a second avenue leading from glutamate to TCA cycle anaplerosis or to gluconeogenesis.
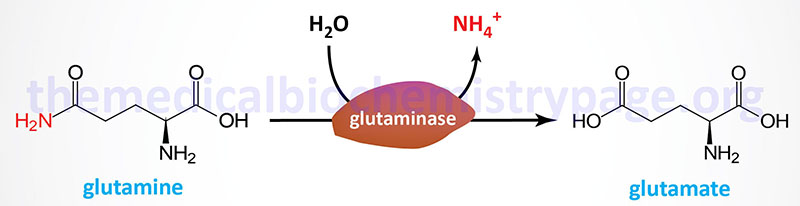
Glutaminase is an important kidney tubule enzyme involved in the process of renal ammoniagenesis. Glutaminase converts glutamine (predominantly derived from the liver but also from many other tissues) to glutamate and NH4+, with the NH4+ being excreted in the urine. Glutaminase activity is present in many other tissues in addition to the kidney, such as the liver, small intestine, and neurons where its role is nearly as significant as it is within the kidney tubule. The glutamate produced from glutamine is converted to 2-oxoglutarate (α-ketoglutarate) via the action of glutamate dehydrogenase (making glutamine a glucogenic amino acid) and yielding another mole of NH4+.
There are two distinct glutaminase genes in humans identified as GLS (encoding the GLS1 enzyme) and GLS2 (encoding the GLS2 enzyme). The GLS gene is located on chromosome 2q32.2 and is composed of 20 exons that generate two alternatively spliced mRNAs generating proteins of 669 amino acids (isoform 1) and 598 amino acids (isoform 2). These two GLS-derived isoforms are often referred to as glutaminase C (GAC) and kidney-type glutaminase (KGA) but are collectively the glutaminase 1 (GLS1) enzymes.
The GLS encoded isoforms of glutaminase are primarily expressed in the kidneys. GLS encoded kidney-type glutaminase is the 669 amino acid isoform 1 protein and GLS encoded glutaminase C is the 598 amino acid isoform 2 protein. The GLS2 gene encoded glutaminase was originally thought to be liver specific but is in fact expressed in numerous tissues and is important in the glutamate-glutamine cycle in the brain.
The GLS2 encoded glutaminase was originally characterized as dependent on inorganic phosphate (Pi) for activity and is, therefore, also referred to as phosphate-activated glutaminase, PAG. However, both the GLS gene encoded enzymes and the GLS2 encoded enzymes require phosphate for activity with GLS enzymes being more sensitive. The GLS2 gene is located on chromosome 12q13.3 and is composed of 19 exons that generate four alternatively spliced mRNAs that encode three different isoforms of the enzyme. The GLS encoded enzymes are inhibited by glutamate but the GLS2 encoded enzyme is not. The GLS2 encoded enzyme is activated by ammonia but the GLS encoded enzymes are not.
Glycine Catabolism
Glycine is classified as a glucogenic amino acid, since it can be converted to serine by serine hydroxymethyltransferase, and serine can be converted back to the glycolytic intermediate, 3-phosphoglycerate or to pyruvate by serine/threonine dehydratase, although the latter reaction (serine to pyruvate) appears not be occur in human tissues.
Nevertheless, the main glycine catabolic pathway leads to the production of CO2, ammonia, and one equivalent of N5,N10-methyleneTHF by the mitochondrial enzyme, glycine dehydrogenase (decarboxylating) which is also called the glycine cleavage complex, GCC.
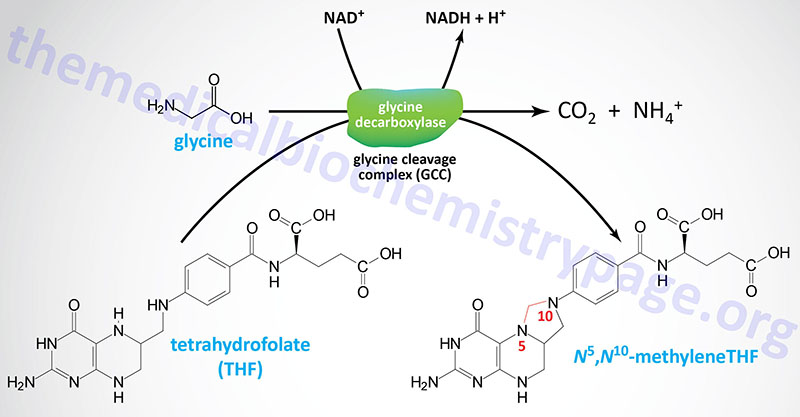
The GCC is composed of four mitochondrial proteins encoded by four genes. The protein components of the GCC are the actual glycine decarboxylase subunit (identified as the P subunit: pyridoxal phosphate-dependent), a lipoic acid-containing subunit (the H subunit), a tetrahydrofolate-requiring enzyme called aminomethyltransferase (the T subunit), and dihydrolipoamide dehydrogenase (the L subunit).
The P subunit is encoded by the GLDC gene. The GLDC gene is located on chromosome 9p24.1 and is composed of 25 exons that encode a precursor protein of 1020 amino acids.
The H subunit is encoded by the GCSH gene. The GCSH gene is located on chromosome 16q23.2 and is composed of 6 exons that generate encode a protein of 173 amino acids.
The T subunit (aminomethyltransferase) is encoded by the AMT gene. The AMT gene is located on chromosome 3p21.31 and is composed of 9 exons that generate four alternatively spliced mRNAs generating four isoforms of the enzyme.
The L subunit is encoded by the DLD gene. The DLD gene is located on chromosome 7q31.1 and contains 14 exons that generate four alternatively spliced mRNAs, each of which encode a distinct protein isoform. The DLD encoded protein is also found as a subunit of several other important dehydrogenase complexes: the pyruvate dehydrogenase complex (PDHc), the 2-oxoglutarate (α-ketoglutarate) dehydrogenase complex, and the branched-chain keto-acid dehydrogenase (BCKD) complex.
Glycine Encephalopathy
Deficiencies in the H, P, or T proteins of glycine decarboxylase result in glycine encephalopathy (GCE) which is characterized by nonketotic hyperglycinemia (NKH). Hyperglycinemia, not associated with defects in glycine catabolism is commonly termed variant nonketotic hyperglycinemia, the most common cause being associated with defects in the biosynthesis of lipoic acid.
Several forms of GCE have been characterized with the neonatal phenotype being the most common.
In the neonatal form of GCE, symptoms appear in the first few days of life and include lethargy, hypotonia, myoclonic jerks, and often death. Neonates that regain spontaneous respiration will develop intractable seizures and profound intellectual impairment.
In the infantile form of GCE, patients are seemingly normal for the first 4-6 months of life and then develop seizures and varying degrees of intellectual impairment.
In the mild-episodic form of GCE, patients present in childhood with mild intellectual impairment. These children will also develop episodes of delirium, chorea, and vertical gaze palsy during a febrile illness.
In the late-onset form of GCE, patients present in childhood with progressive spastic diplegia and optic atrophy, but intellectual function is preserved.
Histidine Catabolism
Histidine catabolism begins with release of the α-amino group catalyzed by histidine ammonia-lyase (also called histidase), introducing a double bond into the molecule. As a result, the deaminated product, urocanate, is not the usual α-keto acid associated with loss of α-amino nitrogens.
Histidine ammonia-lyase is encoded by the HAL gene. The HAL gene is located on chromosome 12q23.1 and is composed of 22 exons generating three alternatively spliced mRNAs, each of which encode a distinct protein isoform. The end product of histidine catabolism is glutamate, making histidine one of the glucogenic amino acids.
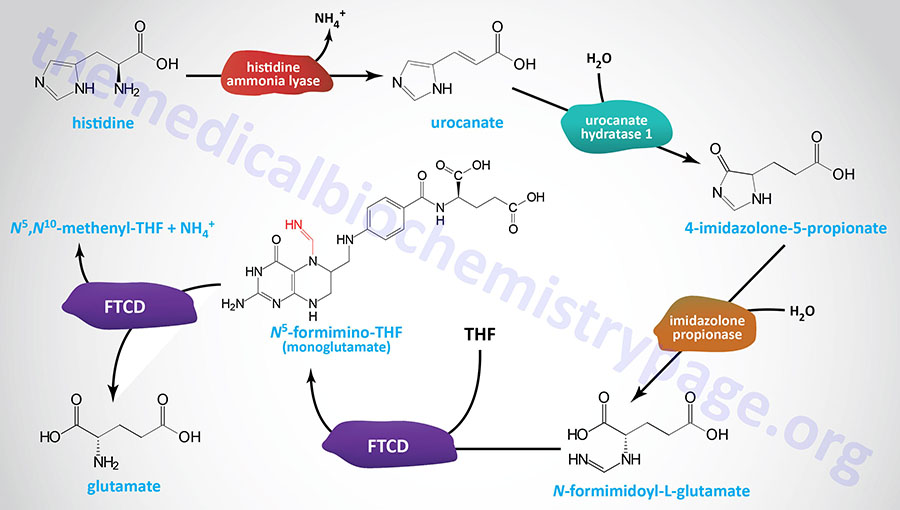
Another key feature of histidine catabolism is that it serves as a source of ring nitrogen to combine with tetrahydrofolate (THF), producing the 1-carbon THF intermediate known as N5-formimino-THF. The latter reaction is one of two routes to N5-formimino-THF.
Urocanate is converted to 4-imidazolone-5-propionate via the action of urocanate hydratase 1 (encoded by the UROC1 gene). The UROC1 gene is located on chromosome 3q21.3 and is composed of 21 exons that generate two alternatively spliced mRNAs encoding proteins of 676 amino acids (isoform 1) and 736 amino acids (isoform 2).
The 4-imidazolone-5-propionate is then converted to N-formimidoyl-L-glutamate via the action of an imidazolone propionase activity. The imidazolone propionase activity is encoded by the amidohydrolase domain-containing 1 gene (symbol: AMDHD1). The AMDHD1 gene is located on chromosome 12q23.1 and is composed of 9 exons that encode a 426 amino acid protein.
The bifunctional enzyme, formimidoyltransferase cyclodeaminase (also called glutamate formiminotransferase), then transfers the formimino group from N-formimidoyl-L-glutamate (commonly referred to as FIGLU) to THF yielding N5-formimino-THF (5-fominino-THF) and glutamate. Formimidoyltransferase cyclodeaminase functions as a homooctameric complex. The 5-formimino-THF is then converted to N5,N10-methenylene-THF (5,10-methenylene-THF) via the cyclodeaminase activity of the complex with glutamate and ammonium ion as the other products.
Formimidoyltransferase cyclodeaminase is encode by the FTCD gene. The FTCD gene is located on chromosome 21q22.3 and is composed of 16 exons that generate three alternatively spliced mRNAs, two of which encode the same 541 amino acid protein (isoform A) and the other encodes a protein of 572 amino acids (isoform C).
Disorders Associated with Defective Histidine Catabolism
The principal genetic deficiencies associated with histidine metabolism are associated with mutations in the HAL, UROC1, and FTCD gene.
Mutations in the HAL gene result in histidinemia which is relatively benign. The disease, which is of relatively high incidence (1 in 10,000), is most easily detected by the absence of urocanate from skin and sweat, where it is normally found in relative abundance.
UROC1 mutations have only been reported in a few individuals who inherited compound heterozygous missense mutations. These individuals exhibited intellectual impairment, short stature, and episodic aggressiveness and affection-seeking behaviors.
Inherited defects in the FTCD gene represent the second most common cause of inborn errors in folate metabolism. Deficiency in formimidoyltransferase cyclodeaminase (glutamate formiminotransferase) is associated with elevated levels of the enzyme substrate, N-formimidoyl-L-glutamate (FIGLU) in the urine. Potential for severe intellectual impairment is the main clinical feature associated with FTCD deficiency. Patients with FTCD defects will also present with megaloblastic anemia due to the role of folate derivatives in the synthesis of purine and thymine nucleotides.
Histamine Synthesis and Functions
Decarboxylation of histidine in the intestine by bacteria gives rise to histamine. Similarly, histamine is synthesized in numerous tissues by the decarboxylation of histidine catalyzed by histidine decarboxylase which is a pyridoxal phosphate-requiring (B6) enzyme.
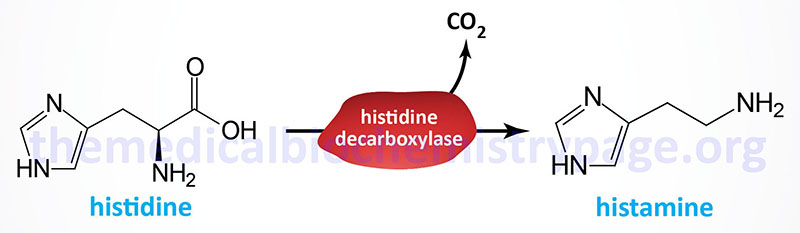
The histidine decarboxylase is encoded y the HDC gene. The HDC gene is located on chromosome 15q21.2 and is composed of 14 exons that generate two alternatively spliced mRNAs that encode proteins of 662 amino acids (isoform 1) and 629 amino acids (isoform 2).
Histamine exerts multiple activities (see the Biochemistry of Nerve Transmission page) but the two most significant are its roles in immunologic response to antigen and inducing the release of hydrogen ion (H+) by parietal cells of the stomach to generate the HCl necessary for gastric digestion. The largest amount of histamine produced in the body is that made by mast cells and basophils. If these cells are sensitized by the presentation of IgE on their surfaces they will degranulate in response to antigen binding, releasing histamine into the circulation. The response to this release is bronchoconstriction and vasodilation which are the general symptoms associated with asthma and various allergic reactions.
Within the gastrointestinal system, histamine is produced in the enterochromaffin-like cells of the stomach. When histamine is released from these cells, in response to vagal nerve stimulation, it binds to specific receptors on parietal cells triggering the mobilization of the proton pump of the parietal cell to the apical membrane allowing for increased H+ transport into the lumen of the stomach.
All of the actions of histamine are the result of binding to one of four cell surface receptors (H1R–H4R), all of which are G-protein coupled receptors (GPCR). The H1 receptor is coupled to a Gq-type G-protein and is responsible for triggering bronchoconstriction and vasodilation. The H2 receptor is a Gs-type G-protein and is present on parietal cells and vascular smooth muscle cells.
Leucine, Isoleucine, and Valine Catabolism: Branched-Chain Amino Acids (BCAA)
This group of essential amino acids is identified as the branched-chain amino acids, BCAA. Because this arrangement of carbon atoms cannot be made by humans, these amino acids are essential in the diet. Indeed, the three BCAA represent 35%-40% of the total of the nine essential amino acids in the human diet and account for approximately 15% of the total of the amino acid composition of skeletal muscle.
The catabolism of all three amino acids occurs in most cells but the highest rates of catabolism takes place in skeletal muscle. BCAA catabolism yields both NADH and FADH2 which can be utilized for ATP generation which is a primary reason for their high rates of catabolism in skeletal muscle. The catabolism of all three of these amino acids uses the same enzymes in the first two steps. The first step in each case is a transamination using a pyridoxal phosphate-dependent BCAA aminotransferase (termed a branched-chain aminotransferase, BCAT), with 2-oxoglutarate (α-ketoglutarate) as amine acceptor.
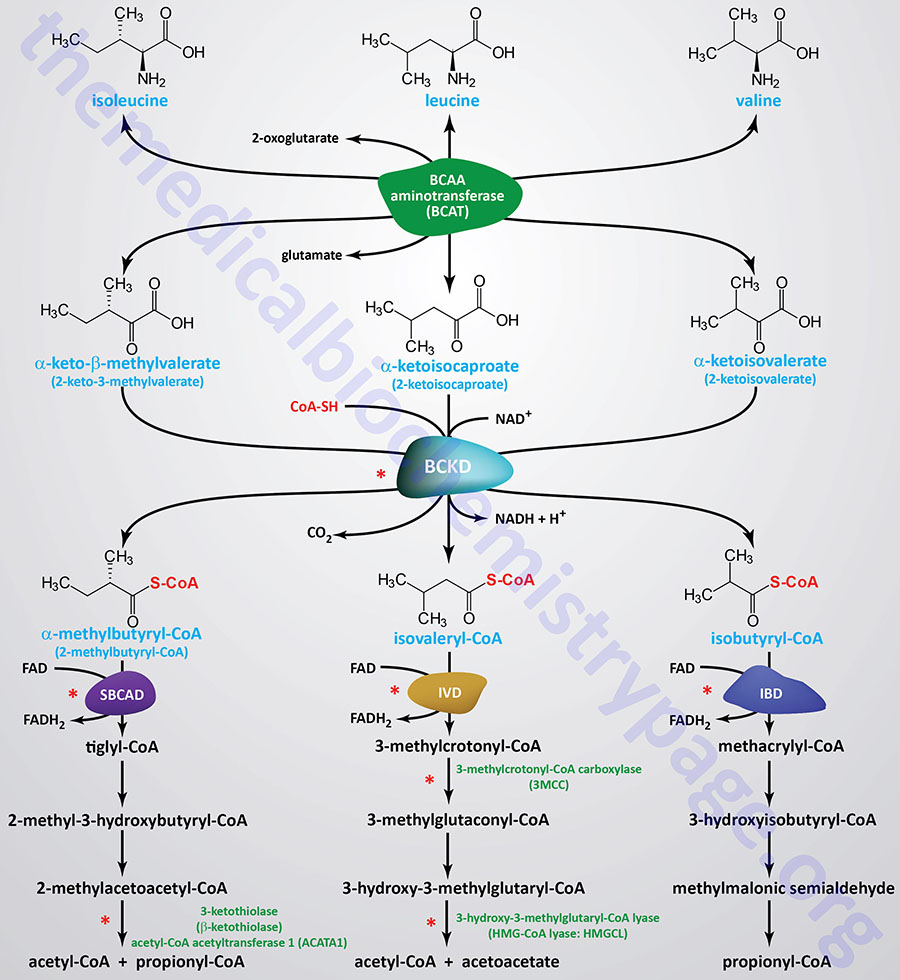
Branched-Chain Aminotransferase: BCAT
Humans express two genes that encode BCAT activity. These two genes are identified as BCAT1 and BCAT2. The primary protein encoded by the BCAT1 gene is a cytosolic version of the enzyme and the protein is identified as BCATc. The primary protein encoded by the BCAT2 gene is a mitochondrial version of the enzyme and this protein is designated BCATm.
The BCAT1 gene is located on chromosome 12p12.1 and is composed of 14 exons that generate five alternatively spliced mRNAs, each of which encode a distinct protein isoform. BCAT1 isoform 1 is a 386 amino acid protein. BCAT1 isoform 2 is a 349 amino acid protein. BCAT1 isoform 3 is a 325 amino acid protein. BCAT1 isoform 4 is a 398 amino acid protein. BCAT1 isoform 5 is a 385 amino acid protein. Expression of the BCAT1 gene is restricted to only a few tissues types. The BCAT1 gene represents the primary BCAT expressing gene in the brain.
The BCAT2 gene is located on chromosome 19q13.33 and is composed of 13 exons that generate three alternatively spliced mRNAs that encode three different isoforms. BCAT2 isoform a is a 392 amino acid protein which is also referred to as PP18a. BCAT2 isoform b is a 300 amino acid protein which is also referred to as PP18b. The isoform b protein is found in the cytosol. BCAT2 isoform c is a 352 amino acid protein. Expression of BCAT2 is widely distributed among numerous tissues.
Although detectable in the fetal liver, the adult liver does not express either BCAT gene. Because BCAT is not present in hepatocytes, dietary BCAA, as well as gut microbiota-derived BCAA, pass through the liver and are deaminated by BCAT in other tissues, predominantly within skeletal muscle. The branched-chain keto acids (BCKA), that are the products of BCAT activity, if not fully oxidized in the tissues in which they are generated, will be delivered, via the blood, to the liver where they are oxidized.
The metabolism of the branched-chain amino acids is critical to overall nitrogen homeostasis in the brain and to the maintenance of proper levels of the excitatory neurotransmitter, glutamate. The critical enzymes that are necessary for this homeostatic process are the BCATc and BCATm isoforms of the BCAA aminotransferases. Within the brain different populations of cells express predominantly BCATc while others express predominantly BCATm and this differential distribution is what is important in overall neuronal nitrogen homeostasis.
As a result of the BCAT reaction, three different α-keto acids are produced and are oxidized using a common branched-chain α-keto acid dehydrogenase (BCKD) complex, yielding the three different CoA derivatives. Subsequently the metabolic pathways diverge, producing many intermediates. The BCKD complex is one of three dehydrogenase complexes whose vitamin-derived cofactor requirements can be remembered by the mnemonic: Tender Loving Care For Nancy, where the T stands for thiamine, the L for lipoic acid, the C for coenzyme A (CoA), the F for riboflavin, and the N for niacin. The other two dehydrogenase complexes are the PDHc and the 2-oxoglutarate dehydrogenase complexes associated with the TCA cycle.
Branched-Chain Keto Acid Dehydrogenase: BCKD
The BCKD complex is a multimeric enzyme composed of three catalytic subunits. The E1 portion of the complex is a thiamine pyrophosphate (TPP)-dependent decarboxylase with a heterotetrameric subunit structure of α2β2. The E2 portion is a dihydrolipoamide branched-chain transacylase composed of 24 lipoic acid-containing polypeptides. The E3 portion is a homodimeric flavoprotein identified as dihydrolipoamide dehydrogenase, DLD.
The E1α gene (symbol: BCKDHA) is located on chromosome 19q13.2 and contains 9 exons that generate two alternatively spliced mRNAs that encode alpha subunit isoform 1 (445 amino acids) and alpha subunit isoform 2 (444 amino acids).
The E1β gene (symbol: BCKDHB) is located on chromosome 6q14.1 and is composed of 21 exons that generate three alternatively spliced mRNAs that collectively encode a 392 amino acid protein (isoform 1 ) and a 322 amino acid protein (isoform 2).
The E2 gene (symbol: DBT) is located on chromosome 1p21.2 and contains 14 exons that that generate three alternatively spliced mRNAs that collectively encode a 482 amino acid precursor protein (isoform 1 ) and a 3301 amino acid protein (isoform 2).
The E3 gene (symbol: DLD) is located on chromosome 7q31.1 contains 14 exons that generate four alternatively spliced mRNAs, each of which encode a distinct protein isoform. The DLD gene encodes the same dihydrolipoamide dehydrogenase subunits found in the PDHc and the 2-oxoglutarate dehydrogenase (α-ketoglutarate dehydrogenase) complexes.
Regulation of BCKD Activity
Regulation of BCKD activity is exerted via phosphorylation and dephosphorylation similarly to the regulation of the activity of the PDHc. The phosphorylation of BCKD inhibits the enzyme, whereas, dephosphorylation activates it.
Phosphorylation of BCKD is catalyzed by the kinase, branched chain keto acid dehydrogenase kinase [commonly referred to as BDK; originally identified as 3-methyl-2-oxobutanoate dehydrogenase (lipoamide) kinase] which is encoded by the BCKDK gene. The BCKDK gene is located on chromosome 16p11.2 and is composed of 13 exons that generate three alternatively spliced mRNAs, each of which encode a distinct protein isoform.
The activity of BCKD kinase is itself regulated by phosphorylation, being phosphorylated on Tyr 246 (Y246) by the SRC tyrosine kinase. Phosphorylation of BCKDK by SRC results in repression of the kinase activity of BCKDK. This effect of SRC leads to increased metabolism of BCAA, an effect that is associated with numerous cancers.
Loss-of-function mutations in the BCKDK gene are associated with a phenotype that is characterized by persistently reduced levels of BCAA in body fluids, developmental delay, microcephaly, and neurobehavioral abnormalities. Conversely, gain-of-function mutations in the BCKDK gene are associated with symptoms reflective of a mild form of maple syrup urine disease, MSUD.
The dephosphorylation of BCKD is catalyzed by the phosphatase identified as protein phosphatase, Mg2+/Mn2+ dependent 1K (also known as PP2Cm) which is encoded by the PPM1K gene. The PPM1K gene is located on chromosome 4q22.1 and is composed of 11 exons that encode a 372 amino acid protein.
Both the BCKDK and PPMK1 gene encoded proteins are localized to the mitochondria as is the BCKD complex. Both the BCKD kinase and phosphatase are tightly associated with the E2 subunit of the BCKD complex. Phosphorylation and dephosphorylation of BCKD occurs on a Ser residue of the α-subunits of the E1 heterotetrameric complex.
Byproducts of Isoleucine Catabolism
The catabolism of isoleucine ultimately yields acetyl-CoA and propionyl-CoA, thus isoleucine is both glucogenic and ketogenic.
The product of the BCKD reaction, 2-methylbutyryl-CoA is metabolized to tiglyl-CoA by short/branched-chain acyl-CoA dehydrogenase, SBCAD. The SBCAD enzyme is encoded by the ACADSB (acyl-CoA dehydrogenase short/branched chain) gene.
Tiglyl-CoA is metabolized to 2-methyl-3-hydroxybutyryl-CoA by the enzyme often identified as crotonase. Crotonase is encoded ECHS1 (enoyl-CoA hydratase, short chain 1; also identified as SCEH) gene.
In the next reaction, 2-methyl-3-hydroxybutyryl-CoA is metabolized to 2-methylacetoacetyl-CoA by hydroxysteroid 17-beta dehydrogenase 10 which is encoded by the HSD17B10 gene. The enzyme encoded by the HSD17B10 gene is also known as 3-hydroxy-2-methylbutyryl-CoA dehydrogenase and as short chain 3-hydroxyacyl-CoA dehydrogenase (SCHAD) and as 3-hydroxyacyl-CoA dehydrogenase type II. The designation of SCHAD for the enzyme encoded by the HSD17B10 can lead to confusion since the enzyme encoded by the HADH (hydroxyacyl-CoA dehydrogenase) gene, which is involved in medium- and short-chain fatty acid oxidation, is also identified as short-chain L-3-hydroxyacyl-CoA dehydrogenase (SCHAD).
Mutations in the HSD17B10 gene are the cause of a rare X-linked disorder referred to as 2-methyl-3-hydroxybutyric aciduria as well as HSD10 disease.
Mutations in the HSD17B10 gene are the cause of a rare X-linked disorder referred to as 2-methyl-3-hydroxybutyric aciduria as well as HSD10 disease.
The final step in isoleucine metabolism involves the hydrolysis of 2-methylacetoacetyl-CoA to acetyl-CoA and propionyl-CoA. This reaction is catalyzed by acetyl-CoA acetyltransferase 1 which is encoded by the ACAT1 gene. Mutations in the ACAT1 gene result in the autosomal recessive disorder somewhat inappropriately identified as β-ketothiolase or 3-ketothiolase deficiency. Due to the common usage of the term, β-ketothiolase, in the context of the final step in mitochondrial β-oxidation of long-chain fatty acids, this disorder is often misconstrued as a disorder of fatty acid oxidation. However, the thiolase activity of long-chain fatty acid β-oxidation is associated with the β-subunit (encoded by the HADHB gene) of the mitochondrial trifunctional protein, MTP. The oxidation of medium- and short-chain fatty acids, and the utilization of the ketone, acetoacetate, involves the mitochondrial matrix-associated β-ketothiolase encoded by the ACAA2 gene.
As pointed out above for the catabolism of methionine, the resulting propionyl-CoA is converted, via a mitochondrially-localized, three reaction, ATP-dependent pathway, to succinyl-CoA. The succinyl-CoA can then enter the TCA cycle for further oxidation or diverted, via malate, into the gluconeogenesis pathway. The enzymes required for this conversion are propionyl-CoA carboxylase, methylmalonyl-CoA epimerase, and the B12-dependent enzyme, methylmalonyl-CoA mutase. This propionyl-CoA conversion pathway is often remembered by the mnemonic as the VOMIT pathway, where V stands for valine, O for odd-chain fatty acids, M for methionine, I for isoleucine, and T for threonine.
Propionyl-CoA carboxylase functions as a heterododecameric enzyme (subunit composition: α6β6) and the two different subunits are encoded by the PCCA and PCCB genes, respectively. The PCCA gene is located on chromosome 13q32.3 and is composed of 32 exons that generate eleven alternatively spliced mRNAs, each of which encode a unique protein isoform. The PCCB gene is located on chromosome 3q22.3 and is composed of 17 exons that generate two alternatively spliced mRNAs encoding proteins of 539 amino acids (isoform 1) and 559 amino acids (isoform 2). Mutations in either the PCCA or PCCB gene result in the organic acidemia, propionic acidemia.
Methylmalonyl-CoA epimerase is encoded by the MCEE gene. The MCEE gene is located on chromosome 2p13.3 and is composed of 4 exons that encode a 176 amino acid precursor protein.
Methylmalonyl-CoA mutase is encoded by the MMUT gene. The MMUT gene is located on chromosome 6p12.3 and is composed of 13 exons that encode a precursor protein of 750 amino acids. Mutations in the MMUT gene are one cause of the methylmalonic acidemias.
Byproducts of Leucine Catabolism
The catabolism of leucine gives rise to acetyl-CoA and acetoacetyl-CoA, and therefore, leucine is classified as strictly ketogenic.
The product of the BCKD reaction, isovaleryl-CoA, is oxidized to 3-methylcrotonyl-CoA by the enzyme isovaleryl-CoA dehydrogenase, IVD. Deficiencies in IVD result in the disorder identified as isovaleric acidemia.
In the next reaction of leucine catabolism 3-methylcrotonyl-CoA is carboxylated by 3-methylcotonyl-CoA carboxylase (3MCC) generating 3-methylglutaconyl-CoA. 3MCC is composed of two subunits in a heterododecameric configuration composed of six α-subunits and six β-subunits (α6β6). The activity of 3MCC is dependent on biotin. The α-subunit covalently binds biotin while the carboxyltransferase activity is encoded by the β-subunit. The structure of 3MCC and its dependence on biotin make it highly similar in structure and catalytic activity to propionyl-CoA carboxylase, PCC. The α-subunit of 3MCC is encoded by the MCCC1 gene and the β-subunit is encoded by the MCCC2 gene. Mutations in the MCCC1 gene cause 3-methylcrotonylglycinuria type I, while mutations in the MCCC2 gene cause 3-methylcrotonylglycinuria type II.
In the next reaction, the 3-methylglutaconyl-CoA is converted to 3-hydroxy-3-methylglutaryl-CoA (HMG-CoA) via the action of the bifunctional enzyme, 3-methylglutaconyl-CoA hydratase. The 3-methylglutaconyl-CoA hydratase enzyme possesses both RNA-binding and hydratase activities accounting for the name of the gene encoding this enzyme, AU RNA-binding methylglutaconyl-CoA hydratase (AUH). Mutations in the AUH gene result in the extremely rare autosomal recessive disorder referred to as 3-methylglutaconic aciduria, type 1 (MGCA1).
The HMG-CoA derived from the catabolism of leucine can be transported to the cytosol where is can contribute to the synthesis of isoprenoid compounds and cholesterol. Conversion of the mitochondrial HMG-CoA, derived from leucine catabolism, to acetoacetyl-CoA and acetoacetate is catalyzed by HMG-CoA lyase (encoded by the HMGCL gene). This is the same enzyme involved in the synthesis of acetoacetate during ketone body synthesis in the liver. Mutations in the HMGCL gene result in a disorder referred to as 3-hydroxy-3-methylglutaric aciduria.
Byproducts of Valine Catabolism
The principal catabolic by-product from valine is propionyl-CoA, the glucogenic precursor of succinyl-CoA. The conversion of propionyl-CoA to succinyl-CoA is described above in the Byproducts of Isoleucine Catabolism.
The product of the isobutyryl-CoA dehydrogenase (IBD) reaction, methacrylyl-CoA, is hydrolyzed to 3-hydroxyisobutyryl-CoA by the enzyme often identified as methacryly-CoA hydratase. This enzyme is actually the same enzyme involved in fatty acid β-oxidation and isoleucine catabolism which is encoded by the ECHS1 (enoyl-CoA hydratase, short chain 1; also identified as SCEH) gene. The ECHS1 encoded enzyme is also identified as crotonase.
In the next step of valine catabolism 3-hydroxyisobutyryl-CoA is metabolized to methylmalonate semialdehyde. This reaction is catalyzed by 3-hydroxyisobutyryl-CoA hydrolase which is encoded by the HIBCH gene.
The final step of valine catabolism involves the conversion of methylmalonate semialdehyde to propionyl-CoA by the enzyme commonly identified as methylmalonate semialdehyde dehydrogenase, MMSDH. The MMSDH enzyme is encoded by the ALDH6A1 (aldehyde dehydrogenase 6 family member A1) gene.
Disorders of Branched-Chain Amino Acid Metabolism
Details of the various diseases resulting from mutations in the enzymes of branched-chain amino acid catabolism are presented in the Branched-Chain Amino Acid Metabolism Disorders page as well as in specific pages for individual diseases, for example Maple Syrup Urine Disease, MSUD.
Leucine Signaling and Metabolic Regulation
Numerous studies have shown that diets high in protein increase fatty acid oxidation and overall energy expenditure and thus, promote weight loss. In addition, high protein diets are known to improve glucose homeostasis and therefore, can have a positive impact on insulin sensitivity and diabetes. In animal models of obesity and diabetes, increasing the protein to carbohydrate ratio in a high-fat diet results in a delay in the development of obesity while simultaneously improving glucose tolerance. In studies on obese humans, subjects that consumed a milk whey-protein-enriched diet exhibited demonstrable improvement in fatty liver symptoms (hepatic steatosis) and reduced plasma lipid profiles. The composition of whey protein is approximately 65% β-lactoglobulin, 25% α-lactalbumin, and 8% serum albumin. In studies examining the effects of dietary protein on energy expenditure, appetite suppression, and weight loss, whey protein is the more beneficial source when comparing the effects of consumption of whey proteins, soy proteins, or casein.
Whey proteins are enriched in the branched-chain amino acids (BCAA) leucine, isoleucine, and valine and are thus, excellent sources of energy production in skeletal muscle as well as serving as building blocks for muscle protein synthesis. Leucine has been proposed to be the primary mediator of the metabolic changes that occur when consuming a high protein diet. At the molecular level, leucine has been shown to activate the metabolic regulatory kinase known as mammalian target of rapamycin, mTOR. For more information on the activities and regulation of mTOR go to the Insulin Function, Insulin Resistance, and Food Intake Control of Secretion page or to the Protein Synthesis (Translation): Processes and Regulation page.
Activation of skeletal muscle mTOR results in increased protein synthesis and thus, increased energy expenditure. Hypothalamic mTOR activation is also involved in the regulation of feeding behaviors. Of note is the fact that direct injection of leucine into the hypothalamus results in increased mTOR signaling leading to decreased feeding behavior and body weight. This effect is unique to leucine, as direct injection of valine, another BCAA, does not result in hypothalamic mTOR activation nor reductions in food intake or body weight.
The effects of leucine supplementation, on the above described parameters, are not as pronounced as the effects observed when consuming a high-protein diet. This suggests that additional factors are likely involved in the effects of high-protein diets. One of these factors may be that consuming a high protein diet is associated with a reduction in total carbohydrate intake. The reduced carbohydrate intake would thus, be associated with a reduction in hepatic lipogenesis and an increase in adipose tissue lipolysis. Although leucine alone is not as effective as high-protein diets at reducing body weight and food intake it cannot be discounted as an important dietary supplement.
However, it must be pointed out that some controversy surrounds the role of high protein consumption, and in particular leucine intake, in overall metabolic homeostasis. This is due to the fact that some studies in laboratory animals have shown that leucine supplementation results in insulin resistance. This latter effect would certainly lead to an increased likelihood for development of type 2 diabetes.
In the typical Western diet consisting of high dairy and meat, the role of leucine in the pathogenesis of type 2 diabetes is suggested by the consequent over activation of mTOR. With respect to diabetes, activation of mTOR results in phosphorylation and activation of the kinase, p70S6K, which in turn phosphorylates the insulin receptor substrate 1 (IRS-1). Phosphorylation of IRS-1 by p70S6K results in reduced insulin-mediated signaling via the insulin receptor, thereby, increasing the metabolic burden on pancreatic β-cells. In addition, increased mTOR activation leads to adipogenesis which can lead to obesity-mediated insulin resistance.
Most of the work in laboratory animals demonstrates that the positive effects of increased leucine intake or high-protein diets are most pronounced when the animals are also consuming a high-fat diet. Therefore, it is suggestive that there are definite benefits for overweight and obese individuals to increase their intake of leucine and/or total protein as a means for appetite and weight control. However, although high-protein diets or leucine supplementation are important considerations in a healthy diet, the total amount consumed must be taken into account so as not to lead to excess mTOR activation.
Lysine Catabolism
Lysine, Arginine, and Ornithine Transporters
Like all amino acids, catabolism of lysine can initiate from uptake of dietary lysine or from the breakdown of intracellular protein. Intestinal uptake of lysine involves specific transporter proteins. In most tissues, cationic amino acids are transported principally by a Na+-independent system, specific for L-isomers of lysine, arginine, and ornithine. This transport system is known as the y+ system and these transporters are members of the SLC7 family of membrane transporters where this Na+-independent transporter is encoded by the SLC7A1 gene.
There are actually at least three transporter mechanisms for lysine transport. One is the aforementioned y+ system that functions as a uniporter. The other two lysine transporters are Na+-dependent antiporter systems that are heterodimeric transporters composed of a heavy chain and a light chain forming transporters of the y+L system.
The common heavy chain of these two different transporters is encoded by the SLC3A2 gene (often referred to as 4F2hc for 4F2 cell-surface antigen heavy chain). The light chains are encoded by the SLC7A6 and SLC7A7 genes which encode the y+LAT2 and y+LAT1 proteins, respectively. Once taken up by the intestines, dietary lysine can be incorporated into protein or catabolized.
The SLC7A1 encoded transporter is the primary transporter for the cellular uptake of arginine whereas the antiporter composed of the SLC7A6 encoded protein is the major arginine efflux transporter for transport of arginine out of cells.
Lysine Catabolism via the Saccharopine Pathway
There are several, at least three, pathways for lysine catabolism but the primary pathway utilized within the liver of humans is one that begins with the formation of an adduct between lysine and 2-oxoglutarate (α-ketoglutarate) called saccharopine.
Lysine catabolism is unusual in the way that the ε-amino group is transferred to 2-oxoglutarate and into the general nitrogen pool. The reaction is a transamination in which the ε-amino group is transferred to the α-keto carbon of 2-oxoglutarate (α-ketoglutarate) forming the metabolite, saccharopine. Unlike the majority of transamination reactions, the enzyme catalyzing the formation of saccharopine does not employ pyridoxal phosphate as a cofactor.
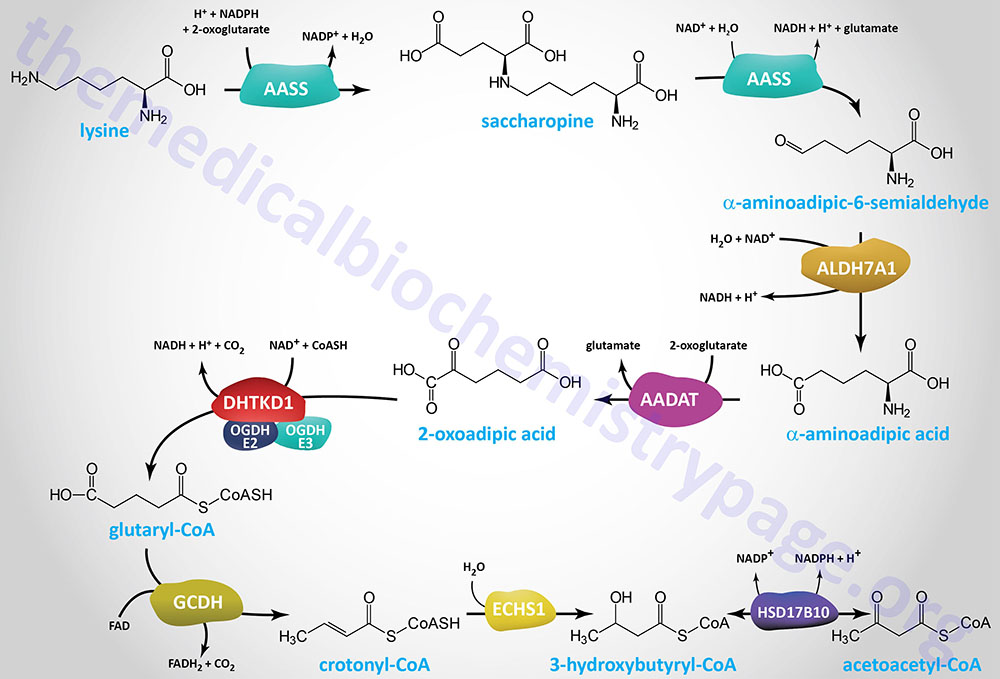
The formation of saccharopine and its hydrolysis to α-aminoadipic-6-semialdehyde (2-aminoadipic-6-semialdehyde; also identified as allysine) is catalyzed by the bifunctional enzyme α-aminoadipic semialdehyde synthase (2-aminoadipic semialdehyde synthase). This reaction results in the amino nitrogen remaining with the α-carbon of 2-oxoglutarate, producing glutamate and α-aminoadipic-6-semialdehyde. Because this transamination reaction is not reversible, lysine is an essential amino acid. Mammalian α-aminoadipic semialdehyde synthase is an NADPH-dependent mitochondria-localized enzyme encoded by the AASS gene.
The AASS gene is located on chromosome 7q31.32 and is composed of 25 exons encoding a mitochondrially localized protein of 926 amino acids. The N-terminal half of the AASS protein harbors the lysine:2-oxoglutarate reductase activity and the C-terminal half harbors the saccharopine dehydrogenase activity.
The α-aminoadipic-6-semialdehyde is catabolized to α-aminoadipic acid via the action of the NAD+-dependent enzyme, aldehyde dehydrogenase 7 family member A1 (encoded by the ALDH7A1 gene), originally identified as α-aminoadipic semialdehyde dehydrogenase (AASA dehydrogenase: AASADH). This enzyme is also known as antiquitin (ATQ1).
The ALDH7A1 gene is located on chromosome 5q23.2 and is composed of 18 exons that generate three alternatively spliced mRNAs. Two of these ALDH7A1 derived mRNAs each encode two proteins through the use of alternative in-frame translation initiation codons. One of the resultant proteins is localized to the mitochondria the other to the cytosol. It is the mitochondrial enzyme that is involved in lysine catabolism.
The ALDH7A1 encoded enzyme is also involved in the pathway of betaine synthesis from choline.
The α-aminoadipic acid is then deaminated to 2-oxoadipic acid (2-OAA; also called α-ketoadipic acid or 2-ketoadipic acid) via the transaminase, aminoadipate aminotransferase (encoded by the AADAT gene). Aminoadipate aminotransferase is highly similar to the rodent kynurenine aminotransferase II (KATII) and the AADAT encoded enzyme is also involved in the conversion of kynurenine to kynurenic acid in the tryptophan catabolism pathway.
The AADAT gene is located on chromosome 4q33 and is composed of 18 exons that generate four alternatively spliced mRNAs that collectively encode two proteins isoforms of 429 amino acids (isoform a) and 425 amino acids (isoform b)
The 2-oxoadipic acid then undergoes oxidative decarboxylation to glutaryl-CoA via the actions of the 2-oxoadipate dehydrogenase complex. This enzyme complex is composed of the E1 component protein encoded by the DHTKD1 (dehydrogenase E1 and transketolase domain containing 1) gene and shares the E2 (dihydrolipoamide S-succinyltransferase: DLST) and E3 (dihydrolipoamide dehydrogenase: DLD) components of the 2-oxoglutarate complex of the TCA cycle (commonly called α-ketoglutarate dehydrogenase).
The DHTKD1 gene is located on chromosome 10p14 and is composed of 17 exons that encode a 919 amino acid protein.
Glutaryl-CoA is oxidized to crotonyl-CoA by the FAD-dependent enzyme, glutaryl-CoA dehydrogenase (GCDH). Glutaryl-CoA dehydrogenase is a member of the acyl-CoA dehydrogenase (ACAD) family of enzymes that includes members of the mitochondrial fatty acid β-oxidation pathway and as such the enzyme is also known as acyl-CoA dehydrogenase 5 (ACAD5). Mutations in the GCDH gene result in the disorder identified as glutaric aciduria/acidemia type 1.
The crotonyl-CoA is converted to β-hydroxybutyryl-CoA (3-hydroxybutyryl-CoA) by the ECHS1 (enoyl-CoA hydratase, short chain 1; also identified as SCEH) gene encoded enzyme. The ECHS1 encoded enzyme is also identified as crotonase. Finally, β-hydroxybutyryl-CoA is converted to acetoacetyl-CoA, a reaction catalyzed by hydroxysteroid 17-beta dehydrogenase 10 (encoded by the HSD17B10 gene) which is also known as 3-hydroxybutyryl-CoA dehydrogenase.
Crotonyl-CoA can also serve as the substrate for the post-translational modification of lysine residues, a process termed protein crotonylation.
Disorders Associated with Defective Lysine Catabolism
Serious disorders associated with lysine metabolism can result from a failure of the transport systems for lysine and the other dibasic amino acids from the intestinal epithelial cells to the blood. Lysine is essential for protein synthesis and deficiencies of its transport into the body can cause seriously diminished levels of protein synthesis. Probably more significant however, is the fact that arginine is transported by the same dibasic amino acid transporter, and resulting arginine deficiencies limit the quantity of ornithine available for the urea cycle. The result is severe hyperammonemia after a meal rich in protein. The addition of citrulline to the diet prevents the hyperammonemia.
Hyperlysinemias
Genetic deficiencies in either of the first two reactions of the saccharopine pathway of lysine catabolism result in hyperlysinemia. Since these two reactions are catalyzed by the bifunctional enzyme, AASS, defects can be found in either the N-terminal lysine:2-oxoglutarate reductase activity or the C-terminal saccharopine dehydrogenase activity. These deficiencies are observed in individuals who excrete large quantities of urinary lysine, pipecolic acid, and some saccharopine.
The most severe form of hyperlysinemia is the type I form which is caused by mutations in the AASS gene that affect both activities of the enzyme. Hyperlysinemia type I is an autosomal recessive disorder that manifests in infancy with nonspecific seizures, hypotonia, or mildly delayed psychomotor development. Many individuals are asymptomatic and the hyperlysinemia is considered a benign condition in these individuals.
Hyperlysinemia type II results when mutations in the AASS gene affect only the C-terminal saccharopine dehydrogenase activity. Hyperlysinemia type II is also termed saccharopinuria. Hyperlysinemia type II is an autosomal recessive disorder exhibiting variable phenotypes that may include intellectual disability, seizures, and delayed speech and language development.
ALDH7A1 Deficiency
Deficiencies in the ALDH7A1 encoded enzyme are associated with epileptic encephalopathy that classically presents within the first weeks to months of life. These deficiencies are inherited in an autosomal recessive manner. Most patients with ALDH7A1 deficiency will suffer from convulsions at birth and some will exhibit convulsions in utero. Some cases of ALDH7A1 deficiency have been associated with death in status epilepticus (prolonged seizure that lasts for more than five minutes or multiple seizures without regaining consciousness between each seizure).
Since the ALDH7A1 encoded enzyme is responsible for the oxidation of α-aminoadipic-6-semialdehyde (2-AMAS; also identified as α-AASA) to α-aminoadipic acid, loss of enzyme activity is associated with the accumulation of 2-AMAS and its cyclic form, Δ1-piperideine-6-carboxylate (P6C). P6C complexes with pyridoxal phosphate (PLP) resulting in the depletion of PLP and thus, affecting the many reactions whose enzymes require PLP as co-factor. The depletion of PLP in ALDH7A1 deficiency makes treatment of the epileptic encephalopathy dependent on vitamin B6 supplementation.
Deficiency in ALDH7A1 can be assessed through measurement of the levels of 2-AMAS and P6C in urine, plasma, cerebrospinal fluid (CSF), and in dried blood spots. However, since these compounds are very unstable sample preparation and storage is critical. As such, the measurement of 6-oxopipecolic acid in these same samples is a more suitable biomarker for ALDH7A1 deficiency since this compound is much more stable.
Minor Pathways of Lysine Catabolism
One of the minor pathways of lysine catabolism is the pipecolic acid pathway. L-pipecolic acid (2-carboxypiperdine) was known to be a widely distributed non-protein amino acid in plants and to be derived from lysine catabolism in mammals. Originally it was thought that the degradation of lysine to pipecolate was the major catabolic pathway for this amino acid. Although it is now known that this product of lysine catabolism reflects a minor catabolic pathway, there is clinical significance to this pathway.
In addition, lysine catabolism via the pipecolic acid pathway may play a significant role in the central nervous system. Lysine catabolism via this pathway involves conversion to α-keto-ε-amino-caproic acid and then to pipecolic acid. Pipecolic acid then is ultimately converted to α-aminoadipic semialdehyde which is also a product of the saccharopine pathway of lysine catabolism.
Formation of α-aminoadipic semialdehyde from pipecolic acid is catalyzed by the dehydrogenase encoded by the aldehyde dehydrogenase 7 family member A1 gene (ALDH7A1). As indicated above this enzyme is also known as α-aminoadipic semialdehyde dehydrogenase (AASA dehydrogenase: AASADH) and is also known as antiquitin (ATQ1).
Carnitine Synthesis from Lysine
Lysine is also important as a precursor for the synthesis of carnitine (γ-trimethyl-β-hydroxybutyrobetaine; also identified as β-hydroxy-γ-N-trimethylamino-butyric acid), required for the transport of fatty acids into the mitochondria for oxidation. Free lysine does not serve as the precursor for this reaction, rather the modified lysine (6-N-trimethyllysine, TML), found in certain proteins, is the precursor.
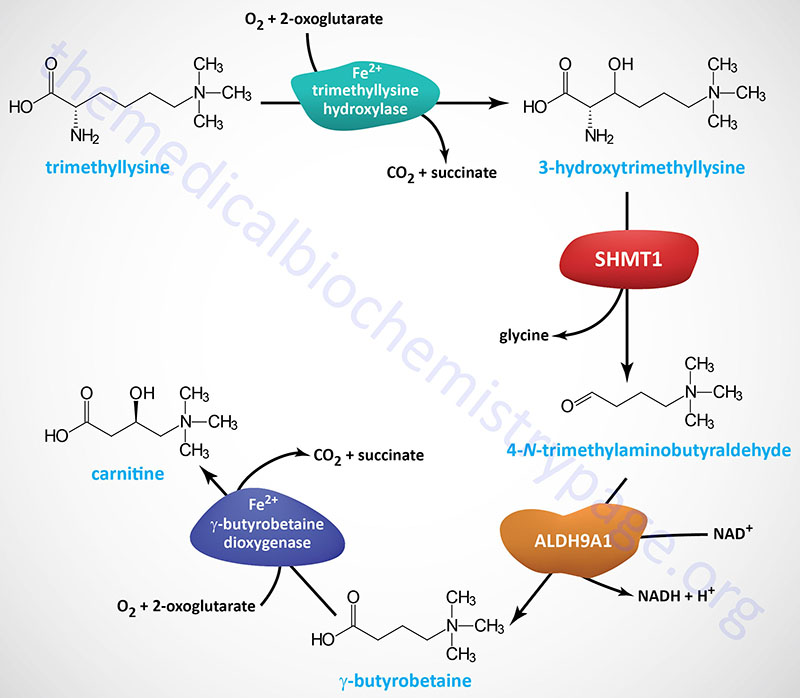
Protein lysine methylation is catalyzed by one of a family of 27 lysine methyltransferases (KMT) expressed in humans. Some of the most critical proteins that are methylated on lysine residues are the histone proteins in chromatin, the consequences of which are regulation of gene expression. The methyl donor for KMT proteins is S-adenosylmethionine (AdoMet of SAM).
Hydrolysis of proteins containing trimethyllysine provides the substrate for the subsequent conversion to carnitine. Once trimethyllysine is released from proteins within the lysosomal compartment, the overall pathway to carnitine synthesis requires four separate reactions.
Trimethyllysine (TML) is transported into the mitochondria via the SLC25A45 encoded transporter. Within the mitochondria the first reaction of carnitine synthesis is catalyzed by the trimethyllysine hydroxylase, epison (also called trimethyllysine dioxygenase) enzyme encoded by the TMLHE gene. The product of the TMLHE reaction, 3-hydroxtrimethyllysine (HTML; also identified as 3-hydroxy-6-N-trimethyllysine) is then transported out of the mitochondria.
The TMLHE gene is located on the X chromosome (Xq28) and is composed of 13 exons that generate two alternatively spliced mRNAs that encode precursor proteins of 421 amino acids (isoform 1) and 376 amino acids (isoform 2). Mutations in the TMLHE gene are associated with a male-specific form of a neurodevelopmental disorder originally identified as autism, X-linked 6 (AUTS6X).
The genes encoding the serine hydroxymethyltransferases, suspected to catalyze the second reaction of carnitine synthesis, are described in the Serine Biosynthesis section of the Amino Acid Biosynthesis page.
The third step in carnitine synthesis is catalyzed by aldehyde dehydrogenase 9 family, member A1 (ALDH9A1). The ALDH9A1 encoded enzyme was formerly known as 4-N-trimethylaminobutyraldehyde dehydrogenase (TMABADH).
The ALDH9A1 gene is located on chromosome 1q24.1 and is composed of 11 exons that generate two alternatively spliced mRNAs that encode proteins of 518 amino acids (isoform 1) and 424 amino acids (isoform 2).
The gene encoding γ-butyrobetaine dioxygenase (also called γ-butyrobetaine 2-oxoglutarate dioxygenase or γ-butyrobetaine hydroxylase) is identified as BBOX1. The BBOX1 gene is located on chromosome 11p14.2 and is composed of 11 exons that generate five alternatively spliced mRNAs, all of which encode the same 387 amino acid protein.
Most human tissues can convert trimethyllysines to γ-butyrobetaine (the compound prior to carnitine) but only the liver, kidney, brain, and testes express the γ-butyrobetaine dioxygenase enzyme that hydroxylates γ-butyrobetaine forming carnitine. The γ-butyrobetaine dioxygenase enzyme, as well as the first enzyme of carnitine synthesis (trimethyllysine hydroxylase, encoded by the TMLHE gene), are members of the large family of 2-oxoglutarate and ferrous (Fe2+) iron-dependent dioxygenases. Although the liver, kidney, brain, and testes can synthesize carnitine, only the liver and the kidneys can transport carnitine to the blood.
Functions of Carnitine
The major function of carnitine is in the activation of long-chain fatty acids, generating fatty acyl-carnitines, a process that is required for the transport of long-chain fatty acids into the mitochondria for β-oxidation. However, carnitine is also involved in the metabolism of ketones, and the catabolism of the branched-chain amino acids. Carnitine is also important for the removal of fatty acyl-CoA metabolites so as to reduce the likelihood for the accumulation of toxic intermediates.
The major carnitine derivatives, that are not the long-chain fatty acylcarnitines involved in fatty acid acid oxidation, are acetyl-carnitine (acetyl-L-carnitine, ALC), propionyl-L-carnitine (PLC), and isovaleryl-L-carnitine (iso-VLC). Both acetyl-carnitine and propionyl-carnitine have been shown to improve insulin sensitivity (reduce insulin resistance) most likely as a result of inhibiting the transport of long-chain fatty acids into cells. Several studies have also demonstrated that propionyl-carnitine is able to lessen the development of atherosclerosis and lessen endothelial cell dysfunction. Acetyl-carnitine also crosses the blood brain barrier and in the brain modifies CNS acetylcholine production as well as improving energy production by neurons.
In addition to these three major carnitine derivates, human cells produce short and medium-chain acylcarnitines such as acetoacetyl-L-carnitine, butyryl-L-carnitine, hydroxybutyryl-L-carnitine, hexanoyl (caproic acid)-L-carnitine (six carbons), octanoyl-L-carnitine (eight carbons), decanoyl (capric acid)-L-carnitine (ten carbons).
The production of hydroxybutyryl-L-carnitine (HB-carnitine) increases in skeletal muscle during periods of fasting to enhance the transport of ketones into the mitochondria where they can be oxidized for ATP production.
Role of de novo Carnitine Synthesis in Fuel Adaptation
The significance of de novo carnitine synthesis to overall metabolism was demonstrated in SLC25A45 knock-out mice. The SLC25A45-mediated transport of trimethyllysine (TML) into the mitochondria was shown to contribute to overall fuel choice adaptation. In SLC25A45 negative mice there is an observable increase in glucose oxidation with a concomitant decrease in mitochondrial fatty acid β-oxidation. In addition, the loss of SLC25A45 results in suppression of fatty acid β-oxidation in brown adipose tissue (BAT) resulting in decreased adaptive thermogenesis.
These results demonstrate that, whereas carnitine can acquired from the diet, and dietary carnitine supplementation is correlated to increased fatty acid β-oxidation capacity, de novo carnitine synthesis is critical to the process that dictate cellular fuel choices under different physiological conditions.
Methionine Catabolism
The principal fates of the conditionally essential amino acid methionine are incorporation into polypeptide chains and use in the synthesis of cysteine and the synthesis of the important methyl donor, S-adenosylmethionine, SAM (also designated AdoMet). Synthesis of cysteine, which is a transulfuration reaction, also yields α-ketobutyrate (also referred to as 2-ketobutyrate or 2-oxobutyrate). The catabolism of methionine is therefore, related to the catabolism of cysteine since it is first used to produce cysteine.
The α-ketobutyrate that is generated during the conversion of methionine to cysteine is converted to propionyl-CoA via the action of branched-chain keto acid dehydrogenase, BCKD. The propionyl-CoA is converted to succinyl-CoA via a series of three reactions taking place in the mitochondria. The succinyl-CoA can then enter the TCA cycle for further oxidation. The enzymes required for this conversion are propionyl-CoA carboxylase, methylmalonyl-CoA epimerase, and methylmalonyl-CoA mutase, respectively. Propionyl-CoA carboxylase is called an ABC enzyme due to the requirements for ATP, Biotin, and CO2 for the reaction.
The clinical significance of methylmalonyl-CoA mutase in this pathway is that it is one of only two enzymes that requires a vitamin B12-derived co-factor for activity. The other B12-requiring enzyme is methionine synthase as described in the the Amino Acid Biosynthesis page. This propionyl-CoA conversion pathway is also required for the metabolism of the amino acids valine, isoleucine, and threonine and fatty acids with an odd number of carbon atoms. For this reason this three-step reaction pathway is often remembered by the mnemonic as the VOMIT pathway, where V stands for valine, O for odd-chain fatty acids, M for methionine, I for isoleucine, and T for threonine.
Propionyl-CoA carboxylase functions as a heterododecameric enzyme (subunit composition: α6β6) and the two different subunits are encoded by the PCCA and PCCB genes, respectively. The PCCA gene is located on chromosome 13q32.3 and is composed of 32 exons that generate eleven alternatively spliced mRNAs, each of which encode a unique protein isoform. The PCCB gene is located on chromosome 3q22.3 and is composed of 17 exons that generate two alternatively spliced mRNAs encoding proteins of 539 amino acids (isoform 1) and 559 amino acids (isoform 2). Mutations in either the PCCA gene or the PCCB gene result in the organic acidemia known as propionic acidemia.
Methylmalonyl-CoA epimerase is encoded by the MCEE gene located on chromosome 2p13.3 and is composed of 4 exons that encode a 176 amino acid precursor protein. Methylmalonyl-CoA mutase is encoded by the MMUT gene located on chromosome 6p12.3 and is composed of 13 exons that encode a precursor protein of 750 amino acids. Mutations in the MMUT gene are one cause of the methylmalonic acidemias.
Regulation of the methionine metabolic pathway is based on the availability of methionine and cysteine. If both amino acids are present in adequate quantities, SAM accumulates and is a positive effector on cystathionine β-synthase, encouraging the production of cysteine and α-ketobutyrate (also referred to as 2-ketobutyrate or 2-oxobutyrate), both of which are glucogenic. However, if methionine is scarce, SAM will form only in small quantities, thus limiting cystathionine β-synthase activity. Under these conditions accumulated homocysteine is remethylated to methionine, using N5-methyl-THF as the methyl donor.
Phenylalanine and Tyrosine Catabolism
Phenylalanine normally has only two fates: incorporation into polypeptide chains, and hydroxylation to tyrosine via the tetrahydrobiopterin-requiring phenylalanine hydroxylase (PAH) reaction. Thus, phenylalanine catabolism always ensues in the pathway of tyrosine biosynthesis followed by tyrosine catabolism. Tyrosine is equally important for protein biosynthesis as well as an intermediate in the biosynthesis of the catecholamines: dopamine, norepinephrine, and epinephrine.
The pathway of tyrosine degradation involves conversion to fumarate and acetoacetate, allowing phenylalanine and tyrosine to be classified as both glucogenic and ketogenic. The catabolism of tyrosine involves five reactions, four of which have been shown to be associated with inborn errors in metabolism and three of these result in clinically significant disorders.
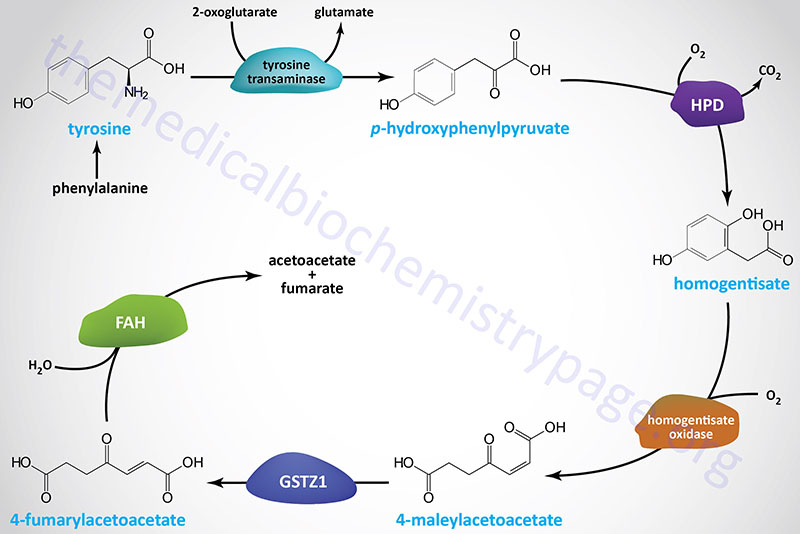
The first reaction of tyrosine catabolism involves the nuclear genome encoded mitochondrial enzyme tyrosine transaminase (also identified as tyrosine aminotransferase) and generates the corresponding ketoacid, p-hydroxyphenylpyruvic acid. Like most aminotransferase reaction, tyrosine aminotransferase utilizes 2-oxoglutarate (α-ketoglutarate) as the amino acceptor with the consequent generation of glutamate.
Tyrosine aminotransferase is encoded by the TAT gene. The TAT gene is located on chromosome 16q22.2 and is composed of 12 exons that generate a protein of 454 amino acids.
The second reaction of tyrosine catabolism is catalyzed by 4-hydroxyphenylpyruvate dioxygenase which is encoded by the HPD gene. The HPD gene is located on chromosome 12q24.31 which is composed of 17 exons that generate two alternatively spliced mRNAs encoding proteins of 393 amino acids (isoform 1) and 354 amino acids (isoform 2).
The product of the HPD reaction is homogentisic acid (homogentisate). Homogentisate is oxidized by the second dioxygenase enzyme of tyrosine catabolism, homogentisate oxidase. Homogentisate oxidase is encoded by the homogentisate 1,2-dioxygenase gene, HGD. The HGD gene is located on chromosome 3q13.33 and is composed of 16 exons that encode a protein of 445 amino acids.
Oxidation of homogentisate yields 4-maleylacetoacetate which is isomerized to 4-fumarylacetoacetate by the enzyme glutathione S-transferase zeta (ζ) 1 which is encoded by the GSTZ1 gene. Glutathione S-transferase zeta 1 was formerly called 4-maleylacetoacetate isomerase or maleylacetoacetate cis–trans-isomerase. The GSTZ1 gene is located on chromosome 14q24.3 and is composed of 9 exons that generate four alternatively spliced mRNAs, each of which encode a distinct protein isoform. One GSTZ1 encoded protein lacks the glutathione binding site and is, therefore, unlikely to exhibit the enzymatic activity of the other three isoforms.
Fumarylacetoacetate is hydrolyzed to fumarate and acetoacetate by the enzyme fumarylacetoacetate hydrolase which is encoded by the FAH gene. The FAH gene is located on chromosome 15q25.1 and is composed of 15 exons that generate three alternatively spliced mRNAs, each of which encode the same 419 amino acid protein.
The fumarate end product of tyrosine catabolism feeds directly into the TCA cycle for further oxidation. The acetoacetate is activated to acetoacetyl-CoA via the action of the mitochondrial ketone body utilization enzyme, succinyl-CoA:3-oxoacid-CoA transferase (SCOT) which is encoded by the OXCT1 (3-oxoacid-CoA transferase 1) gene. Acetoacetate can also be activated in the cytosol by the cytosolic enzyme, acetoacetyl-CoA synthetase (AACS).
Disorders Associated with Defective Tyrosine Metabolism
Defects in three of the enzymes of tyrosine catabolism result in increased concentrations of tyrosine and metabolites in the blood and urine. These disorders are referred to as the tyrosinemias and are also often referred to as the hypertyrosinemias. Because of the involvement of mutations in three distinct genes in the etiology of tyrosinemia, there are three distinct forms identified as tyrosinemia type 1, type 2, and type 3.
Mutations in the FAH gene result in life-threatening disorder, tyrosinemia type 1 (TYRSN1). TYRSN1 is an autosomal recessive disorder characterized by hypertyrosinemia and progressive liver disease. This disorder also leads to a secondary renal tubular dysfunction resulting in hypophosphatemic rickets. Defects in the FAH gene result in the accumulation of the upstream tyrosine metabolites, maleylacetoacetate and fumarylacetoacetate, as well as their metabolic by-products succinylacetone and succinylacetoacetate. The compounds are toxic to cells, particularly liver and kidney, explaining the pathology associated with this disease. Succinylacetone is also a potent inhibitor of δ-aminolevulinic acid dehydratase (also called porphobilinogen synthase) leading to accumulation of δ-aminolevulinic acid (ALA). ALA is neurotoxic and thought to be the cause of the acute porphyria-like pathology in this TYRSN1.
Inherited mutations in the TAT gene lead to tyrosinemia type 2 (TYRSN2; also called Richner-Hanhart syndrome) which, as the name implies, is associated with elevated tyrosine levels in the blood and, consequently the urine. This form of tyrosinemia is associated with intellectual impairment, painful corneal eruptions, photophobia, keratitis, and painful palmoplantar hyperkeratosis.
Mutations in the HPD gene result in tyrosinemia type 3 (TYRSN3). TYRSN3 is an autosomal recessive disease that, in addition to hypertyrosinemia, is associated with mild intellectual impairment and/or convulsions but these patients do not display hepatic damage as is characteristic of tyrosinemia type 1.
The first inborn error in metabolism ever recognized, alkaptonuria, was demonstrated to be the result of a defect in phenylalanine and tyrosine catabolism. Alkaptonuria is caused by defective homogentisic acid oxidase which is the third enzyme in the tyrosine catabolic pathway. Homogentisic acid accumulation is relatively innocuous, causing urine to darken on exposure to air, but no life-threatening effects accompany the disease. The only untoward consequence of alkaptonuria is ochronosis (bluish-black discoloration of the tissues) and arthritis of indeterminate etiology.
Serine Catabolism
The catabolism of serine in humans occurs in both the cytosol and the mitochondria. In both cellular compartments serine catabolism involves the conversion to glycine and then glycine oxidation to CO2 and NH3, with the production of two equivalents of N5,N10-methyleneTHF. This reaction process is also discussed in the context of glycine synthesis in the Amino Acid Biosynthesis page.
Serine can also be catabolized back to the glycolytic intermediate, 3-phosphoglycerate, by a pathway that is essentially a reversal of serine biosynthesis, however, the enzymes are different.
Serine (and glycine) can also be oxidized to oxalate (see also the Lipolysis and the Oxidation of Fatty Acids page) through a series of reactions proceeding to 3-hydroxypyruvate which involves transamination of pyruvate. The 3-hydroxypyruvate can then converted to glycoaldehyde via the action of glyoxylate and hydroxypyruvate reductase (encoded by the GRHPR gene). The glycoaldehyde is ultimately metabolized to glyoxylate. Glyoxylate can be metabolized to oxalate by hydroxyacid oxidase 1 (encoded by the HAO1 gene; also called glycolate oxidase). Alternatively, glyoxylate and alanine can be converted to glycine and pyruvate, respectively via the action of the vitamin B6-dependent enzyme, alanine–glyoxylate and serine–pyruvate aminotransferase (encoded by the AGXT gene).
Although it has been demonstrated in mammals, such as rodents and dogs, that serine can be converted to pyruvate through a deamination reaction catalyzed by the vitamin B6-dependent enzyme, serine/threonine dehydratase (encoded by the SDS gene), the serine deamination activity of the enzyme appears to be lacking in humans even though the activity is involved in the catabolism of threonine in humans as described in the Threonine Catabolism section below.
Role of Serine Catabolism in NADH Production
Serine catabolism in the mitochondria provides a mechanism for the generation of NADH independent of the TCA cycle. Serine is first converted to glycine via the mitochondrial serine hydroxymethyltransferase 2 (SHMT2) catalyzed reaction. The methylene-THF that is generated is oxidized to 10-formyl-THF via the methylene tetrahydrofolate dehydrogenase 2 (MTHFD2) catalyzed reaction which simultaneously reduces NAD+ to NADH. The MTHFD2 enzyme is resistant to inhibition by NADH and so this reaction can contribute to significant mitochondrial NADH production under increased serine catabolism as well as during periods where the major NADH producing catabolic pathways are inhibited by increasing NADH levels.
Accumulation of NADH is toxic to mitochondria via its contribution to the production of reactive oxygen species (ROS), superoxide anion, and hydrogen peroxide. Serine catabolism can, therefore, contribute to mitochondrial ROS production which can precipitate mitochondrial apoptosis.
During hypoxic conditions serine catabolism contributes to NADH production and this may contribute to tumor progression. However, there are limits to this benefit as enhanced serine metabolism-mediated NADH production contributes to ROS-mediated toxicity in these cells. Indeed, this pathway is currently being studied as a means to pharmacologically enhance serine catabolism in cancer cells to mediate their programmed death. Conversely, pharmacologic inhibition of mitochondrial serine catabolism could prove to be beneficial in chronic ischemia conditions such as cardiovascular disease.
Threonine Catabolism
Threonine Catabolism via Serine Dehydratase
There are at least three pathways for threonine catabolism that have been identified in yeasts, insects, and vertebrates including mammals. The principal threonine catabolizing pathway in humans involves a glycine-independent serine/threonine dehydratase (also known as serine dehydratase/threonine deaminase) yielding ammonium ion (NH4+) and α-ketobutyrate (also referred to as 2-ketobutyrate or 2-oxobutyrate). The 2-ketobutyrate is converted to propionyl-CoA via the addition of Co-A catalyzed via branched-chain keto acid dehydrogenase (BCKD).
Serine/threonine dehydratase is derived from the SDS gene. The SDS gene is located on chromosome 12q24.13 and is composed of 8 exons that encode a protein of 328 amino acids. Serine/threonine dehydratase is expressed at high levels only in the liver. It appears that in newborn infants catabolism of threonine occurs exclusively via the action of the serine/threonine dehydratase. Therefore, it is presumed that this is the predominant threonine catabolizing pathway in humans.
The resulting propionyl-CoA is converted, via a series of three reactions, to succinyl-CoA. These reactions take place within the mitochondria. The enzymes required for this conversion are propionyl-CoA carboxylase, methylmalonyl-CoA epimerase, and methylmalonyl-CoA mutase, respectively. Propionyl-CoA carboxylase is called an ABC enzyme due to the requirements for ATP, Biotin, and CO2 for the reaction. This propionyl-CoA conversion pathway is also required for the metabolism of the amino acids valine, isoleucine, and methionine, and odd-chain length fatty acids. For this reason this three-step reaction pathway is often remembered by the mnemonic as the VOMIT pathway, where V stands for valine, O for odd-chain fatty acids, M for methionine, I for isoleucine, and T for threonine.
Propionyl-CoA carboxylase functions as a heterododecameric enzyme (subunit composition: α6β6) and the two different subunits are encoded by the PCCA and PCCB genes, respectively. The PCCA gene is located on chromosome 13q32.3 and is composed of 32 exons that generate eleven alternatively spliced mRNAs, each of which encode a unique protein isoform. The PCCB gene is located on chromosome 3q22.3 and is composed of 17 exons that generate two alternatively spliced mRNAs encoding proteins of 539 amino acids (isoform 1) and 559 amino acids (isoform 2). Mutations in either the PCCA gene or the PCCB gene result in the organic acidemia known as propionic acidemia.
Methylmalonyl-CoA epimerase is encoded by the MCEE gene located on chromosome 2p13.3 and is composed of 4 exons that encode a 176 amino acid precursor protein. Methylmalonyl-CoA mutase is encoded by the MMUT gene located on chromosome 6p12.3 and is composed of 13 exons that encode a precursor protein of 750 amino acids. Mutations in the MMUT gene are one cause of the methylmalonic acidemias.
Threonine Catabolism via Serine Hydroxymethyltransferase (SHMT)
]The second pathway of threonine catabolism utilizes serine hydroxymethyltransferase (SHMT). As indicated in the Glycine Biosynthesis section of the Amino Acid Biosynthesis page, humans express two serine hydroxymethyltransferase genes, SHMT1 and SHMT2. The encoded enzymes belong to the family of one-carbon transferases. These enzymes are also identified as glycine hydroxymethyltransferase (was also identified as threonine aldolase). The products of SHMT action on threonine are acetaldehyde and glycine. The glycine can be converted to serine via the same enzyme and the serine is then catabolized as described above yielding pyruvate and NH4+. Thus, via this catabolic pathway threonine yields ketogenic and glucogenic byproducts.
In humans it appears that threonine aldolase is actually encoded by a non-functional pseudogene, whereas in other mammals and vertebrates (e.g. mice, zebrafish, and clawed frogs) the threonine aldolase gene encodes a functional threonine catabolizing enzyme.
Threonine Catabolism via Threonine Dehydrogenase (non-functional in humans)
An additional pathway of threonine catabolism in mammals occurs in the mitochondria and is initiated by threonine dehydrogenase (TDH) yielding α-amino-β-ketobutyrate (2-amino-3-ketobutyrate). The 2-amino-3-ketobutyrate is either converted to acetyl-CoA and glycine, via the action of 2-amino-3-ketobutyrate coenzyme A ligase (also called glycine C-acetyltransfease), or it can spontaneously degrade to aminoacetone which is converted to pyruvate.
The threonine dehydrogenase gene in humans appears to be an expressed non-functional pseudogene due to the incorporation of three inactivating mutations. Due to these mutations there are only truncated proteins produced that would be non-functional since they have lost part of the NAD+ binding motif and the C-terminal domain that would be responsible for binding L-threonine. Thus, whereas, this enzyme is a major threonine catabolizing enzyme in other mammals such as mice, it is the serine/threonine dehydratase enzyme that is most important for threonine catabolism in humans.
Tryptophan Catabolism
The major tryptophan catabolic pathway in humans results in the formation of acetoacetyl-CoA. This pathway is referred to as the kynurenine pathway and occurs predominantly in the liver. However, it is important to note that overall metabolism of tryptophan includes not just its catabolism but also its use in the synthesis of serotonin and melatonin, the synthesis of indole derivatives such as the trace amine, tryptamine, and the synthesis of NAD+.
The first enzyme of the kynurenine pathway of tryptophan catabolism in the liver opens the indole ring. This enzyme is tryptophan 2,3-dioxygenase which is encoded by the TDO2 gene. The product of the TDO2 reaction is N-formyl-kynurenine.
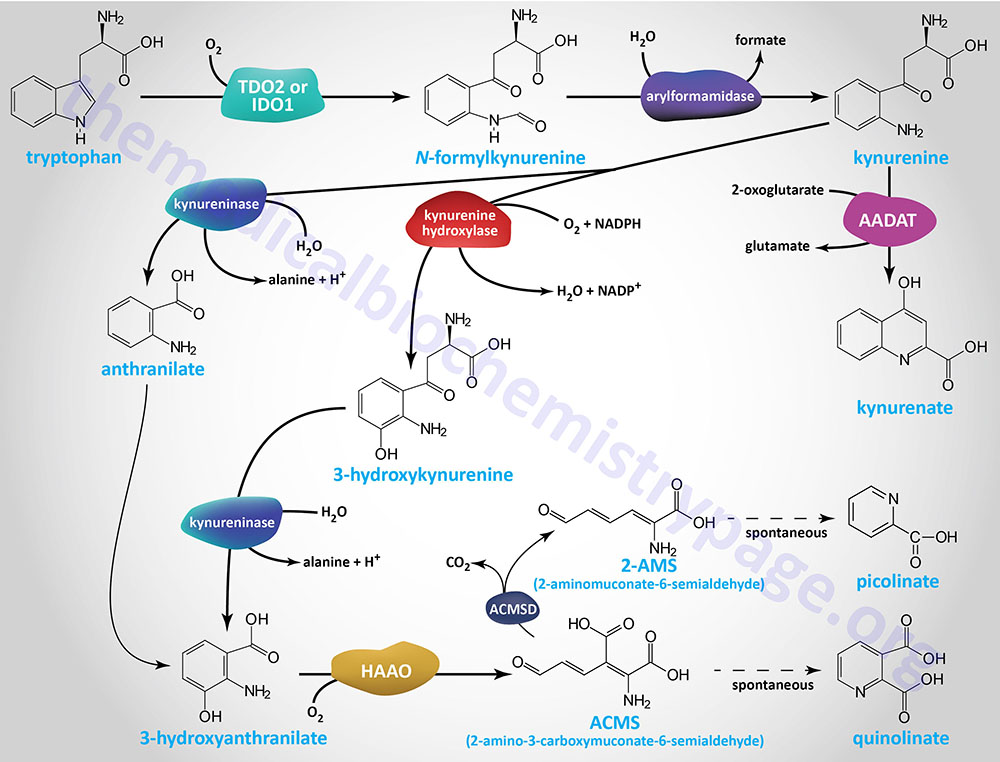
The TDO2 gene is located on chromosome 4q32.1 and is composed of 12 exons that encode a protein of 406 amino acids. Expression of the TDO2 gene is very nearly exclusive to the liver. The TDO2 gene is highly inducible, leading to increasing concentrations of the enzyme, of up to 10-fold, on a diet high in tryptophan.
Tryptophan catabolism can also be initiated within non-hepatic tissues by the enzyme indoleamine 2,3-dioxygenase 1 encoded by the IDO1 gene. The activity of the IDO1 enzyme also results in the generation of N-formyl-kynurenine.
The IDO1 gene is located on chromosome 8p11.21 and is composed of 10 exons that encode a protein of 403 amino acids. Unlike the TDO2 gene, whose expression is restricted to the liver, the IDO1 gene is expressed in numerous tissues but at levels much lower than the level of liver expression of the TDO2 gene. In addition to tryptophan, IDO1 can metabolize 5-hydroxytryptophan, tryptamine, and serotonin.
N-formyl-kynurenine is then converted to kynurenine by the enzyme, arylformamidase (also called kynurenine formamidase), encoded by the AFMID gene. The AFMID gene is located on chromosome 17q25.3 and is composed of 12 exons that generate two alternatively spliced mRNAs that generate 11 alternatively spliced mRNAs, each of which encode distinct protein isoforms.
Kynurenine is the first key branch point intermediate in the catabolic pathway leading to three distinct catabolic fates for tryptophan. These branches involve conversion of kynurenine to anthranilic acid, to kynurenic acid, or to 3-hydroxyanthranilate. The latter compound can be fully catabolized or serve as the precursor for NAD+ synthesis.
Kynurenine can undergo deamination to kynurenic acid in a transamination reaction. The transamination reaction can be catalyzed by several different transaminases. One of these enzymes is the human homolog of mouse kynurenine aminotransferase II, yielding kynurenic acid. The human kynurenine aminotransferase II (KAT II or KAT2) enzyme is called aminoadipate aminotransferase which is encoded by the AADAT gene. The enzyme encoded by the AADAT gene has two aminotransferase activities, one of which is utilized in the saccharopine pathway of lysine catabolism, the other is used in the transamination of kynurenine.
Another kynurenine transaminase is cysteine conjugate-beta lyase 2 encoded by the CCBL2 gene. The CCBL2 encoded protein was formerly known as kynurenine aminotransferase III (KAT III or KAT3). Another related enzyme, that was once called kynurenine aminotransferase I (KAT I or KAT1), is cysteine conjugate-beta lyase encoded by the CCBL1 gene. The CCBL1 encoded protein is a cytosolic enzyme that, as its name implies, metabolizes cysteine conjugates.
Conversion of kynurenine to anthranilic acid involves the enzyme, kynureninase. Kynureninase is a pyridoxal phosphate (vitamin B6)-requiring enzyme encoded by the KNYU gene. The KYNU gene is located on chromosome 2q22.2 and is composed of 18 exons that generate three alternatively spliced mRNAs that collectively encode two distinct protein isoform. Expression of the KYNU gene is highest in the liver which is to be expected since the majority of tryptophan catabolism occurs in this organ.
The major step in tryptophan catabolism involves the hydroxylation of kynurenine by kynurenine hydroxylase (correctly identified as kynurenine 3-monooxygenase) forming 3-hydroxykynurenine. Kynurenine 3-monooxygenase is encoded by the KMO gene. The KMO gene is located on chromosome 1q43 and is composed of 15 exons that generate two alternatively spliced mRNAs, both of which encode distinct protein isoforms.
The next step in the pathway produces 3-hydroxyanthranilic acid and the amino acid alanine from 3-hydroxykynurenine. This reaction is catalyzed by kynureninase. It is the production of alanine that allows tryptophan to be classified among the glucogenic amino acids.
As indicated above kynureninase, which is encoded by the KNYU gene, is also used for the conversion of kynurenine to anthranilic acid with the release of alanine. The anthranilic acid subsequently is converted to 3-hydroxyanthranilic acid. This latter two-step reaction is a minor component of overall tryptophan catabolism.
The next step in the pathway is the conversion of 3-hydroxyanthranilic acid to quinolinic acid catalyzed by the enzyme 3-hydroxyanthranilate 3,4-dioxygenase which is encoded by the HAAO gene. The product of the HAAO reaction is α-amino-β-carboxymuconate-ε-semialdehyde (ACMS: 2-amino-3-carboxymuconate-6-semialdehyde). ACMS can spontaneously cyclize to quinolinic acid.
The HAAO gene is located on chromosome 2p21 and is composed of 11 exons that encode a 286 amino acid protein. Expression of the HAAO gene is highest in the liver with expression in the small intestine and kidney being the next highest locations.
Catabolism of α-Amino-β-Carboxymuconate-ε-Semialdehyde: ACMS
The α-amino-β-carboxymuconate-ε-semialdehyde (ACMS: 2-amino-3-carboxymuconate-6-semialdehyde) product of the HAAO catalyzed reaction is ultimately catabolized to acetoacetyl-CoA.
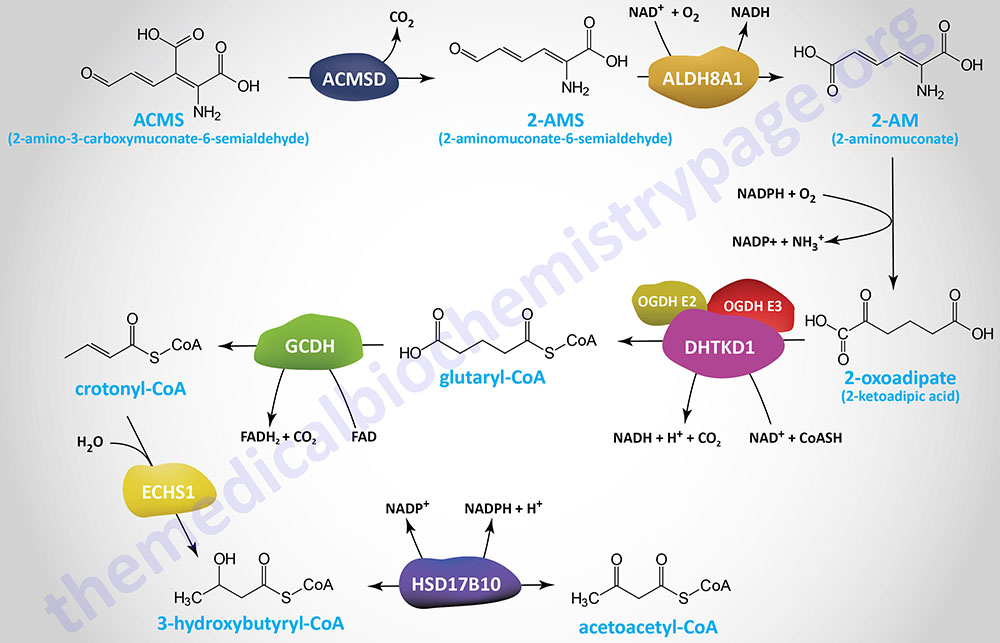
In the initial step, ACMS is decarboxylated to α-aminomuconate-ε-semialdehyde (AMS). AMS is also called 2-aminomuconate-6-semialdehyde (2-AMS) and is also called more simply, 2-aminomuconate semialdehyde. The decarboxylation is catalyzed by the enzyme, α-amino-β-carboxymuconate-ε-semialdehyde decarboxylase (encoded by the ACMSD gene). The ACMSD encoded enzyme is also identified as 2-amino-3-carboxymuconate-6-semialdehyde decarboxylase.
The ACMSD gene is located on chromosome 2q21.3 and is composed of 13 exons that generate two alternatively spliced mRNAs. Only one mRNA encodes a functional protein as the other is likely degraded via the nonsense-mediated decay (NMD) pathway. The functional ACMSD encoded protein (isoform 1) is 336 amino acids.
AMS is ultimately converted to 2-oxoadipic acid (2-OAA; also called α-ketoadipic acid or 2-ketoadipic acid) through a series of two reactions and then to glutaryl-CoA. The first reaction of AMS metabolism, which converts 2-AMS to 2-aminomuconate (2-AM) is catalyzed by the enzyme encoded by the ALDH8A1 (aldehyde dehydrogenase 8 family member 1) gene. The enzyme encoded by the ALDH8A1 gene is commonly referred to as 2-aminomuconate-6-semialdehyde (2-AMS) dehydrogenase (AMSDH).
AMS, like ACMS, can spontaneously cyclize. Whereas this produces quinolinic acid from ACMS, the cyclization of AMS yields picolinic acid (pyridine-2-carboxylic acid), a compound very similar to nicotinic acid (pyridine-3-carboxylic acid). Picolinic acid functions as a natural chelator of minerals such as iron, zinc, and chromium. Purported functions of picolinic acid in humans include neuroprotection, anti-proliferative effects, and immunological effects.
The lysine catabolic pathway merges with the tryptophan catabolic pathway with the production of 2-OAA and thus, the remainder of both pathways are the same. The 2-oxoadipic acid undergoes oxidative decarboxylation to glutaryl-CoA via the actions of the 2-oxoadipate dehydrogenase complex whose E1 subunits are encoded by the DHTKD1 (dehydrogenase E1 and transketolase domain containing 1) gene. The E2 and E3 subunits of this complex are shared with the 2-oxoglutarate dehydrogenase (α-ketoglutarate dehydrogenase) complex of the TCA cycle.
The DHTKD1 gene is located on chromosome 10p14 and is composed of 17 exons that encode a 919 amino acid protein.
Glutaryl-CoA is oxidized to crotonyl-CoA by the FAD-dependent enzyme, glutaryl-CoA dehydrogenase (GCDH). Glutaryl-CoA dehydrogenase is a member of the acyl-CoA dehydrogenase (ACAD) family of enzymes that includes members of the mitochondrial fatty acid β-oxidation pathway and as such the enzyme is also known as acyl-CoA dehydrogenase 5 (ACAD5).
Glutaryl-CoA dehydrogenase is encoded by the GCDH gene. The GCDH gene is located on chromosome 19p13.13 and is composed of 12 exons that generate two alternatively spliced mRNAs, both of which encode distinct precursor proteins, isoform a is 438 amino acids and isoform b is 428 amino acids, The isoform b protein is not functional as a glutaryl-CoA dehydrogenase. The isoform a precursor protein undergoes cleavage by mitochondrial processing peptidase complex to form the mature glutaryl-CoA dehydrogenase enzyme. Functional glutaryl-CoA dehydrogenase is a homotetrameric complex. Mutations in the GCDH gene result in the organic acidemia called glutaric acidemia type 1.
As in the lysine catabolic pathway, the crotonyl-CoA derived from tryptophan catabolism is converted to β-hydroxybutyryl-CoA (3-hydroxybutyryl-CoA) by the ECHS1 (enoyl-CoA hydratase, short chain 1; also identified as SCEH) gene encoded enzyme. The ECHS1 encoded enzyme is also identified as crotonase. Finally, β-hydroxybutyryl-CoA is converted to acetoacetyl-CoA, a reaction catalyzed by hydroxysteroid 17-beta dehydrogenase 10 (encoded by the HSD17B10 gene) which is also known as 3-hydroxybutyryl-CoA dehydrogenase.
Crotonyl-CoA can also serve as the substrate for the post-translational modification of lysine residues, a process termed protein crotonylation.
Tryptophan as Precursor for NAD+
Quinolinic acid is ultimately converted, via three steps, to the nicotinamide adenine dinucleotide co-factors. The three steps in the conversion of quinolinic acid (quinolinate) to NAD+ first involves the conversion to nicotinate mononucleotide (NAMN), then to nicotinate adenine dinucleotide (NAAD), and then to NAD+.
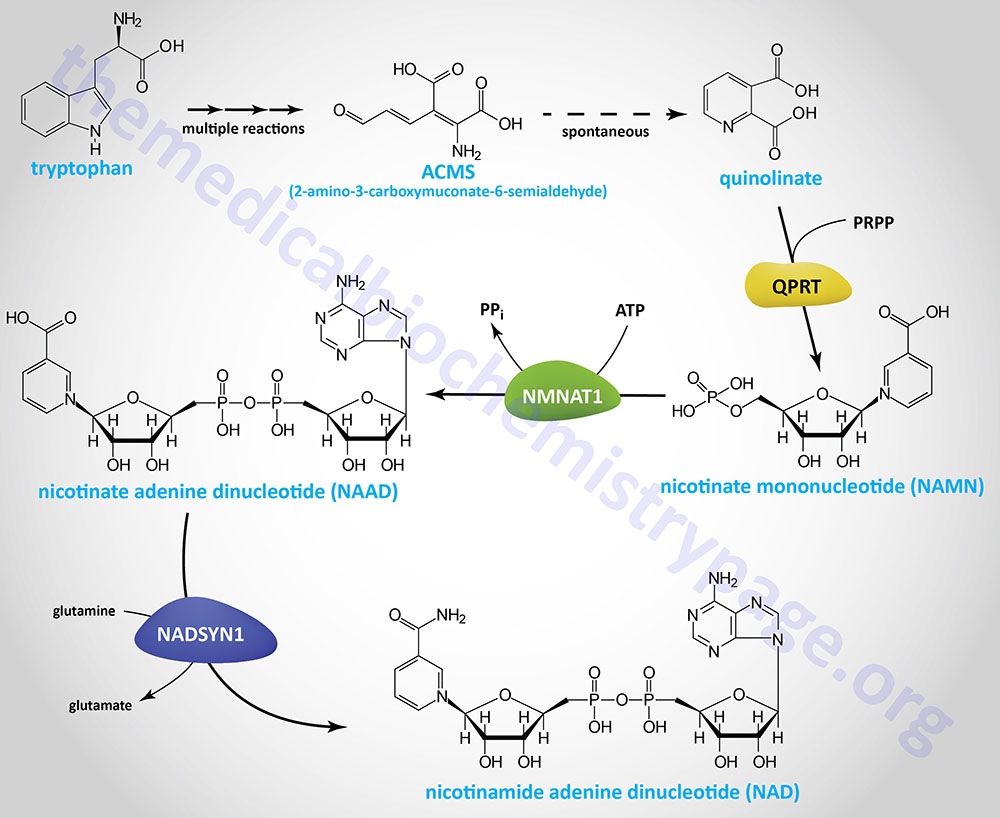
The first reaction is catalyzed by quinolinate phosphoribosyltransferase which is encoded by the QPRT gene. The QPRT encoded enzymes utilizes the activated form of ribose-5-phosphate (PRPP) to generate nicotinate mononucleotide, NAMN (nicotinic acid mononucleotide).
The QPRT gene is located on chromosome 16p11.2 and is composed of 4 exons that generate three alternatively spliced mRNAs, each of which encode a distinct protein isoform.
The second reaction is catalyzed by one of the nicotinamide nucleotide adenylyltransferase (NMNAT) family enzymes. Humans express three NMNAT encoding genes. The NMNAT enzyme adds AMP from ATP to NAMN with the release of pyrophosphate (PPi). The product of the NMNAT reaction is nicotinate adenine dinucleotide, NAAD (nicotinic acid dinucleotide).
The final reaction is catalyzed by NAD synthase (encoded by the NADSYN1 gene) which utilizes the NH4+, released from glutamine, to generate NAD from NAAD. Evidence suggests that the NADSYN1 encoded enzyme can also utilize free NH4+ as the substrate for the generation of NAD from NAAD.
The details of NAD+ synthesis and salvage are also described in the Vitamin B3: Metabolism and Functions page.
Clinical Significance of Kynurenine and Kynurenate
The levels of kynurenine have been shown to increase with the aging process as well as in conditions of inflammation. The aging related increases in kynurenine have been shown to be linked to increased frailty and mortality. In organisms such as the nematode (roundworm), Caenorhabditis elegans, and the fruit fly, Drosophila melanogaster, experiments have shown that elevated levels of kynurenine are directly correlated to reduced life span. The mechanism by which kynurenine contributes to these processes is due to the fact that it is an agonist of the aryl hydrocarbon receptor (AhR).
The AhR was originally identified as a protein that bound, and was activated by, the highly toxic chemicals of the dioxin family. Kynurenine was the first endogenous ligand identified as binding to and activating the AhR. In addition to kynurenine the AhR binds additional endogenous ligands such as tryptophan and the tryptophan-derived bioactive trace amine, tryptamine.
There are numerous AhR agonists, both exogenous and endogenous, and their effects upon activation of the receptor can be pathological or beneficial. Naturally occurring plant derived phytochemicals, such as quercetin, exert anti-aging effects via the AhR. In contrast kynurenine exerts pathological effects related to aging. These differences are due to ligand-specific downstream signaling processes. Kynurenine activation of AhR leads to enhancement of oxidative stress and inflammation as a result of increased expression of the cytochrome P450 enzymes encoded by the CYP1A1 and CYP1B1 genes, the interleukin-6 (IL-6) gene, and the transforming growth factor beta 1 (TGFB1) gene. Quercetin on the other hand appears to induce AhR activation of an antioxidant pathway via activation of the basic leucine zipper (bZIP) transcription factor, nuclear factor erythroid 2–related factor 2 (NRF2).
Kynurenic acid has been shown exert protective effects within the central nervous system (CNS) in part by acting as an anti-excitotoxic and anti-convulsive. Conversely, high levels of kynurenic acid have been found in the urine of individuals suffering from schizophrenia. Kynurenic acid has been shown to act as a non-competitive antagonist at the glycine binding site of the NMDA receptor (NMDA: N-methyl-D-aspartate) which is an ionotropic (ligand-gated ion channel) receptor for glutamate. The NMDA receptor is a key component of the glutamatergic neurotransmission system believed to be involved in the pathophysiology of schizophrenia, thus explaining the potential role of kynurenic acid in schizophrenia.
Hepatic Tryptophan Metabolism and Hepatocellular Carcinoma
Chronic physiological stress has been shown to contribute to numerous disease states including cancers such as hepatocellular carcinomas. Contributing to stress-induced cancers are perturbations of normal immunological responses. The liver is somewhat unique that it has specialized immune system functions and is occupied by several different types of immune cells such as B- and T-cells and NK (natural killer) cells that exist throughout the body, but also liver specific immune system cells such the myeloid lineage Kupffer cells.
During normal stressful circumstances the postganglionic sympathetic nervous system neurotransmitter, norepinephrine, binds to adrenergic receptors on hepatocytes. Hepatocytes express both α1– and β2-adrenergic receptors to which sympathetic norepinephrine (as well as epinephrine) binds. The consequences of adrenergic stimulation of hepatocytes is the activation of the normal responses of gluconeogenesis, glycogen breakdown, ketogenesis, ammonia uptake, and ureagenesis. However, chronic stress-induced stimulation of adrenergic receptors in the liver, particularly the β2-adrenergic receptor, is associated with disturbances in the inflammatory and metabolic processes of the liver.
Recent experiments (2026) have found that chronic stress-mediated activation of the β2-adrenergic receptor results in suppression of expression of the gene (QPRT) encoding quinolinate phosphoribosyltransferase. The QPRT encoded enzyme functions in the de novo pathway of NAD+ synthesis from tryptophan. Quinolinate phosphoribosyltransferase converts quinolinic acid (quinolinate) to nicotinate mononucleotide, NAMN (nicotinic acid mononucleotide). The inhibition of this pathway results in increased diversion of tryptophan catabolism to the kynurenine pathway yielding increased production of kynurenic acid.
Accumulation of kynurenic acid, accompanied by reduced de novo NAD+ synthesis, has a negative effect on mitochondrial function in liver resident T-cells which impairs their anti-tumor activity. These effects of chronic stress, on hepatocyte tryptophan metabolism, are highly contributory to the development of hepatocellular carcinoma.
Tryptophan Metabolism and Neurotransmission
Quinolinic acid, predominantly produced by microglial cells, is also an excitatory neurotoxic compound that functions via its ability to activate the glutamate NMDA receptors, and effect that is in opposition that of kynurenic acid. Quinolinic acid contributes to increased release of glutamate from glutamatergic neurons while simultaneously inhibiting its uptake by astrocytes. This effect of quinolinic acid results in excessive activation of NMDA receptors accounting for the neurotoxicity of quinolinic acid.
An intermediate in the HAAO catalyzed reaction is α-amino-β-carboxymuconate-ε-semialdehyde (or simply aminocarboxymuconate semialdehyde: ACMS). The ACMS intermediate can be metabolized via the action of the enzyme ACMS decarboxylase encoded by the ACSMD gene.
The ACMSD gene is located on chromosome 2q21.3 and is composed of 13 exons that generate two alternatively splice mRNAs. The isoform 2 mRNA is a likely candidate for nonsense-mediated decay, NMD. The isoform 1 mRNA encodes a protein of 336 amino acids.
The ACMSD catalyzed reaction is important for the prevention of over accumulation of quinolinic acid from ACMS. The product of the ACMSD reaction goes on to be further metabolized to acetoacetyl-CoA, the ultimate end product of complete tryptophan catabolism.
In addition to its role as an amino acid in protein biosynthesis and as a precursor for the synthesis of NAD+, tryptophan also serves as a precursor for the synthesis of the neurotransmitters serotonin and melatonin. These products are discussed in the Amino Acid Derivatives: Neurotransmitters, Nitric Oxide, and More page.