
Last Modified: April 3, 2026
Introduction to Amino Acid Biosynthesis
All tissues have some capability for synthesis of the non-essential amino acids as well as amino acid remodeling that generates other amino acids such as glutamate conversion to glutamine. In addition, there are several non-protein amino acids that contribute to various amino acid biosynthesis pathways such as ornithine, an amino acid critical to the function of the urea cycle, which can be converted to proline as well as to glutamate.
One of the major sources of non-essential amino acids is dietary, either as free amino acids or from the digestion of dietary proteins. Protein digestion and intestinal amino acid uptake is briefly reviewed in the following section but the details of the processes are discussed in the Digestion and Digestive Processes page.
Intestinal and Cellular Amino Acid Uptake
Dietary amino acids can be consumed as free amino acids or, more often, they are acquired from digested dietary proteins that are hydrolyzed through the concerted actions of gastric and pancreatic peptidases. The details of protein digestion are covered in the Digestion and Digestive Processes page.
Dietary protein digestion begins in the stomach via the actions of the pepsins, and continues within the lumen of the duodenum. Within the small intestine there are two principal pancreatic enzymes involved in protein digestion; trypsin and chymotrypsin. Several additional pancreatic peptidases play a lesser role in peptide digestion and include the carboxypeptidases and the elastases.
The absorption of most amino acids from the intestine, as well as uptake into cells of the various tissues of the body, requires an active transport process that is dependent upon Na+ or H+ co-transport. There are numerous amino acid transporters encompassing at least six major families of transport systems which are sometimes grouped into three broad categories. These three broad categories are the neutral amino acid (monoamino monocarboxylic) transporters, the dibasic (and cysteine) amino acid transporters, and the acidic (dicarboxylic) amino acid transporters. The details of amino acid transporters are covered in the Digestion and Digestive Processes page.
Essential vs. Nonessential Amino Acids
| Nonessential | Alanine, Asparagine, Aspartate, Cysteine, Glutamate, Glutamine, Glycine, Proline, Serine, Tyrosine |
| Essential | Arginine*, Histidine, Isoleucine, Leucine, Lysine, Methionine*, Phenylalanine*, Threonine, Tryptophan, Valine |
*The amino acids arginine, methionine and phenylalanine are considered essential (actually conditionally essential) for reasons not directly related to lack of synthesis. Arginine is synthesized by mammalian cells but at a rate that is insufficient to meet the growth needs of early development and during periods infection or inflammation. Methionine is required in large amounts to produce cysteine if the latter amino acid is not adequately supplied in the diet. Similarly, phenylalanine is needed in large amounts to form tyrosine if the latter is not adequately supplied in the diet.
Alanine Biosynthesis and the Glucose-Alanine Cycle
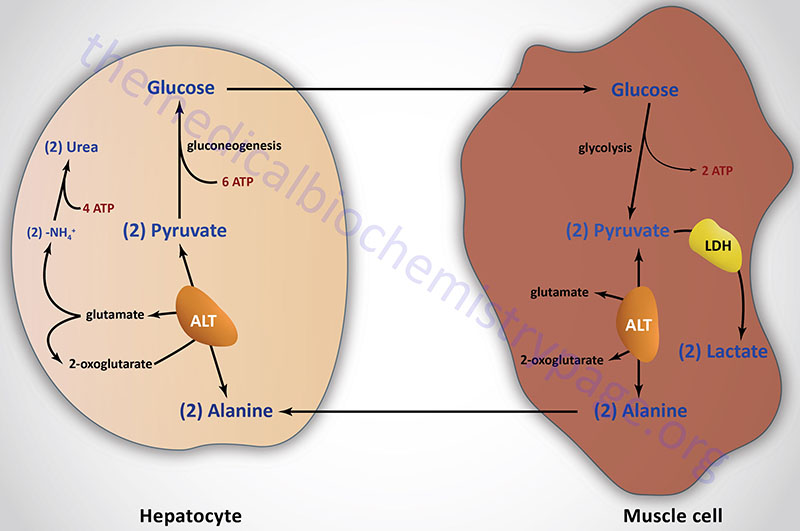
Aside from its role in protein synthesis, alanine is second only to glutamine in prominence as a circulating amino acid. In this capacity it serves a unique role in the transfer of nitrogen from peripheral tissue to the liver. Alanine is transferred to the circulation by many tissues, but mainly by skeletal muscle, in which alanine is formed from pyruvate at a rate proportional to intracellular pyruvate levels. Liver accumulates plasma alanine, reverses the transamination that occurs in muscle, and proportionately increases urea production. The pyruvate is either oxidized or converted to glucose via gluconeogenesis.
When alanine transfer from muscle to liver is coupled with glucose transport from liver back to muscle, the process is known as the glucose-alanine cycle. The key feature of the cycle is that in 1 molecule, alanine, peripheral tissue exports pyruvate and ammonia (which are potentially rate-limiting for metabolism) to the liver, where the carbon skeleton is recycled and most nitrogen eliminated.
There are two main pathways to production of muscle alanine: directly from protein degradation, and via the transamination of pyruvate by alanine transaminase, ALT (historically referred to as serum glutamate-pyruvate transaminase, SGPT).
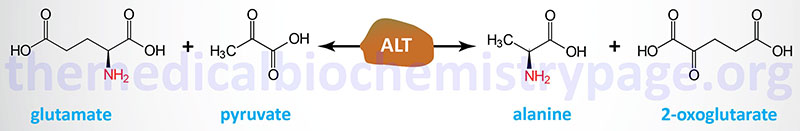
Humans express two genes encoding alanine transaminase. These genes are identified as GPT (also designated GPT1) and GPT2. The enzyme encoded by the GPT gene is localized to the cytosol, whereas the GPT2 encoded enzyme is localized to the mitochondria.
The GPT gene is located on chromosome 8q24.3 and is composed of 12 exons that generate three alternatively spliced mRNAs, all of which encode the same 496 amino acid protein. The tissues with the highest levels of GPT expression are the liver, the intestines, the kidneys, and adipose tissue.
The GPT2 gene is located on chromosome 16q11.2 and is composed of 13 exons that generate two alternatively spliced mRNAs that encode proteins of 523 amino acids (isoform 1) and 423 amino acids (isoform 2). The tissues with the highest levels of GPT2 expression are the liver, the intestines, the kidneys, adipose tissue, brain, and pancreas.
Arginine Biosynthesis
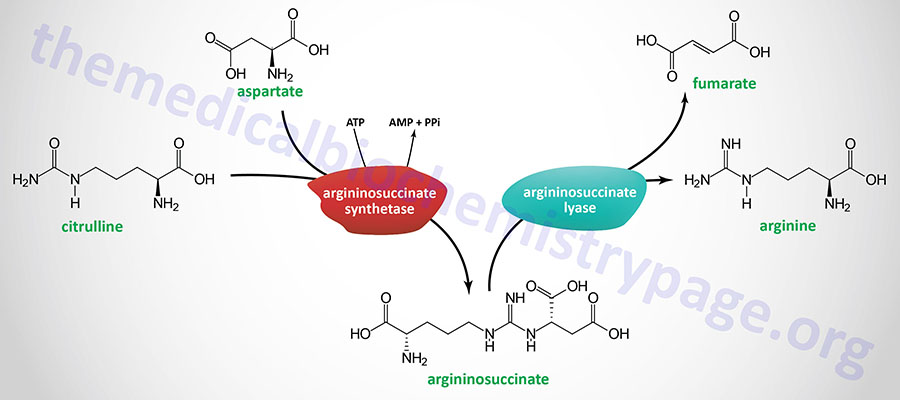
Humans cannot synthesize arginine at levels that are sufficient for cellular functions during periods of infection and inflammation as well as growth needs of the body during early development, thus the amino acid is considered a conditionally essential amino acid. Arginine can be synthesized from citrulline and aspartate in the context of the urea cycle. In this pathway citrulline and aspartate are condensed by argininosuccinate synthetase (also identified as argininosuccinate synthase) forming argininosuccinate. Argininosuccinate is subsequently hydrolyzed by argininosuccinate lyase forming fumarate and arginine.
Central Role of Arginine in Overall Metabolism
Arginine is a critical amino acid in multiple metabolic processes that includes amino acid biosynthesis, protein biosynthesis, neurotransmitter biosynthesis, synthesis of many regulatory molecules, and, like all amino acids, energy generation.
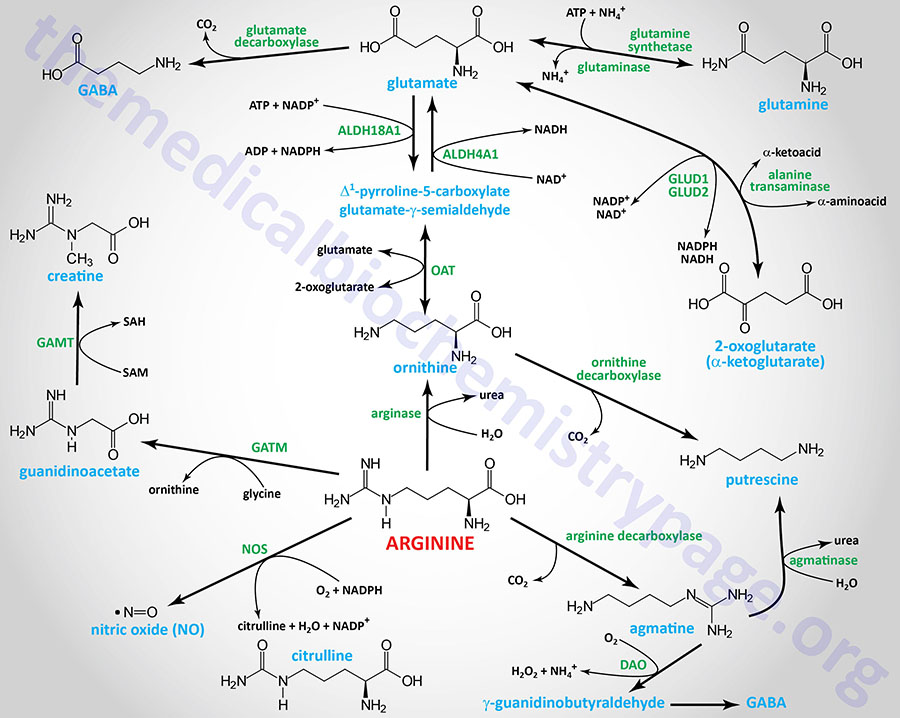
As indicated, although arginine is an essential amino acid, limited quantities can be derived via enzymes of the urea cycle. Indeed, the urea cycle represents one of the many critical metabolic functions of arginine, serving as an intermediate in the process of waste nitrogen disposal.
Arginine is the direct precursor for the synthesis of creatine, an energy storage molecule as creatine phosphate, and the critical gasotransmitter, nitric oxide, NO. These important pathways are discussed in detail in the Amino Acid Derivatives: Neurotransmitters, Nitric Oxide, and More page.
Through its conversion to ornithine, arginine is centrally involved in the synthesis of a class of biological molecules called the polyamines. Polyamines serve functions in DNA replication, are involved in the processes of protein synthesis, ion channel function (particularly the inwardly rectifying potassium channels), regulation of gene expression, cell proliferation, and apoptosis (programmed cell death). The synthesis of the polyamines is covered in detail in the Amino Acid Derivatives: Neurotransmitters, Nitric Oxide, and More page.
Arginine is converted to the neurotransmitter, agmatine, via decarboxylation. Agmatine, a member of the polyamine family of molecules, exerts numerous effects in the CNS that include inhibition of nociception (sensation of pain) and reduction in anxiety (anxiolytic) as well as anticonvulsive and anti-depressive effects.
Whereas arginine decarboxylase (ADC) has been characterized in plants, bacteria, and other mammals, the definitive ADC encoding gene in humans has yet to be characterized. An original assessment that ADC activity was associated with one of the antizyme inhibitor proteins, specifically the AZIN2 encoded protein, has subsequently been shown to be erroneous. Nonetheless, agmatine synthesis from arginine in humans is known to occur, particularly within the brain.
Asparagine Biosynthesis
Asparagine is synthesized from aspartate via an amidotransferase reaction catalyzed by asparagine synthetase. Asparagine synthetase is encoded by the ASNS gene. The ASNS gene is located on chromosome 7q21.3 and is composed of 16 exons that generate seven alternatively spliced mRNAs that collectively encode three protein isoforms.
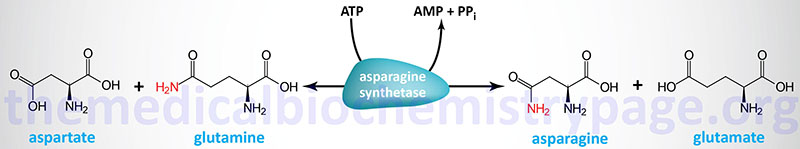
Clinical significance of asparagine synthesis is demonstrated by the fact that the level of expression of the ASNS gene is correlated to metastatic potential in numerous forms of cancer. Additionally, treatment of several cancer types with the enzyme asparaginase, or dietary restriction of asparagine, reduces metastasis but does not alter the growth of primary tumors. Conversely, enforced expression of the ASNS gene strongly promotes metastatic potential.
Aspartate Biosynthesis
Aspartate can be formed in a transamination reaction or via the deamination of asparagine. The transamination reaction is catalyzed by aspartate transaminase, AST. This reaction uses the α-keto acid, oxaloacetate, as the amino acceptor and glutamate as the primary amino group donor.

Humans express two AST enzymes, one that is localized in the cytosol and the other is localized to the mitochondria.
The cytosolic AST enzyme is encoded by the glutamate oxaloacetic transaminase 1 gene (GOT1). The GOT1 gene is located on chromosome 10q24.2 and is composed of 9 exons that encode a 413 amino acid protein.
The mitochondrial AST enzyme is encoded by the GOT2 gene which is located on chromosome 16q21 and is composed of 10 exons that generate two alternatively spliced mRNAs encoding precursor proteins of 430 amino acids (isoform 1) and 387 amino acids (isoform 2).
Aspartate is also formed by deamination of asparagine, a reaction catalyzed by asparaginase. Humans express at least three enzymes that can catalyze the deamination of asparagine to aspartate. However, the true primary physiologic substrates for these enzymes may not be asparagine.
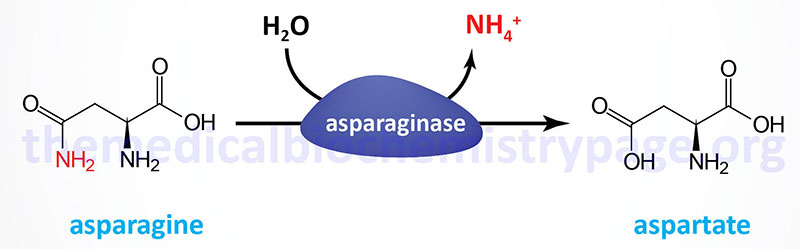
One enzyme was originally identified as a lysophospholipase due to its ability to hydrolyze lysophospholipids in addition to it being subsequently shown to harbor asparaginase activity. This enzyme is encoded by the asparaginase (ASPG) gene. The ASPG gene is located on chromosome 14q32.33 and is composed of 20 exons that generate two alternatively spliced mRNAs encoding proteins of 573 amino acids (isoform 1) and 555 amino acids (isoform 2).
A second human enzyme with asparaginase activity is the enzyme N-aspartyl-β-glucosaminidase (also called aspartylglucosaminidase) which is encoded by the AGA gene. The AGA gene is located on chromosome 4q34.3 and is composed of 9 exons that generate two alternatively spliced mRNAs. These mRNAs encode a 346 amino acid protein (isoform 1) and a 336 amino acid protein (isoform 2). The 346 amino acid protein exhibits an approximate mass of 35 kDa. Following synthesis the protein is cleaved to generate two subunits. The α-subunit is 27 kDa and the β-subunit is 17 kDa and these subunits form a heterotetrameric enzyme. The AGA encoded protein has as its primary function the hydrolysis of carbohydrates linked to asparagine residues in glycoproteins. Defects in the AGA gene result in aspartylglucoasminuria which is a lysosomal storage disease (lysosomal storage disorder).
The third human enzyme with asparaginase activity is called asparaginase like 1 (also called asparaginase and isoaspartyl peptidase 1) which is encoded by the ASRGL1 gene. In addition to being able to hydrolyze asparagine to aspartate, the ASRGL1 encoded enzyme hydrolyzes isoaspartyl peptide linkages. The ASRGL1 gene is located on chromosome 11q12.3 and is composed of 10 exons that generate two alternatively spliced mRNAs that express the same 308 amino acid protein.
Asparaginases from bacteria have significant clinical utility as they are used in the treatment of several forms of hematopoietic malignancies that express low levels of the ASNS gene and, therefore, rely on extracellular asparagine for their rapid growth. Leukemias that are asparaginase resistant have elevated levels of expression of the ASNS gene. Also, cancers that are capable of synthesizing asparagine de novo via asparagine synthetase are less responsive to asparaginase therapy.
Glutamate Biosynthesis
Glutamate can be synthesized by two distinctly different reaction pathways. In one reaction glutamate is synthesized by the reductive amination of 2-oxoglutarate (α-ketoglutarate) catalyzed by glutamate dehydrogenase (GDH); it is thus a nitrogen-incorporating reaction. In the second type of reaction, glutamate is formed from 2-oxoglutarate by aminotransferase reactions, with the amino nitrogen being donated by a number of different amino acids. Thus, glutamate is a general collector of amino nitrogen, a critical reaction in overall nitrogen homeostasis as discussed in detail in the Nitrogen Metabolism and the Urea Cycle page.
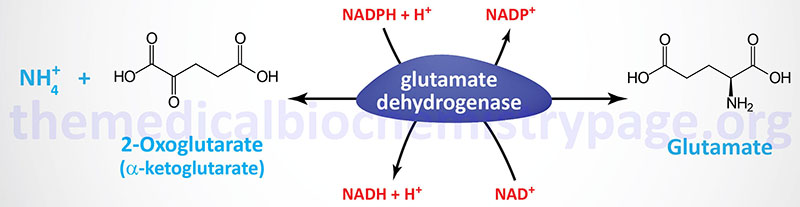
Humans express two distinct glutamate dehydrogenase genes identified as GLUD1 and GLUD2.
The GLUD1 gene encoded enzyme is the primary glutamate dehydrogenase (GDH1) activity in most tissues. This enzyme is localized to the mitochondrial matrix and functions as a homohexameric complex. The GLUD1 gene is located on chromosome 10q23.2 and is composed of 18 exons that generate seven alternatively spliced mRNAs that collectively encode three distinct protein isoforms.
The GLUD2 gene is thought to have arisen as a result of a retrotranspostitional event to the X chromosome. The GLUD2 gene is located at Xq24 and is an intronless gene encoding a precursor protein of 558 amino acids. The GLUD2 encoded protein (GDH2) also forms a homohexameric complex in the mitochondrial matrix. Expression of the GLUD2 gene is highest in neural tissues and the regulatory controls over this form of the enzyme are distinct from the GLUD1 encoded enzyme. The GDH2 complex is not inhibited by GTP which allows astrocyte GDH2 to continue to function under conditions of intense excitatory neurotransmission allowing these cells to handle the increased loads of the neurotransmitter glutamate.
Glutamate can also be derived from glutamine via the action of glutaminase. Indeed, the interconversion of glutamine and glutamate represents a critical pathway in overall nitrogen, acid-base, and energy homeostasis in the body as described in greater detail below. Glutaminase activity is present in many tissues such as the liver, kidney, small intestine, adipose tissue, and neurons. The primary subcellular site of glutaminase localization is the mitochondria.
There are two distinct glutaminase genes in humans identified as GLS (encoding the GLS1 enzyme) and GLS2 (encoding the GLS2 enzyme).
The GLS gene is located on chromosome 2q32.2 and is composed of 22 exons that undergo alternative splicing to yield two mRNAs generating two isoforms of the enzyme. These two GLS-derived isoforms are often referred to as glutaminase C (GAC) and kidney-type glutaminase (KGA) but are collectively the glutaminase 1 (GLS1) enzymes. The GLS encoded isoforms of glutaminase are primarily expressed in the kidneys. GLS encoded kidney-type glutaminase is a protein of 669 amino acids and the GLS encoded glutaminase C is a protein of 598 amino acids.
The GLS2 gene encoded glutaminase was originally thought to be liver specific but is in fact expressed in numerous tissues and is important in the glutamate-glutamine cycle in the brain. The GLS2 encoded glutaminase was originally characterized as dependent on inorganic phosphate (Pi) for activity and is, therefore, also referred to as phosphate-activated glutaminase, PAG.
The GLS2 gene is located on chromosome 12q13.3 and is composed of 19 exons that undergo alternative splicing to yield four mRNAs. The two major GLS2-derived mRNAs have been identified as LGA and GAB. The GAB mRNA contains all of the coding exons and the LGA mRNA lacks exon 1. The LGA mRNA results from the use of an alternative promoter present in intron 1 of the GLS2 gene.
As a result of the use of alternative promoters and through alternative splicing there are four major isoforms of glutaminase enzymes. These four isoforms exhibit different molecular, kinetic, protein interacting partners, and regulatory properties. As indicated above, one of the first characterized differences was the dependence on Pi for activity. Both the GLS gene encoded enzymes and the GLS2 encoded enzymes require phosphate for activity. With GLS gene derived enzymes the GAC isoform is the most responsive to phosphate. This difference in sensitivity is believed to be the result of the differences in the C-terminus of the KGA and GAC isoforms encoded by the GLS gene. The GLS encoded enzymes are inhibited by glutamate but the GLS2 encoded enzyme is not. The GLS2 encoded enzyme is activated by ammonia but the GLS encoded enzymes are not.
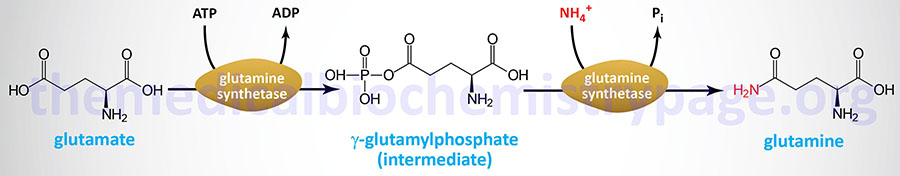
Glutamine Biosynthesis
Glutamine is synthesized from glutamate via the action of glutamine synthetase. The glutamine synthetase enzyme is encoded by the glutamate-ammonia ligase (GLUL) gene. The GLUL gene is located on chromosome 1q25.3 and is composed of 9 exons that generate three alternatively spliced mRNAs, each of which encode the same 373 amino acid protein.
Ultimately, however, glutamine is derived from 2-oxoglutarate (α-ketoglutarate) via the sequential actions of amino transferases, or glutamate dehydrogenase, that yield glutamate from 2-oxoglutarate and then via the glutamine synthetase reaction acting on glutamate.
The interrelationships between glutamate and glutamine, particularly the role of glutaminase in converting glutamine to glutamate, is collectively referred to as the process of glutaminolysis. Through the processes of glutaminolysis, glutamine and glutamate demonstrate their essential roles in numerous cellular processes that includes the TCA cycle and energy generation, nucleotide biosynthesis, glutathione biosynthesis, and epigenetic modifications of histone proteins.
The whole body ratio of glutamine to glutamate is also predictive of disease, particularly type 2 diabetes. The higher the glutamine to glutamate ratio, the more protected is an individual from a risk for type 2 diabetes. Adipose tissue plays a major role in the overall regulation of the circulating glutamine to glutamate ratio. This is accomplished by the release of glutamine and the uptake of glutamate. The significance of the role of adipose tissue in glutaminolysis, and its effects on metabolic dysfunction, has been demonstrated in studies of pharmacological inhibition of glutaminase in white adipose tissue. Under these conditions there is an increase in glucose tolerance and an increase in energy expenditure.
Glutamine Metabolism in Growth and Cancer
Glutamine represents the most abundant amino acid in the blood with levels that are in the range of 500μM. This level of glutamine accounts for over 20% of the total pool of free amino acids in the blood. Although briefly outlined here, the details of the significance of glutamine metabolism in cancer are covered in the Metabolic Alterations Associated with Cancer page.
Glutamine is also critical in the biosynthesis of nucleotides for RNA and DNA synthesis as is required in rapidly proliferating cells and cancer cells. Glutamine can serve as a precursor for aspartate through the concerted actions of glutaminase, yielding glutamate, followed by transfer of the nitrogen from glutamate to oxaloacetic acid (OAA) forming aspartate. The oxaloacetic acid can also be derived from glutamine, thus indicating this pivotal role of glutamine in overall nucleotide homeostasis. The aspartate that can be derived from glutamine is required as a source of carbon for both purine and pyrimidine nucleotide synthesis.
In cancer cells, the expression of enzymes involved in glutamine metabolism varies widely and is related to factors such as tissue of origin and the overall genotype of the specific cancer. As indicated in the previous section, humans express two genes encoding glutaminase enzymes, GLS encoding the GLS1 enzyme and GLS2 encoding the GLS2 enzyme. The GLS gene is more widely expressed and is the glutaminase encoding gene believed to be of more significance in cancer cell metabolism.
Glutamate dehydrogenase activity is also altered in cancer cells. Glutamate dehydrogenase is allosterically activated by leucine, the levels of which will be higher in cancer cells due to increased activity of various amino acid transporters. In the direction of glutamate deamination, glutamate dehydrogenase is activated by ADP. Therefore, low cellular energy states will lead to induction of 2-oxoglutarate production via the glutamate dehydrogenase reaction allowing for increased ATP synthesis from the TCA cycle-derived NADH and FADH2.
Glycine Biosynthesis
The main pathway to glycine is a one-step reversible reaction catalyzed by serine hydroxymethyltransferase (SHMT). This enzyme is a member of the family of one-carbon transferases and is also known as glycine hydroxymethyltransferase. This reaction involves the transfer of the hydroxymethyl group from serine to the cofactor tetrahydrofolate (THF), producing glycine and N5,N10-methylene-THF.
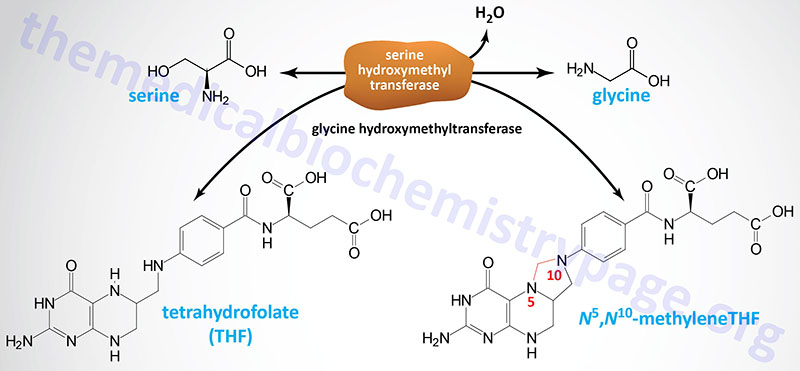
As pointed out in the previous section, there are mitochondrial and cytosolic versions of serine hydroxymethyltransferase. The cytosolic enzyme is encoded by the SHMT1 gene on chromosome 17 and the mitochondrial enzyme is encoded by the SHMT2 gene on chromosome 12. The major glycine biosynthetic enzyme is the cytosolic form of SHMT.
Role of Choline, Glyoxalate, and 4-Hydroxyproline in Glycine Synthesis
Glycine can be derived from sources other than serine but these alternative pathways do not represent significant sources of the amino acid.
Choline (a five-carbon quaternary ammonium cation) is a minor source for the synthesis of glycine. Choline is primarily acquired in the diet and is transported into cells via the FLVCR1 (feline leukemia virus subgroup C receptor 1) and FLVCR2 encoded transporters. The transporters are members of the SLC family of transporters and as such are also identified as SLC49A1 and SLC49A2, respectively. Two isoforms of FLVCR1 have been characterized, FLVCR1a and FLVCR1b. Once inside cells, free choline is phosphorylated by choline kinase-α (encoded by the CHKA gene) which activates it for phospholipid synthesis. As a result of phospholipid synthesis choline for glycine synthesis can also be derived from phosphatidylcholine.
Choline synthesis takes place in the liver and kidney and involves the conversion of phosphatidylethanolamine (PE) to phosphatidylcholine (PC) via the actions of the methyltransferase, phosphatidylethanolamine N-methyltransferase (PEMT). The choline can be released from PC through the actions of enzymes of the phospholipase D (PLD) family as well as in the conversion of PC to phosphatidylserine (PS) catalyzed by phosphatidylserine synthase 1 (encoded by the PTDSS1 gene).
The pathway of choline conversion to glycine takes place in the mitochondria following its transfer into the matrix of this organelle by the inner membrane-localized transporter encoded by the SLC25A48 gene. The pathway of choline conversion to glycine involves several distinct enzyme catalyzed reactions.
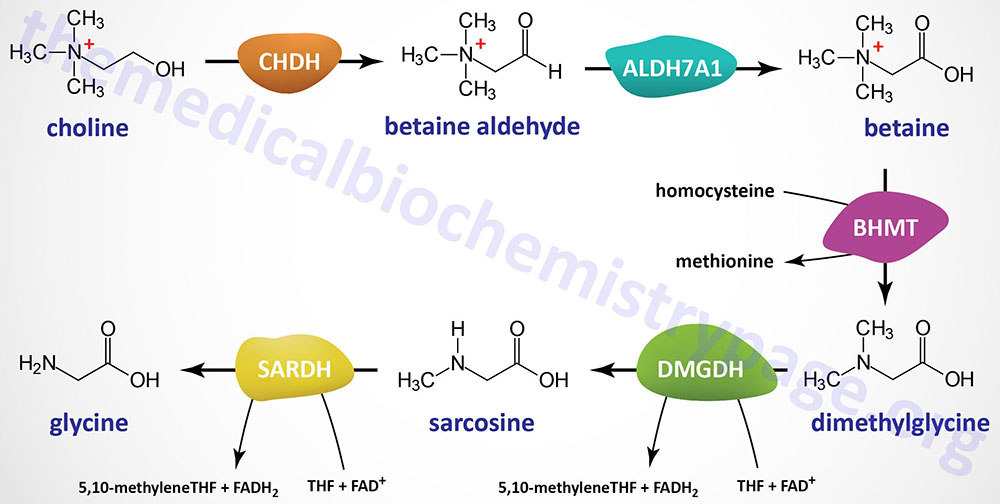
Choline dehydrogenase, encoded by the CHDH gene, converts choline to betaine (trimethylglycine) aldehyde. Conversion of choline to betaine aldehyde may also be catalyzed by butyrylcholinesterase (encoded by the BCHE gene) which is also referred to as choline oxidase. Betaine aldehyde is then oxidized to betaine by aldehyde dehydrogenase 7 family member A1 (encoded by the ALDH7A1 gene). The ALDH7A1 encoded enzyme has also been referred to as betaine aldehyde dehydrogenase. The ALDH7A1 encoded enzyme is also known as α-aminoadipic semialdehyde dehydrogenase (AASA dehydrogenase: AASADH) and is also known as antiquitin (ATQ1)
Betaine is then converted to dimethylglycine in a transmethylation reaction involving a methyl acceptor, most significantly homocysteine, and the enzyme betaine-homocysteine S-methyltransferase (BHMT) as depicted in The Methionine Cycle section. Dimethylglycine is oxidatively demethylated to sarcosine (N-methylglycine) by the FAD- and THF-dependent enzyme, dimethylglycine dehydrogenase (encoded by the DMGDH gene). Sarcosine is then converted to glycine in another FAD- and THF-dependent reaction catalyzed by sarcosine dehydrogenase (encoded by the SARDH gene).
Glyoxalate and 4-hydroxyproline (which is metabolized to glyoxalate and pyruvate) can also serve as precursors for glycine. Within the cytosol of hepatocytes, glyoxylate is reduced to glycolate via the action of glyoxylate and hydroxypyruvate reductase (encoded by the GRHPR gene). Glycolate then is transferred to the peroxisomes where it is converted back to glyoxylate and the resulting glyoxylate, along with alanine, serve as substrates, for the AGXT encoded enzyme (see Figure below) which releases glycine and pyruvate.
The conversion of 4-hydroxyproline to glyoxylate and ultimately to glycine or oxalate is detailed in the following Figure:
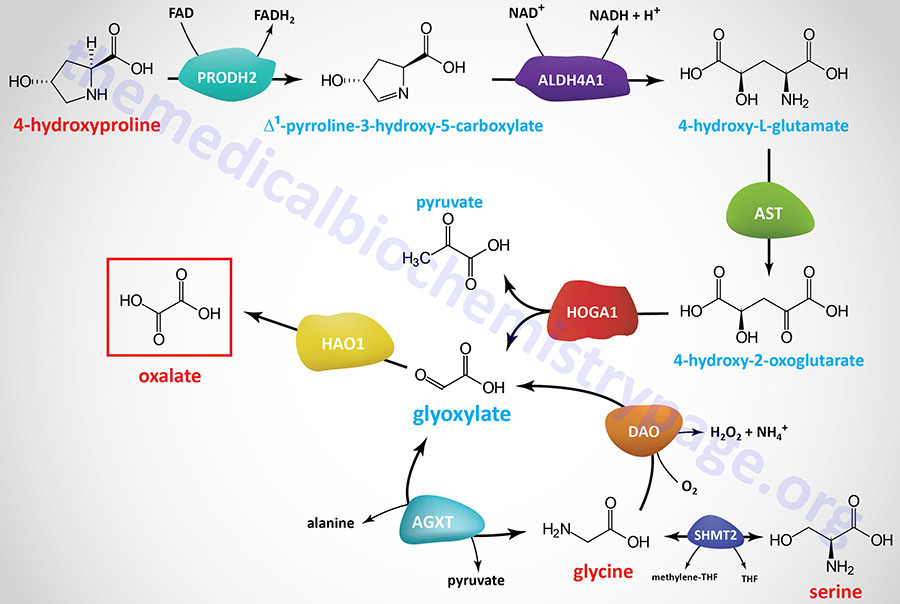
The final reaction in this five step metabolic pathway involves the amino transferase encoded by the AGXT gene, alanine–glyoxylate and serine–pyruvate aminotransferase.
Although there have been reports that demonstrate that glycine can be derived from threonine in mammals via a pathway involving the enzyme L-threonine dehydrogenase, this pathway is of no functional significance in the adult human. The human gene (TDH) encoding L-threonine dehydrogenase is an expressed pseudogene that does not encode a functional enzyme. Due to mutations in the TDH gene there are only truncated proteins that would be non-functional since they have lost part of the NAD+ binding motif and the C-terminal domain that would be responsible for binding L-threonine.
Glycine as a Neurotransmitter
Glycine is involved in many anabolic reactions other than protein synthesis including the synthesis of purine nucleotides, heme, glutathione, creatine and serine. In addition, glycine functions in the central nervous system as an inhibitory neurotransmitter where it participates in regulating signals that process motor and sensory information that permit movement, vision and audition. Glycine is co-released with GABA which is the primary inhibitory neurotransmitter.
Glycine action as a neurotransmitter is a function of the amino acid binding to a specific receptor, GlyR. GlyR is a member of the nicotinicoid receptor superfamily that includes the GABAA receptor (GABAAR), the excitatory nicotinic acetylcholine receptors (nAChR) and the serotonin type 3 receptor (5HT3). GlyR is a heteromeric (pentameric) complex consisting of a complex of either three or four α-subunits and one β-subunit. There are four different α-subunit genes (GLRA1, GLRA2, GLRA3, GLRA4) and a single β-subunit gene (GLRB) in the human genome. The GlyR is a ligand-gated ionotropic receptor that is a chloride channel. In addition to glycine, the GlyR can be activated by several other small amino acids such as alanine and taurine.
Glycine is also involved in the modulation of excitatory neurotransmission exerted via glutamate binding to N-methyl-D-aspartate (NMDA) type glutamate receptors.
Methionine, S-Adenosylmethionine, and Cysteine Biosynthesis
The essential amino acid methionine plays many crucial roles in the body, most of which are not related to its function in the synthesis of proteins. In the context of the role of methionine in protein synthesis it also serves as the precursor for the synthesis of the amino acid cysteine. The sulfur for cysteine synthesis comes from methionine through a series of interrelated reaction pathways. In addition to its role in the synthesis of cysteine, methionine is critical in the synthesis of the methyl donor, S-adenosylmethionine (SAM, SAMe, or AdoMet).
Synthesis of S-Adenosylmethionine
In the synthesis of S-adenosylmethionine (SAM), methionine and the adenosine from ATP are condensed via the action of methionine adenosyltransferase, MAT. In the production of SAM all phosphates of an ATP are lost: one as Pi and two as PPi, which itself is converted to two moles of Pi via the action of either of two enzymes (PPA1 or PPA2) of the pyrophosphatase (inorganic) family. In the MAT catalyzed reaction it is adenosine which is transferred to methionine and not AMP. MAT is also called S-adenosylmethionine synthetase.
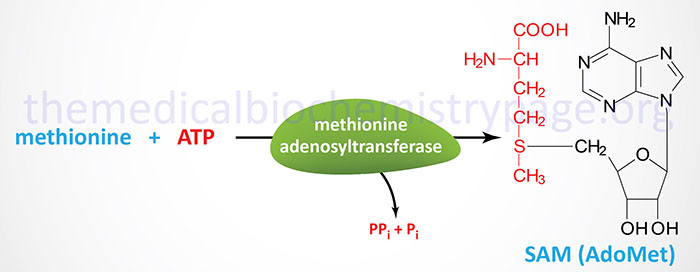
There are three genes encoding MAT enzymes in humans identified as MAT1A, MAT2A, and MAT2B. The MAT enzymes encoded by the MAT1A gene function as either homotetramers identified as MAT I (called the alpha form), or as homodimers identified as MAT III (called the beta form). The MAT I and MAT III isoforms are only expressed in the liver. The MAT enzyme encoded by the MAT2A gene is identified as MAT II (called the gamma form). The MAT II enzyme is expressed in several non-hepatic tissues. The MAT2B encoded protein is the non-catalytic regulatory subunit.
The MAT1A gene is located on chromosome 10q22.3 and is composed of 9 exons that encode a protein of 395 amino acids. Expression of the MAT1A gene is very nearly exclusive to the liver.
The MAT2A gene is located on chromosome 2p11.2 and is composed of 9 exons that encode a protein of 395 amino acids. Expression of the MAT2A gene is ubiquitous.
The MAT2B gene is located on chromosome 5q34 and is composed of 8 exons that generate two alternatively spliced mRNAs encoding the MAT 2 beta isoform 1 (334 amino acids) and the MAT 2 beta isoform 2 (323 amino acids) enzymes. Expression of the MAT2B gene is ubiquitous.
Mutations in the MAT1A gene are associated with the disorder referred to as methionine adenosyltransferase I/III deficiency. This disorder can be inherited as an autosomal recessive disease or as an autosomal dominant disease. In most individuals the disorder is associated with a benign phenotype, with pathology only seen in patients with plasma methionine levels exceeding 800μM. Symptoms in these patients result from demyelination in the CNS.
Metabolism of S-Adenosylmethionine
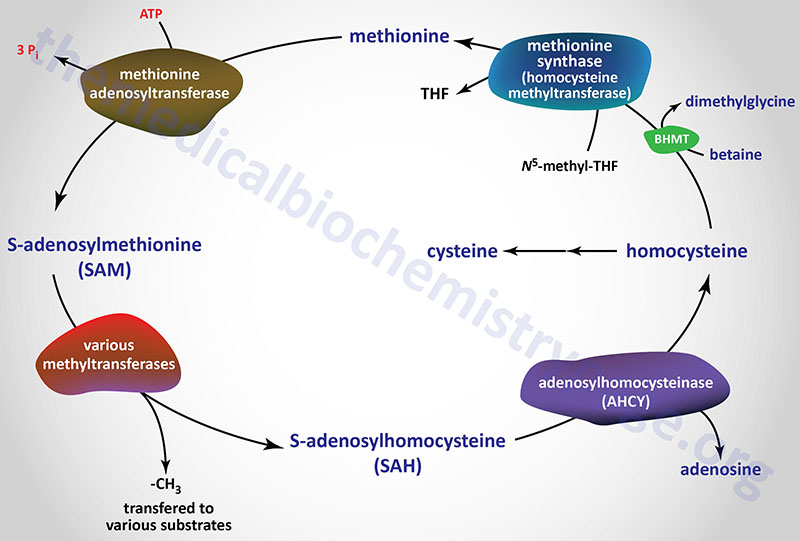
SAM serves as a precursor for numerous different methyl transfer reactions where one of the most physiologically relevant reactions is the conversion of norepinephrine to epinephrine (see Amino Acid Derivatives: Neurotransmitters, Nitric Oxide, and More page). The result of methyl transfer is the conversion of SAM to S-adenosylhomocysteine (SAH or AdoHcy). S-adenosylhomocysteine is then cleaved by adenosylhomocyteinase (also called S-adenosylhomocysteine hydrolase) to yield homocysteine and adenosine.
Adenosylhomocysteinase is encoded by the AHCY gene. The AHCY gene is located on chromosome 20q11.22 and is composed of 17 exons that generate six alternatively spliced mRNAs encoding three distinct proteins isoforms of 432 amino acids (isoform 1), 404 amino acids (isoform 2), and 434 amino acids (isoform 3). Mutations in the AHCY gene are one of the inherited causes of hypermethioninemia.
Transmethylation reactions employing SAM are extremely important, but in the context of cysteine biosynthesis, the role of S-adenosylmethionine in transmethylation is secondary to the production of homocysteine (essentially a by-product of transmethylase activity).
SAM can also be converted to SAH and sarcosine (N-methylglycine, also referred to as monomethylglycine) via the actions of the enzyme, glycine N-methyltransferase. Glycine N-methyltransferase is a cytosolic enzyme that functions as a homotetrameric complex. Glycine N-methyltransferase is encoded by the GNMT gene. The GNMT gene is located on chromosome 6p21.1 and is composed of 6 exons that generate two alternatively spliced mRNAs encoding proteins of 295 amino acids (isoform 1) and 276 amino acids (isoform 2). Highest levels of GNMT expression are seen in the liver, pancreas, and prostate.
Sarcosine has numerous function in humans including regulation of glycine transport, protein synthesis, and DNA methylation. However, despite the generation of sarcosine, the primary function of the GNMT encoded enzyme is the in the regulation of the levels of SAM.
Mutations in the GNMT gene are extremely rare but are one of the inherited causes of hypermethioninemia. In addition to hypermethioninemia, patients with GNMT deficiency experience hepatomegaly, elevated plasma AST and ALT as well as elevated levels of SAM with normal levels of SAH (AdoHcy) and homocysteine.
Methionine Synthesis from Homocysteine
The primary pathway for the conversion of homocysteine back to methionine is catalyzed by methionine synthase (also called homocysteine methyltransferase). This reaction was also discussed in the context of vitamin B12-requiring enzymes in the Vitamins: Water and Fat Soluble page. The methionine synthase reaction represents the clinically most significant of only two vitamin B12-requiring enzymes in human cells. Methionine synthase, in addition to B12, requires the folate-derived vitamin co-factor, N5-methyltetrahydrofolate (N5-methylTHF; or simply 5-methylTHF of just methylTHF).
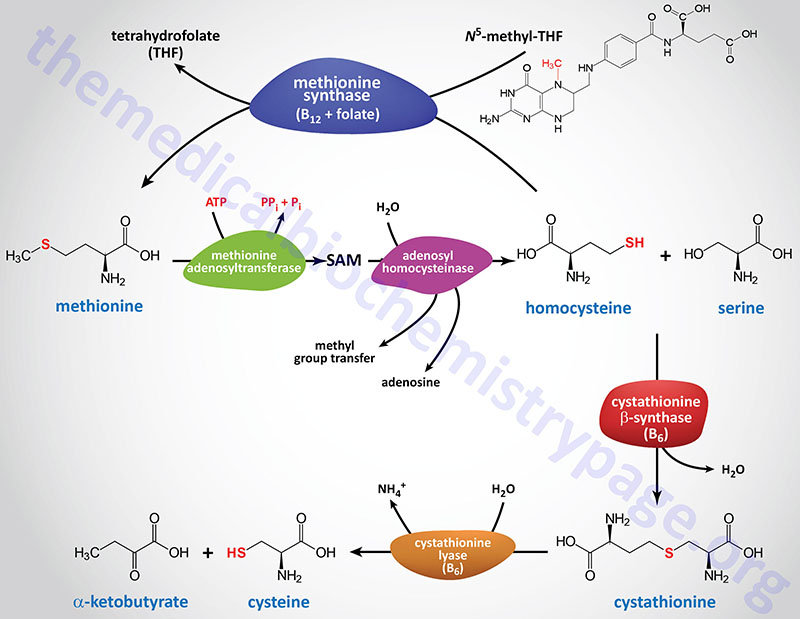
Methionine synthase is encoded by the MTR gene (5-methyltetrahydrofolate-homocysteine methyltransferase). The MTR gene is located on chromosome 1q43 and is composed of 33 exons that generate four alternatively spliced mRNAs, each of which encode a distinct protein isoform.
Mutations in the MTR gene are a rare cause of homocysteinemia/homocystinuria, commonly termed homocystinuria type 2.
During the conversion of homocysteine to methionine the cobalt within the methyl-cobalamin cofactor required of methionine synthase can become oxidized. The cobalt of cobalamin can exist in three oxidation states, +1 [cob(I)alamin], +2 [cob(II)alamin], and +3 [cob(III)alamin]. Within the context of vitamin B12 function the cobalamin is in the +1 or +2 state. The oxidation of cobalt within the context of the methionine synthase reaction converts cob(I)alamin to cob(II)alamin. The reduction of the cob(II)alamin state to the cob(I)alamin state is required to allow methionine synthase to continue to convert homocysteine to methionine.
The reduction of cobalt in cobalamin of methionine synthase is catalyzed by methionine synthase reductase. Methionine synthase reductase is encoded by the MTRR (5-methyltetrahydrofolate-homocysteine methyltransferase reductase) gene. The MTRR gene is located on chromosome 5p15.31 and is composed of 21 exons that generate five alternatively spliced mRNAs, each of which encode the same 698 amino acid protein.
Mutations in the MTRR gene are another rare cause of homocysteinemia/homocystinuria.
Role of Betaine in Methionine Synthesis
Homocysteine can be converted back to methionine via a folate-independent pathway involving the enzyme betaine-homocysteine S-methyltransferase, BHMT.
There are many compounds that are defined as being a betaine, but the most common, and important, is trimethylglycine which is commonly just called betaine. Any betaine is defined as neutral zwitterionic compound possessing a positively charged cationic functional group, such as a quaternary ammonium, that has no hydrogen atoms, along with a negatively charged anionic functional group, such as a carboxylate group. The negatively charged anionic group cannot reside adjacent to the cationic site.
Betaine, the trimethylglycine form, is an abundant amino acid that, although it is not found in proteins, plays a critical biochemical roles in humans. The majority of betaine is obtained from the diet and can also be synthesized from choline. The major source of choline for betaine synthesis comes from phosphatidylcholine.
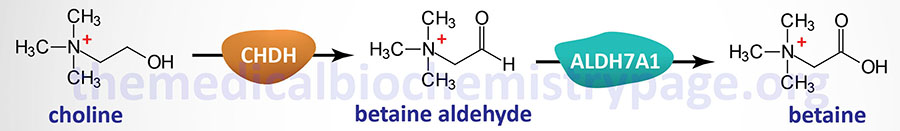
In order to synthesize betaine, choline is first transported into the mitochondria via the action of the transporter encoded by the SLC25A48 gene. In the mitochondria choline is oxidized to betaine aldehyde by choline dehydrogenase (encoded by the CHDH gene). Betaine aldehyde is then oxidized to betaine by aldehyde dehydrogenase 7 family member A1 (encoded by the ALDH7A1 gene) which is also known as betaine aldehyde dehydrogenase and as antiquitin (ATQ1). Following its synthesis, betaine is transported out of the mitochondria where is can function as a methyl donor in the synthesis of methionine from homocysteine.
The CHDH gene is located on chromosome 3p21.1 and is composed of exons 9 that encode a 594 amino acid protein. Befitting the role of betaine as an important osmolyte, the highest levels of expression of the CHDH gene are seen in the kidneys.
The ALDH7A1 gene is located on chromosome 5q23.2 and is composed of 18 exons that generate three alternatively spliced mRNAs. Two of these ALDH7A1 derived mRNAs each encode two proteins through the use of alternative in-frame translation initiation codons. One of the resultant proteins is localized to the mitochondria, the other to the cytosol. Highest levels of expression of the ALDH7A1 gene are seen in the liver and kidneys.
The ALDH7A1 encoded enzyme is also involved in the catabolism of lysine. It is the mitochondrial form of the enzyme that is involved in lysine catabolism.
The role of choline in betaine synthesis, and the subsequent principal role of betaine as a methyl donor primarily in the context of the methionine cycle in the liver, makes this function of choline one of its most important. Betaine is also important as an osmolyte in the kidney.
In the methionine cycle the methyl group of betaine is transferred to homocysteine via the action of betaine-homocysteine S-methyltransferase encoded by the BHMT gene. Betaine-homocysteine S-methyltransferase is a cytosolic enzyme whose expression is restricted to the liver and kidneys.
The BHMT gene is located on chromosome 5q14.1 and is composed of 8 exons that encode a 406 amino acid protein. Humans express a second betaine-homocysteine S-methyltransferase enzyme (BHMT2) that is encoded by the BHMT2 gene located in the same region of chromosome 5 (5q14.1) as the BHMT gene.
Given that methionine synthase is ubiquitously expressed, the role of BHMT in homocysteine re-methylation outside of hepatocytes and kidneys is limited.
Cysteine Biosynthesis
In cysteine synthesis (as outlined in the Figure above), homocysteine condenses with serine to produce cystathionine which is catalyzed by cystathionine β-synthase (cystathionine beta synthase: CBS). Cystathionine is subsequently cleaved by cystathionine γ-lyase (also called cystathionase) to produce cysteine and α-ketobutyrate (also referred to as 2-ketobutyrate or 2-oxobutyrate). The sum of the latter two reactions is known as transsulfuration.
Both of these enzymes require pyridoxal phosphate (PLP: derived from vitamin B6) as a cofactor, and both are under regulatory control. Cystathionine γ-lyase is also negatively regulated by cysteine-mediated allosteric control. In addition, cysteine inhibits the expression of the gene encoding cystathionine β-synthase.
Cysteine is used for protein synthesis and other body needs, while the α-ketobutyrate is first converted to propionyl-CoA and then via a 3-step process to the TCA cycle intermediate succinyl-CoA. While cysteine readily oxidizes to form the disulfide cystine, cells contain less cystine than cysteine in the cytosol because the ubiquitous reducing agent, glutathione (which contains cysteine), effectively reverses the formation of cystine by a non-enzymatic reduction reaction.
Cystathionine β-synthase is encoded by the CBS gene. The CBS gene is located on chromosome 21q22.3 and is composed of 23 exons which generate five alternatively spliced mRNAs that collectively encode two distinct proteins of 551 amino acids (isoform 1) and 446 amino acids (isoform 2).
Cystathionine γ-lyase (designated CSE) is encoded by the CTH gene. The CTH gene is located on chromosome 1p31.1 and is composed of 13 exons that undergo alternative splicing generating three mRNAs, each of which encode distinct protein isoforms. The longest protein (isoform 1) is 405 amino acids in length.
Genetic defects have been identified in both the CBS gene (see below) and the CTH gene. Missing or impaired cystathionine γ-lyase leads to excretion of cystathionine in the urine but does not have any other untoward effects. Rare cases are known in which cystathionine γ-lyase is defective and operates at a low level. This genetic disease leads to methioninuria with no other consequences.
Deficiency in B12, or folate, or defects in the MTR gene all will result in the development of megaloblastic anemias. The anemia develops due to the trapping of folate in the reduced form (N5-methyl-THF) which results in depletion of the active folate pool. The active folate pool is required for purine and thymine nucleotide biosynthesis needed for DNA synthesis. Bone marrow erythroblasts begin to enlarge in preparation for cell division but, due to loss of nucleotide synthesis, they remain as megaloblasts.
Other than incorporation into protein, the most important metabolic functions of cysteine are in the synthesis of glutathione (GSH), the synthesis of the bile salt modifying compound, taurine, and as a source of the sulfur for coenzyme-A synthesis.
The catabolism of cysteine, described in detail below, also significantly contributes to metabolic homeostasis, particularly mitochondrial homeostasis, via serving as the substrate for the synthesis of hydrogen sulfide, H2S.
Cystathionine β-Synthase and Cystathionine γ-lyase Activity and Longevity
In addition to their roles in the conversion of methionine to cysteine, cystathionine β-synthase (CBS) and cystathionine γ-lyase (CSE) catalyze the catabolism of cysteine in the context of the production of hydrogen sulfide (H2S). In these latter reactions CBS generates serine and H2S while CTH generates pyruvate, ammonium ion (NH4+), and H2S.
Hydrogen sulfide (H2S) functions as a gasotransmitter (gaseous molecule functioning as a messenger) that exerts numerous biological effects including as a vasodilator through induction of smooth muscle relaxation. Another important gasotransmitter is nitric oxide, NO.
H2S also functions in the process of persulfidation of proteins with more than 300 protein targets having been identified. Protein persulfidation is a post-translational modification of the thiol groups of cysteine residues forming persulfides (RSSH). The consequences of H2S-mediated protein persulfidation (sulfhydration) has been shown to contribute to increased production of NAD+ resulting in increased health span as evidenced by improvements in stress response and reductions in the levels of age-associated biomarkers.
The significance of cystathionine γ-lyase function, leading to health span improvement, was demonstrated by experiments showing that the enzyme was required for the persulfidation of the cytosolic form of glycerol-3-phosphate dehydrogenase (cGAPDH; encoded by the GPD1 gene). The GPD1 encoded enzyme is often designated cGPDH. The cystathionine γ-lyase-mediated persulfidation of cGAPDH directly contributes to the increased production of NAD+ via the reduction of glyceraldehyde-3-phosphate to glycerol-3-phosphate and the oxidation of NADH to NAD+.
The Methionine Cycle
Given that methionine can be recycled in the context of conversion to, and utilization of, S-adenosylmethionine (SAM), the enzymes and intermediates in the recycling of methionine constitute what is referred to as the methionine cycle. Although all of the enzymes and intermediates in this methionine cycle were discussed in the context of the methionine to cysteine conversion, the following Figure outlines the key features of the cycle.
Despite the role of methionine synthase in the synthesis of methionine from homocysteine, the major function of methionine synthase, under normal physiological conditions, is in the recycling of of tetrahydrofolate (THF) and not in the synthesis of methionine.
Role of the Methionine Cycle in Epigenetics
The role of methionine synthesis and regeneration, via the methionine cycle, in the methylation of DNA and histones, events that are central to epigenetic regulation of gene expression, is evident from the fact that this process is required for the synthesis of S-adenosylmethionine (SAM). As indicated in the Functions of S-Adenosylmethionine (SAM or AdoMet) section below, SAM is the required methyl donor for numerous methylation reactions, including those responsible for epigenetic regulation.
Cystine (Cysteine Disulfide) Role in Epigenetics and Oxidative Stress
Cystine is formed from the oxidation of the —SH (sulfhydryl) of the R-groups of two cysteine molecules which generates a disulfide bond. Because this is an oxidation process the formation of cystine occurs most readily in the endoplasmic reticulum (ER), the Golgi, and the extracellular environment. Conversely, within the cytosol the actions of, primarily, glutathione and thioredoxin reduce the disulfide bond generating two molecules of cysteine.
Cystine is an important molecule for proliferating cells, normal and tumorigenic, because these cells, particularly cancer cells, experience increased oxidative stress. One major antioxidant utilized by all cells is glutathione (GSH) which is a tripeptide containing cysteine. Proliferating cells thus require access to free cysteine to deal with oxidative stress. However, because the extracellular environment is highly conducive to oxidizing cysteine to cystine the ability to transport cysteine into cells is limiting. Recent work has shown that proliferating cells and cancer cells increase the expression of the cystine transporter encoded by the SLC7A11 gene. This transporter is commonly called the xCT transporter. The Xc– transporter, a heterodimeric transporter, is composed of the SCL7A11 encoded protein and the SLC3A2 encoded protein identified as 4F2hc (4F2 cell-surface antigen heavy chain).
The increased uptake of cystine by proliferating cells provides a mechanism to increase access to cysteine. The cysteine can be used to synthesize more glutathione (GSH) allowing cells to more efficiently deal with oxidative stress. However, since the reduction of cystine to two cysteines requires NADPH, in the context of GSH activity, the increased cystine uptake can lead to depletion of NADPH which can then lead to exacerbation of oxidative stress. One mechanism to deal with this conundrum is to increase glucose diversion into the pentose phosphate pathway since this is the major NADPH generating pathway. Indeed, in cancer cells, the activation of the PKM gene and the synthesis of the PKM2 isoform of pyruvate kinase promotes glucose diversion into pathways conducive for biomass production.
Increased cystine uptake, via the increased expression of the SCL7A11 transporter, has been shown to contribute to changes in the epigenome. Cystine forms a complex with cystathionine γ-lyase (most often designated CSE) and the a-subunit of the E1 portion of the PDH complex (PDHc) which is encoded by the PDHA1 gene. The PDHA1 encoded enzyme is known to function in the nucleus in addition to its role in the PDHc in the mitochondria. Once the complex of cystine, PDH α-subunit, and cystathionine γ-lyase are transported into the nucleus, cystathionine γ-lyase oxidatively catabolizes cystine into cysteine and pyruvate. The PDH α-subunit then oxidizes pyruvate generating nuclear acetyl-CoA. This acetyl-CoA can be utilized by histone acetyltransferases (HAT) to acetylate histones. The acetylation of histones is an epigenetic process that results in open chromatin and increased transcription.
Hypermethioninemia
There are several causes of hypermethioninemia that include genetic (familial), associated other disorders, and environmental. When hypermethioninemia results from inherited mutations in at least three genes associated with methionine metabolism it is referred to as primary hypermethioninemia. These three genes are MAT1A, AHCY, and GNMT. Inherited mutations in the gene (ADK) encoding adenylate kinase are also associated with hypermethioninemia.
The MAT1A gene encodes the MAT I (called the alpha form) isoform of methionine adenosyltransferase which is involved in the synthesis of S-adenosylmethionine (SAM or AdoMet). MAT1A deficiency can be inherited as an autosomal recessive or dominant disorder. Most cases of MAT1deficiency are associated with elevated serum methionine levels but without any significant intellectual or physical impairment.
The AHCY gene encodes adenosylhomocysteinase. Following the use of SAM in methyl transfer reactions, the resultant S-adenosylhomocysteine (SAH or AdoHcy) is hydrolyzed by adenosylhomocyteinase (also called S-adenosylhomocysteine hydrolase) to yield homocysteine and adenosine. ACHY deficiency is an autosomal recessive disorder associated with hypermethioninemia, failure to thrive, psychomotor impairment, hypotonia, facial dysmorphism, and cardiomyopathy.
The GNMT gene encodes glycine N-methyltransferase. Glycine N-methyltransferase catalyzes a reaction between glycine and SAM generating S-adenosylhomocysteine (SAH) and sarcosine. GNMT deficiency is inherited as an autosomal recessive disorder associated with hypermethioninemia, elevated serum SAM levels, and normal levels of sarcosine. The most common physical abnormality is mild hepatomegaly with chronically elevated levels of the transaminases, AST and ALT.
The ADK gene encodes adenylate kinase, also called adenosine kinase or myokinase. This enzyme catalyzes the transfer of the terminal (gamma) phosphate from ATP to AMP forming two moles of ADP and also transfers the gamma phosphate of ATP to adenosine generating ADP and AMP. At least nine different isoforms of ADK have been identified in humans resulting from multiple alternatively spliced mRNAs. Deficiencies in ADK are associated with characteristic biochemical features of hypermethioninemia, normal levels of homocysteine, and increased SAM and SAH. Infants with ADK deficiency exhibit global developmental delay, early-onset seizures, and mild dysmorphic features.
The primary environmental cause of hypermethioninemia is deficiency in vitamin B6. Deficiency in B6 results in impairment of the cystathionine β-synthase and cystathionine lyase catalyzed reactions, both of which require B6 (pyridoxal phosphate, PLP) as cofactor. The impairment in these two enzymes results in increased levels of homocysteine which in turn increases its conversion to methionine via the methionine synthase catalyzed reaction.
Homocysteinemia / Homocystinuria
Homocysteinemias (commonly referred to as homocystinurias) represent a family of inherited disorders resulting from defects in several of the genes involved in the conversion of methionine to cysteine. As the name implies, these disorders result in elevated levels of homocysteine and homocystine in the blood, where the elevated urine output of the metabolite is referred to as homocysteinurina or homocystinuria. Homocystine is a disulfide-bonded homodimer of two homocysteines. This is similar to the formation of cystine from two cysteines as diagrammed in the Cystinuria page.
In addition to homocysteinuria/homocystinuria, patients excrete elevated levels of methionine and metabolites of homocysteine. The most common causes of homocystinuria (classic homocystinuria) are mutations in the gene (CBS) encoding cystathionine β-synthase.
Homocystinuria is often associated with intellectual impairment, although the complete syndrome is multifaceted and many individuals with this disease are mentally normal, while others experience variable levels of developmental delay along with learning problems. Common symptoms of homocystinuria are dislocated optic lenses (ectopia lentis), osteoporosis, lengthening and thinning of the long bones, and an increased risk of abnormal blood clotting (thromboembolism). The nature of the lens dislocation (subluxation), evident in homocysteinemia patients, can serve as a differential diagnostic tool. Lens dislocation is also found in patients suffering from Marfan syndrome, which is a connective tissue disorder caused by mutations in the fibrillin gene (FBN1). In homocystinuria the lens subluxation occurs in a downward and inward direction, whereas in Marfan syndrome the lens subluxation occurs upward and outward. Some instances of genetic homocysteinemia respond favorably to pyridoxine therapy suggesting that in these cases the defect in CBS is a decreased affinity for the cofactor, pyridoxal phosphate.
Homocysteinemia can also result from vitamin deficiencies due to the role of the co-factor forms of B6, B12, and folate in the overall metabolism of methionine. Vitamin B6 (as pyridoxal phosphate) is required for the activity of CBS and cystathionine γ-lyase and the homocysteinemia that results with B6 deficiency is also associated with elevated methionine levels in the blood. Vitamin B12 and folate (as N5-methyl-THF) are required for the methionine synthase reaction so a deficiency of either vitamin can result in homocysteinemia presenting with reduced levels of plasma methionine. The enzyme methylmalonyl-CoA mutase also requires B12 and so a homocysteinuria resulting from a deficiency in this vitamin is also associated with methylmalonic acidemia. Indeed, the measurement of serum methionine and methylmalonic acid in cases of homocysteinemia (homocystinuria) allows for a differential diagnosis of the nutritional (non-genetic) cause.
Another related disorder, referred to as hyperhomocysteinemia, is most often associated with manifestation of symptoms much later in life. The characteristic pathology of hyperhomocysteinemia is coronary artery disease (CAD) and an increased risk for deep vein thromboses, DVT. In addition, there is an increased risk for mild cognitive impairment and dementia. Evidence suggests a link between hyperhomocysteinemia and Alzheimer disease. Similar to the dietary causes of homocystinuria, hyperhomocysteinemia can result from deficiencies in vitamin B6, vitamin B12, and folate.
An inherited form of hyperhomocysteinemia results from mutations in the methylene tetrahydrofolate reductase (MTHFR) gene. The most common mutation in this gene that results in hyperhomocysteinemia is a C to T change at nucleotide position 677 (C677T). Mutations in the methionine synthase (MTR) gene are also associated with inherited hyperhomocysteinemia. In the case of the MTR gene the most common mutation is a change of an A for a G at nucleotide position 2756 (A2756G).
Elevated levels of homocysteine in the blood have been shown to correlate with cardiovascular dysfunction. The role of homocysteine in cardiovascular disease is related to its ability to induce a state of inflammation. Homocysteine serves as a negatively charged surface that attracts the contact phase of the intrinsic pathway of blood coagulation. Activation of the intrinsic coagulation cascade leads to inappropriate thrombolytic events as well as resulting in increases in inflammatory cytokine release from leukocytes that are activated as a result of the pro-coagulant state. Therefore, it is important to ensure that proper function of the methionine synthase reaction is maintained. Although it would be assumed that increased intake of vitamin B12 should lead to increased conversion of homocysteine to methionine and thus, reduced levels of circulating homocysteine, controlled studies have shown that this does not occur.
Functions of S-Adenosylmethionine (SAM or AdoMet)
S-adenosylmethionine (abbreviated SAM, SAMe, or AdoMet), synthesized as described above, is utilized in a wide array of methyl transfer reactions involving nucleic acids, lipids, proteins, neurotransmitters, and small molecules. During the course of the transfer of the methyl group from SAM to an appropriate acceptor, S-adenosylhomocysteine (abbreviated SAH or AdoHcy) is generated. The resultant SAH is catabolized to adenosine and homocysteine through the action of adenosylhomocysteinase (also called S-adenosylhomocysteine hydrolase) which is encoded by the AHCY gene.
The role of SAM in nucleotide and protein methylation contributes to several epigenetic processes and points to the role of nutritional components, in this case methionine, in the control of gene expression. The DNA methyltransferases encoded by the DNMT1 and DNMT3 genes utilize SAM in the methylation of cytidine residues found in CpG dinucleotides in DNA. The RNA methyltransferase encoded by the RNMT gene utilizes SAM as a substrate for the N7-methylation of the guanine residue present in the mRNA 5′-cap structure. The RNA methyltransferases encoded by the METTL3 and METTL14 genes incorporate methyl groups into adenine and cytidine residues in mRNA molecules utilizing SAM as the methyl donor. These latter mRNA methylation events represent important posttranscriptional mechanisms for the regulation of gene expression.
Numerous SAM-dependent methyltransferases are involved in the methylation of histone proteins which represents another mode of epigenetic regulation of gene expression. These methyltransferases all utilize SAM as the methyl donor and incorporate the methyl group onto lysine residues, arginine residues, and histidine residues in proteins. Several histone lysine and histone arginine N-methyltransferases have been identified including all of the HMT (histone lysine methyltransferase) gene family enzymes and the PRMT (protein arginine methyltransferase) gene family enzymes.
All lysine methyltransferase enzymes belong to the large family of enzymes identified as the methyltransferase family. Humans express six large families of methyltransferases identified as the homocysteine methyltransferase family, the lysine methyltransferase family, the radical S-adenosylmethionine domain containing family, the seven-beta-strand methyltransferase motif containing family, the SET domain containing family, and the SPOUT methyltransferase domain containing family.
Many of the proteins that are targets for enzymes of the PRMT family are involved in the processes of signal transduction or regulation of transcription. In addition to members of the histone protein family and transcription factor family, numerous other proteins are subject to either lysine or arginine methylation.
Additional enzymes that utilize SAM as a methyl donor are involved in the modification of proteins that serve functions in diverse processes such as protein damage repair, protein stability, and protein function. The enzyme identified as protein-L-isoaspartate (D-aspartate) O-methyltransferase (encoded by the PCMT1 gene) is required for the repair of deamidated aspartate and asparagine residues in proteins and it utilizes SAM in these reactions.
The function of the enzyme, isoprenylcysteine carboxyl methyltransferase (encoded by the ICMT gene) is to methylate the cysteine residues in the C-terminus of proteins following their prenylation. Humans express three genes that catalyze the SAM-dependent cysteine methylation reaction on prenylated proteins with the ICMT gene being the most abundantly expressed.
The SAM-dependent protein methyltransferase encoded by the LCMT1 gene (leucine carboxyl methyltransferase 1) catalyzes the methylation of a C-terminal leucine residue in the Ser/Thr phosphatase identified as PP2A, a modification required for its proper function.
The synthesis of the diphthamide residue found on His715 in human translation elongation factor eEF2 requires a methylation step involving SAM as the methyl donor.
Numerous reaction pathways involved in the synthesis of small molecules, as well as the synthesis and catabolism of neurotransmitters involve enzymes that are SAM-dependent methyltransferases. These methyltransferases are classified as either N-methyltransferases or O-methyltransferases dependent on whether the acceptor of the methyl group is nitrogen or an oxygen, respectively.
The synthesis of epinephrine from norepinephrine requires the SAM-dependent enzyme phenylethanolamine N-methyltransferase encoded by the PNMT gene.
The conversion of serotonin to melatonin requires the enzyme acetylserotonin O-methyltransferase encoded by the ASMT gene.
Creatine synthesis also requires SAM-dependent methylation in a reaction catalyzed by guanidinoacetate N-methyltransferase (encoded by the GAMT gene).
The catabolism of the catecholamines, epinephrine, norepinephrine, and dopamine, involves the SAM-dependent enzyme catechol O-methyltransferase (encoded by the COMT gene).
The catabolism of histamine occurs either through oxidation or methylation. The methylation pathway involves the SAM-dependent enzyme histamine N-methyltransferase which is encoded by the HNMT gene.
Lipid metabolism is another important process that involves SAM-dependent methylation. The conversion of phosphatidylethanolamine (PE) to phosphatidylcholine (PC) requires the enzyme phosphatidylethanolamine N-methyltransferase (encoded by the PEMT gene) which carries out three successive SAM-dependent methylation reactions. This reaction is a critically important reaction of membrane lipid homeostasis. Lipid synthesis and remodeling is important in all cell membranes but is particularly critical in the homeostasis of the myelin sheath protecting neurons in the nervous system.
Under fasting conditions numerous adaptive metabolic responses are triggered particularly in the liver, adipose tissue, and the brain. Within the liver, the levels of SAM play a crucial role in the adaptive responses to fasting. Hepatic SAM acts to fine-tune the catabolic responses to fasting by increasing the activity the PEMT encoded enzyme. Increased PEMT activity results in an increase in the PC to PE ratio which in turn leads to enhanced mitochondrial fatty acid β-oxidation, ATP production, and reduces the level of endoplasmic reticulum (ER) stress.
Reduced capacity to carry out the methionine synthase reaction, due to nutritional or disease mediated deficiency of vitamin B12, results in reduced SAM production. In turn, reduced levels of SAM in the brain are a contributor to the neural degeneration (i.e. depression and peripheral neuropathy) seen in chronic B12 deficiency.
The metabolism and detoxification of numerous xenobiotic compounds is also known to require SAM-dependent methyltransferase family enzymes. The anti-cancer drugs of the thiopurine class, such as 6-mercaptopurine, are metabolized by the enzyme thiopurine S-methyltransferase which is encoded by the TPMT gene.
Ornithine and Proline Biosynthesis
Glutamate is the precursor for the endogenous synthesis of both proline and ornithine with Δ1-pyrroline-5-carboxylate serving as a branch point intermediate leading to one or the other of these two amino acids. While ornithine is not one of the 20 amino acids used in protein synthesis, it plays a significant role as the acceptor of carbamoyl phosphate in the urea cycle. Indeed, the urea cycle represents another pathway that can replenish ornithine utilizing arginine. Ornithine serves an additional important role as the precursor for the synthesis of the polyamines.
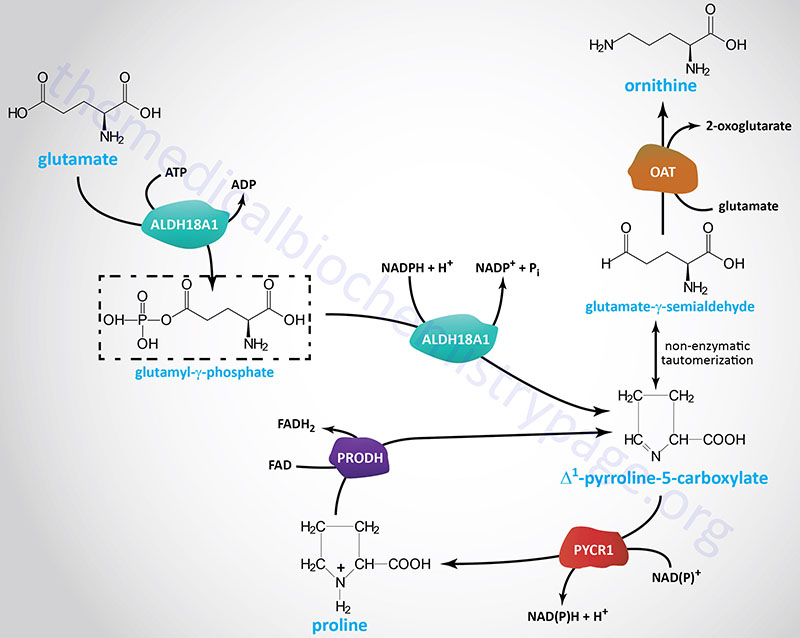
The production of ornithine from glutamate is important when dietary arginine, the other principal source of ornithine, is limited. The synthesis of ornithine from glutamate occurs only in the intestines. The conversion of glutamate to Δ1-pyrroline-5-carboxylate is catalyzed by a bi-functional enzyme that is a member of the aldehyde dehydrogenase family. This enzyme, aldehyde dehydrogenase 18 family, member A1 (encoded by the ALDH18A1 gene), possess both γ-glutamyl kinase activity and an NADPH-dependent γ-glutamylphosphate reductase activity. The ALDH18A1 encoded enzyme is also known as delta-1-pyrroline-5-carboxylate synthase (P5CS).
The Δ1-pyrroline-5-carboxylate intermediate in this pathway can exist as the open-chain tautomeric compound, glutamate γ-semialdehyde, which can be transaminated to ornithine via the action of ornithine aminotransferase. Ornithine aminotransferase utilizes glutamate as the amino donor and releases 2-oxoglutarate (α-ketoglutarate).
The ALDH18A1 gene is located on chromosome 10q24.1 and is composed of 17 exons that generate ten alternatively spliced mRNAs that collectively encode six distinct protein isoforms. Mutations in the ALDH18A1 gene result is a disorder characterized by hyperammonemia, hypoornithinemia, hypocitrullinemia, hypoargininemia and hypoprolinemia. This disorder is also associated with CNS degeneration, connective tissue disruption, and cataract formation.
Ornithine aminotransferase is encoded by the OAT gene. The OAT gene is located on chromosome 10q26.13 and is composed of 13 exons that generate ten alternatively spliced mRNAs that collectively encode four distinct protein isoforms.
The fate of Δ1-pyrroline-5-carboxylate (and its open-chain tautomer, glutamate γ-semialdehyde) depends on prevailing cellular conditions. Ornithine production occurs from the semialdehyde compound via a simple glutamate-dependent transamination, producing ornithine. When arginine concentrations become elevated, the ornithine contributed from the urea cycle plus that from Δ1-pyrroline-5-carboxylate inhibit the aminotransferase reaction, with accumulation of Δ1-pyrroline-5-carboxylate as a result.
The accumulating Δ1-pyrroline-5-carboxylate can then be reduced to proline by the NADP(H)-dependent enzyme pyrroline-5-carboxylate reductase (PYCR or P5CR). Humans express two versions of PYCR encoded by the PYCR1 and PYCR2 genes. The PYCR enzymes are localized to the mitochondria. The NADP(H) required for mitochondrial PYCR1 and PYCR2 activity is derived via the NADK2-mediated phosphorylation of mitochondrial NAD+. The evidence of the necessity for NADP(H) in proline biosynthesis can be demonstrated by the fact that loss of NADK2 activity impairs proline synthesis.
Pyrroline-5-carboxylate reductase 1 is encoded by the PYCR1 gene. The PYCR1 gene is located on chromosome 17q25.3 and is composed of 9 exons that generate six alternatively spliced mRNAs that collectively encode five different isoforms of the enzyme.
Pyrroline-5-carboxylate reductase 2 is encoded by the PYCR2 gene. The PYCR2 gene is located on chromosome 1q42.12 and is composed of 7 exons that generate two alternatively spliced mRNAs that encode proteins of 320 amino acids (isoform 1) and 246 amino acids (isoform 2).
Serine Biosynthesis
The main pathway to de novo biosynthesis of serine starts with the glycolytic intermediate 3-phosphoglycerate. An NADH-linked dehydrogenase (phosphoglycerate dehydrogenase; also commonly called 3-phosphoglycerate dehydrogenase) converts 3-phosphoglycerate into a keto acid, 3-phosphohydroxypyruvate, suitable for subsequent transamination. The aminotransferase, phosphoserine aminotransferase 1, utilizing glutamate as a donor, produces 3-phosphoserine, which is converted to serine by phosphoserine phosphatase.
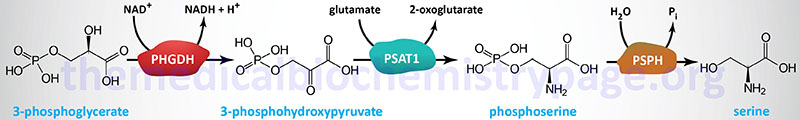
Phosphoglycerate dehydrogenase is encoded by the PHGDH gene which is located on chromosome 1p12 and is composed of 18 exons that generate a protein of 533 amino acids. Phosphoglycerate dehydrogenase is found in a complex with aldolase A and PFK-1 of the glycolytic pathway. The activity of this complex is regulated by the state of ubiquitylation. In the deubiquitylated state the complex enhances glucose metabolism and serine synthesis from glucose, a process that has been shown to be enhanced in cancer cells. The primary deubiquitylating (DUB) enzyme is encoded by the JOSD2 (josephin domain containing 2) gene.
Mutations in the PHGDH gene are associated with a rare autosomal recessive disorder characterized by presentation in infancy with microcephaly, intractable seizures, and little to no psychomotor development. Cranial MRI analysis shows hypomyelination and absence of white matter.
Phosphoserine aminotransferase 1 is encode by the PSAT1 gene located on chromosome 9q21.2 and is composed of 9 exons that generate two alternatively spliced mRNAs encoding isoform 1 (370 amino acids; also called PSAT1 beta: PSAT1β) and isoform 2 (324 amino acids; also called PSAT1 alpha: PSAT1α).
The phosphoserine phosphatase gene (symbol: PSPH) is located on chromosome 7p11.2 and is composed of 13 exons that encode a 225 amino acid protein. The location of the PSPH gene on chromosome 7p11.2 is in a region of chromosome 7 that is deleted in Williams-Beuren syndrome, WBS (also called Williams syndrome, WS). WBS is associated with multiple organ system involvement typically showing supravalvular aortic stenosis (SVAS), intellectual impairment, and distinctive facial features.
Serine Synthesis from Glycine
As indicated in the Glycine Synthesis section, serine can be derived from glycine (and vice versa) by a single step reaction that involves serine hydroxymethyltransferase (SHMT; also called glycine hydroxymethyltransferase) and tetrahydrofolate (THF). Indeed, the interconversion of serine and glycine via the involvement of THF represents the major pathway for the generation of N5,N10-methylene-THF which of a member of the active pool of folate derivatives. N5,N10-methylene-THF is required for purine nucleotide and thymine nucleotide biosynthesis. Humans express two serine hydroxymethyltransferase genes, one is a cytosolic enzyme while the other is located in the mitochondria.
The cytosolic enzyme is derived from the SHMT1 gene located on chromosome 17p11.2 which is composed of 14 exons that generate three alternatively spliced mRNAs, each of which encode a distinct protein isoform.
The location of the SMHT1 gene (17p11.2) resides within the 3.7 Mb region of chromosome 17 that is deleted in Smith-Magenis syndrome, SMS, a disorder associated with behavioral problems, psychomotor impairment, growth delay, speech delay, brachycephaly, midface hypoplasia, and a hoarse voice.
The mitochondrial enzyme is derived from the SHMT2 gene located on chromosome 12q13.3 which is composed of 14 exons that generate five alternatively spliced mRNAs that collectively encode three distinct isoforms of the enzyme.
One of the major functions of the SHMT2 encoded enzyme is in mitochondrial thymidylate synthesis pathway via its role in glycine and tetrahydrofolate metabolism. Mitochondrial thymidylate synthesis is required to prevent uracil accumulation in mitochondrial DNA (mtDNA). Another important function of SHMT2 is in the generation of NADH via metabolism of serine.
Because serine can be generated from glycine it is correct to state that the same alternative precursors for glycine synthesis, choline, 4-hydroxpyruvate, and glyoxylate, are precursors for the synthesis of serine. However, these alternative pathways for glycine synthesis represent minor pathways. The main pathway for glycine synthesis involves serine which is primarily derived from glucose as well as the nitrogen from glutamate.
Serine and Glycine in Oxalate Metabolism
Generally, dicarboxylic acids generated via the ω-oxidation pathway, are degraded in the mitochondria as well as the peroxisomes by their respective fatty acid β-oxidation pathways. Of clinical significance is the short-chain dicarboxylic acid, oxalic acid (oxalate). Oxalate is a dicarboxylic acid composed of two carbon atoms and exits in solution as a dianion of the formula C2O42– (–OOC—COO–).
Oxalate is present in many foods such as spinach, chard, nuts, black pepper, poppy seeds, and also occurs as a result of the metabolism of ascorbic acid, and the amino acids glycine, serine, and 4-hydroxyproline as well as metabolism of ethanolamine. The majority (40%) of daily endogenous oxalate production comes from the metabolism of dehydroascorbate. Only around 0.1% of endogenous oxalate arises from glycine catabolism, whereas approximately 15% of the daily endogenous production of glyoxylate derives from 4-hydroxyproline. The origin of 4-hydroxyproline is either the diet or the degradation of collagen proteins.
Glyoxylate, the endogenous precursor to oxalate, is a byproduct of the metabolism of the amino acids 4-hydroxyproline, serine, and glycine. Conversion of 4-hydroxyproline to oxalate begins with its oxidation to Δ1-pyrroline-3-hydroxy-5-carboxylate by proline dehydrogenase 2 (encoded by the PRODH2 gene). Δ1-pyrroline-3-hydroxy-5-carboxylate is converted to 4-hydroxy-L-glutamate via the action of aldehyde dehydrogenase 4 family member A1 (encoded by the ALDH4A1 gene). The ALDH4A1 encoded enzyme has also been referred to as Δ1-pyrroline-3-hydroxy-5-carboxylate dehydrogenase (1P5CDH). 4-Hydroxy-L-glutamate is a substrate for deamination by aspartate transaminase (AST) which uses 2-oxoglutarate (α-ketoglutarate) as the amino acceptor, generating 4-hydroxy-2-oxoglutarate and glutamate. 4-Hydroxy-2-oxoglutarate is hydrolyzed into pyruvate and glyoxylate by 4-hydroxy-2-oxoglutarate aldolase 1 (encoded by the HOGA1 gene).
Hepatic glycine and pyruvate can be converted to glyoxylate and alanine via the action of the vitamin B6-dependent enzyme, alanine–glyoxylate and serine–pyruvate aminotransferase (encoded by the AGXT gene). The AGXT gene is expressed exclusively in hepatocytes and is localized to the peroxisomes. The AGXT enzyme can also convert glyoxylate and alanine to glycine and pyruvate.
The majority of serine catabolism involves its conversion to glycine, followed by glycine decarboxylase-mediated metabolism as detailed in the Amino Acid Catabolism page. Alternatively, serine can be converted to 3-hydroxypyruvate via transamination of pyruvate catalyzed by alanine–glyoxylate and serine–pyruvate aminotransferase 2 (encoded by the AGXT2 gene). The 3-hydroxypyruvate can then be converted to glycoaldehyde via the action of glyoxylate and hydroxypyruvate reductase (encoded by the GRHPR gene). The glycoaldehyde is ultimately metabolized to glyoxylate.
Glyoxylate can be metabolized to oxalate by hydroxyacid oxidase 1 (encoded by the HAO1 gene; also called glycolate oxidase) as well as by lactate dehydrogenase. Excess glyoxylate will enhance the metabolism of glyoxylate to oxalate. The HAO1 encoded enzyme also converts glycolate to glyoxylate.
Oxalate is excreted in the urine and approximately 50% – 80% is derived from the diet. If levels of oxalate in the glomerular filtrate increase (hyperoxaluria) it can chelate Ca2+ ions forming the insoluble compound, calcium oxalate. Indeed, calcium oxalate crystals are the most common type of compound contributing to renal calculi (kidney stones; nephrolithiasis).
Hyperoxaluria
The hyperoxalurias are divided into two categories: the primary hyperoxalurias (PH) and the secondary hyperoxalurias. Primary hyperoxalurias are rare autosomal recessive disorders of glyoxylate metabolism that are associated with high oxalate production, primarily within the liver. There are three distinct forms of PH identified as PH1, PH2, and PH3 (or PH I, PH II, and PH III). The primary hyperoxalurias are associated with overproduction and excessive urinary excretion of oxalate. The excess urinary oxalate leads to recurrent urolithiasis and nephrocalcinosis. Glomerular filtration rates decline in PH due to progressive renal involvement leading to oxalate accumulation and systemic oxalosis that manifests with metabolic bone disease, anemia, and skin ulcerations.
Primary hyperoxaluria type 1 is the most common inherited form of hyperoxaluria, representing 70%-80% of all cases. PH 1 results from mutations in the peroxisomal specific enzyme alanine–glyoxylate and serine–pyruvate aminotransferase encoded by the AGXT gene. The AGXT gene is located on chromosome 2q37.3 and is composed of 11 exons that encode a 392 amino acid protein.
Primary hyperoxaluria type 2 accounts for around 10% of all cases. PH 2 results from mutations in glyoxylate and hydroxypyruvate reductase encoded by the GRHPR gene. The GRHPR gene is located on chromosome 9p13.2 and is composed of 12 exons that encode a 328 amino acid protein. Expression of the GRHPR gene is seen in most tissues with the highest levels in the liver and then the kidney.
Primary hyperoxaluria type 3 results from mutations in 4-hydroxy-2-oxoglutarate aldolase 1 encoded by the HOGA1 gene. The HOGA1 gene is located on chromosome 10q24.2 and is composed of 7 exons that generate two alternatively spliced mRNAs that encode proteins of 327 (isoform 1) and 164 (isoform 2) amino acids. The HOGA1 gene is predominantly expressed in the kidney and the enzyme is localized to the mitochondria.
Tyrosine Biosynthesis
Tyrosine is produced in cells by hydroxylating the essential amino acid phenylalanine. This reaction is catalyzed by phenylalanine hydroxylase (PAH). This relationship is much like that between cysteine and methionine. Half of the daily requirement for phenylalanine is for the production of tyrosine; if the diet is rich in tyrosine itself, the requirements for phenylalanine can be reduced by about 50%.
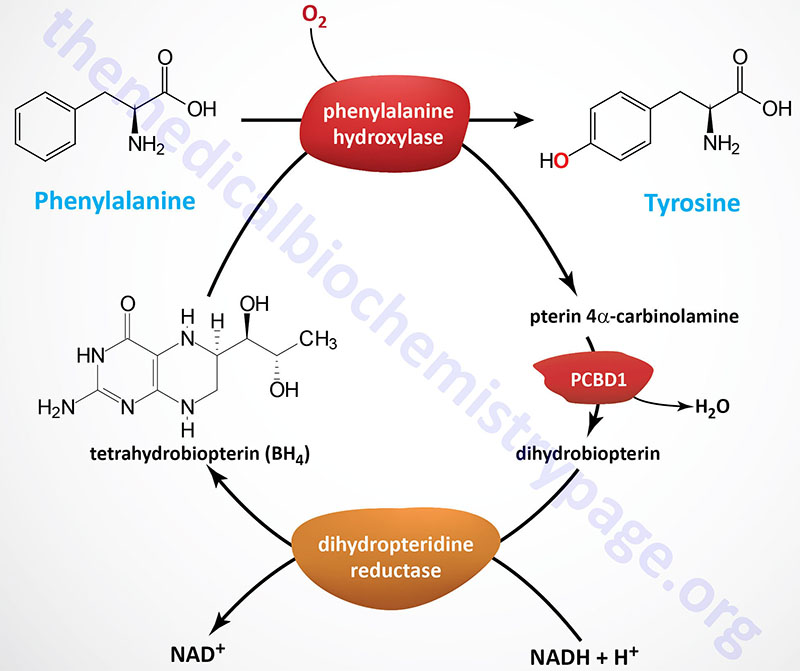
Phenylalanine hydroxylase (PAH: more specifically phenylalanine 4-hydroxylase) is a mixed-function monooxygenase that is one of three enzymes belonging to the biopterin-dependent aromatic amino acid hydroxylase (AAAH) family. Phenylalanine hydroxylase is encoded by the PAH gene. The PAH gene is located on chromosome 12q23.2 and is composed of 15 exons that generate two alternatively spliced mRNAs, both of which encode the same 452 amino acid protein. The required biopterin is in the form of tetrahydrobiopterin (often designated BH4 or H4B).
Phenylalanine hydroxylase functions primarily as a homotetrameric enzyme but exists in cells in equilibrium with homodimeric forms as well. The PAH monomeric protein is composed of three functional domains. The N-terminal domain (amino acids 1-117) comprises the regulatory domain containing the Phe-binding subdomain. The catalytic domain comprises amino acids 118-427. The C-terminal domain (amino acids 428-452) is responsible for the oligomerization of the monomers.
The other two enzymes in the AAAH family are tyrosine hydroxylase and tryptophan hydroxylase. An additional clinically significant group of enzymes that requires tetrahydrobiopterin as cofactor are the nitric oxide synthase (NOS) family of enzymes.
Phenylalanine hydroxylase transfers one atom from molecular oxygen (O2) into the hydroxyl of tyrosine and the other hydroxylates tetrahydrobiopterin forming an intermediate identified as pterin 4α-carbinolamine (also called 4α-hydroxypterin or 4α-hydroxydihydrobiopterin).
Tetrahydrobiopterin can be regenerated in a salvage pathway that involves the enzymes pterin 4α-carbinolamine dehydratase 1 and dihydropteridine reductase.
The pterin 4α-carbinolamine dehydratase 1 enzyme is encoded by the PCBD1 gene. The PCBD1 gene is located on chromosome 10q22.1 and is composed of 6 exons that generate three alternatively spliced mRNA, each of which encode a distinct protein isoform. The PCBD1 gene does not contain the typical TATA-box and CAAT-box basal promoter elements found in the majority of mRNA encoding genes.
The product of the PCBD1 encoded enzyme, acting on pterin 4α-carbinolamine, is the quinoid molecule identified as dihydrobiopterin. In addition to the catalytic function of PCBD1, the protein serves as a transcriptional co-activator for two transcription factors of the hepatocyte nuclear factor-1 family, HNF-1α (encoded by the HNF1A gene) and HNF-1β (encoded by the HNF1B gene).
Dihydrobiopterin is then converted to tetrahydrobiopterin by the NADH-dependent enzyme commonly referred to as dihydropteridine reductase. Human dihydropteridine reductase is encoded by the QDPR (quinoid dihydropteridine reductase) gene. The QDPR gene is located on chromosome 4p15.32 and is composed of 7 exons that generate two alternatively spliced mRNAs encoding proteins of 244 amino acids (isoform 1) and 213 amino acids (isoform 2).
Missing or deficient phenylalanine hydroxylase results in hyperphenylalaninemia. Hyperphenylalaninemia is defined as a plasma phenylalanine concentration greater than 2 mg/dL (120 μM). The most widely recognized hyperphenylalaninemia (and most severe) is the genetic disease known as phenylketonuria (PKU). Patients suffering from PKU have plasma phenylalanine levels >1000 μM, whereas the non-PKU hyperphenylalaninemias exhibit levels of plasma phenylalanine <1000 μM.
Untreated PKU leads to severe intellectual impairment, however, the precise mechanism by which this enzyme deficiency leads to the severe neural degeneration is not fully understood. One theory suggests that the accumulation of phenylalanine interferes with the transport of tyrosine into the brain. A reduction in brain tyrosine levels would then result in reduced synthesis of the neurotransmitters dopamine and norepinephrine. Another theory suggests that the intellectual impairment is caused by the accumulation of phenylalanine in the brain, which becomes a major donor of amino groups in aminotransferase activity and depletes neural tissue of 2-oxoglutarate (α-ketoglutarate). This absence of 2-oxoglutarate in the brain shuts down the TCA cycle and the associated production of aerobic energy. Although both theories are plausible no direct evidence for either has been demonstrated.
The product of phenylalanine transamination, phenylpyruvic acid, is reduced to phenylacetate and phenyllactate, and all three compounds appear in the urine. The presence of phenylacetate in the urine imparts a “mousy” odor. If the problem is diagnosed early, the addition of tyrosine and restriction of phenylalanine from the diet can minimize the extent of intellectual impairment.
Because of the requirement for tetrahydrobiopterin in the function of phenylalanine hydroxylase, mutations in the PCBD1 gene or the QDPR gene can manifest with hyperphenylalaninemia. Mutations in the PCBD1 gene and the QDPR gene are causes of a family of disorders termed tetrahydrobiopterin deficiency syndromes.
Tetrahydrobiopterin Synthesis
Tetrahydrobiopterin (6R-L-erythro-5,6,7,8-tetrahydrobiopterin) is most often abbreviated BH4 but also can be written as HB4. Depending upon cell type, BH4 can be generated by several different (alternative) de novo biosynthesis pathways as well as by two different salvage pathways. The major de novo pathway is diagrammed in the Figure below. The synthesis of BH4 occurs de novo from the nucleotide, GTP. There are three principal enzymes involved in the major de novo pathway of BH4 synthesis. In reaction order these enzymes are GTP cyclohydrolase 1 (GTPCH), 6-pyruvoyltetrahydropterin synthase (PTS; often designated PTPS), and sepiapterin reductase (SPR).
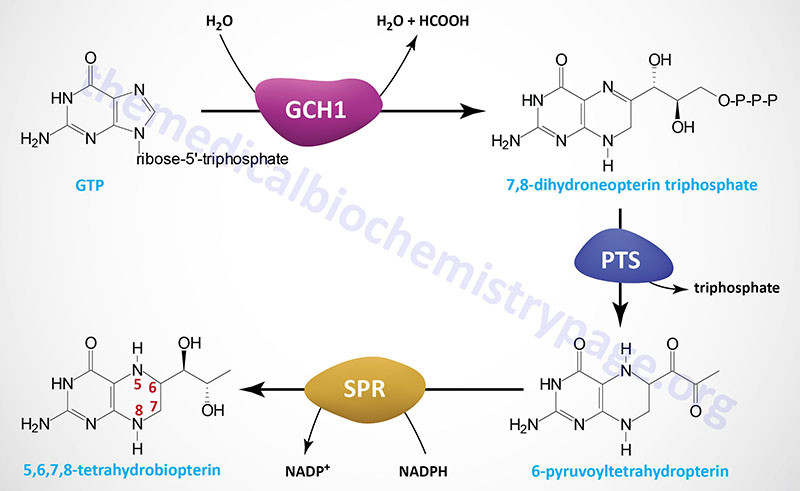
GTP cyclohydrolase 1 is encoded by the GCH1 gene. The GCH1 gene is located on chromosome 14q22.2 and is composed of 9 exons that generate four alternatively spliced mRNAs that collectively encode three distinct protein isoforms.
The 6-pyruvoyltetrahydropterin synthase enzyme is encoded by the PTS gene. The PTS gene is located on chromosome 11q23.1 and is composed of 6 exons that encode a 145 amino acid protein.
Sepiapterin reductase is encoded by the SPR gene. The SPR gene is located on chromosome 2p13.2 and is composed of three exons that encode a 261 amino acid protein.
The reaction catalyzed by GTP cyclohydrolase 1 (GTPCH) is considered the rate-limiting and committed reaction of BH4 synthesis. Functional GTP cyclohydrolase 1 exists as a homodecamer that consists of two tightly associated dimers that each comprise a pentameric complex of the GTP cyclohydrolase 1 monomers. Through the actions of GTP cyclohydrolase 1 GTP is converted to 7,8-dihydroneopterin triphosphate.
The second step in BH4 synthesis, catalyzed by 6-pyruvoyltetrahydropterin synthase, involves the Zn2+– and Mg2+-dependent rearrangement of 7,8-dihydroneopterin triphosphate, with concomitant loss of the triphosphate, to 6-pyruvoyltetrahydropterin. Functional 6-pyruvoyltetrahydropterin synthase is a homoheaxmeric complex that is formed via the head-to-head interaction of a pair of trimers.
The final step of BH4 synthesis actually involves three distinct steps all of which are catalyzed by the NADPH-dependent enzyme, sepiapterin reductase (SR). Functional sepiapterin reductase is a homodimeric enzyme which first converts 6-pyruvoyltetrahydropterin to 1′-hydroxy-2′-oxopropyltetrahydropterin, which is then reduced to 1′-oxo-2′-hydroxypropyltetrahydropterin, and then finally to 5,6,7,8-tetrahydrobiopterin, BH4.
Disorders Associated with Tetrahydrobiopterin Synthesis and Salvage
There are at least four identified disorders that manifest with BH4-deficient hyperphenylalaninemia designated as hyperphenylalaninemia, BH4-deficient type A (HPABH4A), HPABH4B, HPABH4C, and HPABH4D. Each of the four HPABH4 disorders are phenotypically characterized by hyperphenylalaninemia, depletion of the neurotransmitters dopamine and serotonin, and progressive cognitive and motor deficits.
Mutations in the PTS gene are the cause of the autosomal recessive disorder referred to as HPABH4A. HPABH4A represents the most common form of inherited tetrahydrobiopterin deficiency.
Mutations in the GCH1 gene are the cause of the autosomal recessive disorder referred to as HPABH4B.
Mutations in the QDPR gene, that encodes the enzyme commonly referred to as dihydrobiopteridine reductase (DHPR), are the cause of the autosomal recessive disorder referred to as HPABH4C. At least 30 different mutations in the QDPR gene are associated with inherited tetrahydrobiopterin deficiencies and these mutations account for approximately 30% of all the cases of these related disorders. Because the activity of the QDPR gene encoded enzyme is also involved in the functions of the tyrosine hydroxylase and tryptophan hydroxylase enzymes, mutations in the QDPR gene present as atypical hyperphenylalaninemias but are also associated with the potential for microcephaly, hypotonia, intellectual impairment, and convulsions due to neurotransmitter synthesis deficits. In these atypical hyperphenylalaninemias, that result from QDPR mutations, restriction of phenylalanine from the diet has no therapeutic benefits.
Mutations in the PCBD1 gene, that encodes the tetrahydrobiopterin salvage enzyme pterin 4α-carbinolamine dehydratase 1, are the cause of the autosomal recessive disorder referred to as HPABH4D. At least nine different mutations in the PCBD1 gene have been found to be associated with inherited tetrahydrobiopterin deficiency. However, the mutations in PCBD1 are not associated with significant pathology. It is thought that other enzymes can compensate for the reduced activity of PCBD1.
Mutations in the SPR gene are associated with a form of DOPA-responsive dystonia. DOPA is also designated L-DOPA and refers to L-3,4-dihydroxyphenylalanine. Clinically L-DOPA is levodopa. Inheritance of SPR deficiency may be autosomal recessive or autosomal dominant but has not been definitively characterized. Loss of the SPR encoded enzyme results in neurologic deterioration due to severe dopamine and serotonin deficiencies in the central nervous system.