
Last Updated: July 5, 2026
Overview of Metabolism in Cancer
Associations between altered metabolic pathways and cancer were first made nearly a century ago. Since then significant advances have been made in understanding the molecular and cellular basis for these altered metabolic profiles in cancer cells and how these changes facilitate and promote the transformed state. What is very clear now is that metabolic alterations at various stages of tumorigenesis affect metabolite influx through conferring an increased ability to acquire the necessary nutrients and to alter the pathways of nutrient metabolism for the aberrant proliferation of cancer cells.
The metabolic changes observed in cancer cells result in the redistribution of carbon and nitrogen for biomass production and lead to changes in the tumor microenvironment that promote the transformed state. Some of the most significant metabolic alterations involve the redistribution glucose carbons away from complete oxidation into biomass and the carbons and nitrogens from glutamine into pathways that promote incorporation into biomass.
Glutamine Metabolism in Cancer
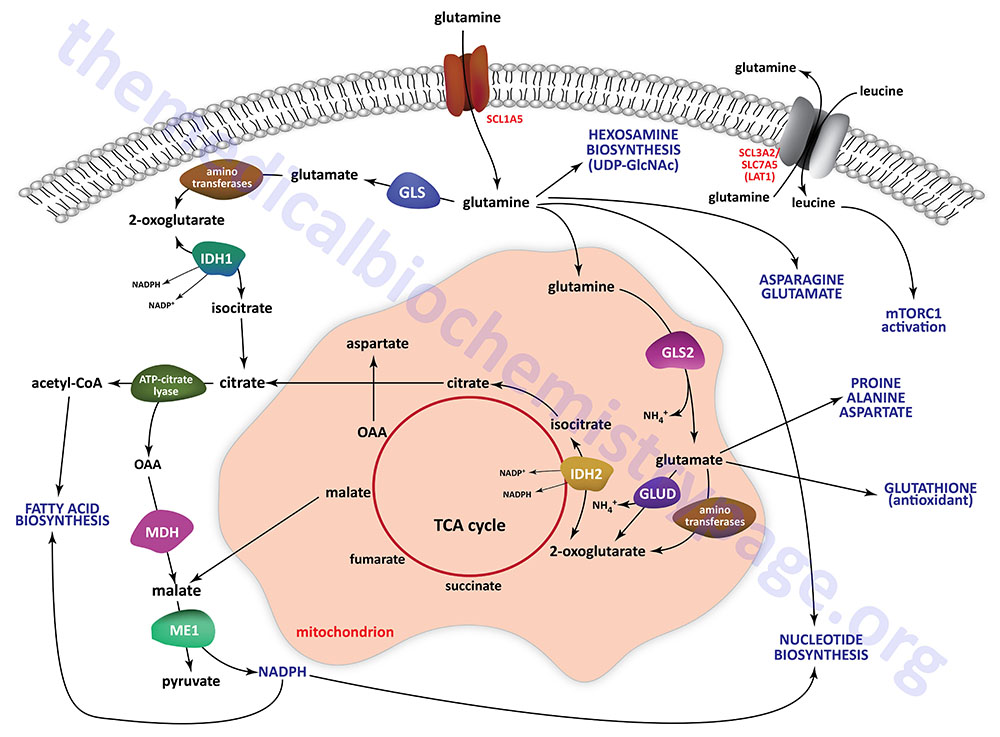
Glutamine represents the most abundant amino acid in the blood with levels that are in the range of 500μM. This level of glutamine accounts for over 20% of the total pool of free amino acids in the blood. Although the diet serves as a principal source of glutamine from digested proteins that is then absorbed through the small intestine, another major source of blood glutamine is the ammonia scavenging reactions that take place in skeletal muscle and liver as well as many other tissues.
By circulating in the plasma glutamine represents a major contributor to the energy needs of cells, as a major molecule for movement of ammonia in a non-toxic form, as a carbon source for the synthesis of glucose via gluconeogenesis in the kidney and small intestine, and as a critical substrate for the hexosamine biosynthesis pathway which synthesizes UDP-GlcNAc. UDP-GlcNAc is added to numerous cytoplasmic and nuclear proteins (referred to as O-GlcNAcylation) altering their activities. O-GlcNAcylation also represents a mode of epigenetic modification of histone proteins. The need for UDP-GlcNAc for glycoprotein synthesis also demonstrates the role of glutamine in endoplasmic reticulum (ER) homeostasis preventing the activation of ER stress pathways.
Glutamine carbons and nitrogen atoms also serve as substrates for the synthesis of other amino acids, nucleotides, and the antioxidant, glutathione. All of these roles of glutamine metabolism, more specifically the deamination of glutamine to glutamate, are collectively referred to as glutaminolysis. As such, the role of glutaminolysis in the generation of biomass is significant to the growth of cancer cells.
Glutamine in Acid-Base Control
The release of ammonia from glutamine, within the proximal tubule, represents a major mechanism linking metabolism to the role of the kidneys in the maintenance of acid-base homeostasis. In rapidly dividing cells such as small intestinal enterocytes, lymphocytes, and especially cancer cells, glutamine is rapidly consumed and used for both energy generation and as a source of carbon and nitrogen for the synthesis of biomass.
The high level of glutamine that is maintained in the plasma provides a readily accessible source of carbon and nitrogen that can support the metabolic demands of cancer cells. In the small intestine and in the kidney the deamination of glutamine to 2-oxoglutarate (α-ketoglutarate) allows the carbon skeleton to be diverted into the TCA cycle and ultimately diverted, via oxaloacetic acid, to the gluconeogenesis pathway for the de novo synthesis of endogenous glucose. The removal of the nitrogens from glutamine within the kidney proximal tubule epithelial cells allows the kidney to release excess acid in the from of ammonium ion (NH4+).
Glutamine Transport in Energy Generation
Glutamine is transported into cells through the actions on one of the several glutamine transporters such as SLC1A5 (also known as ASCT2), SLC38A1, SLC38A3, SLC38A5 (also known as system N isoform 2; SN2), and SLC38A7. Intracellular glutamine can be transported from cells in exchange for other amino acids through the action of several antiporter-type transporters such as the L-type amino acid transporter. This latter transporter is a heterodimer of the SLC3A2 and SLC7A5 encoded proteins forming what is commonly identified as LAT1.
Glutamate, that is formed via glutaminase action on glutamine, can be transported from cells, in exchange for cystine, through the action of the antiporter commonly identified as xCT. The xCT transporter is a heterodimer of the SLC3A2 and SLC7A11 encoded proteins. Within cells cystine is rapidly reduced to two molecules of the amino acid cysteine. This represents a major link between glutamine and the sulfur necessary for biomass synthesis, particularly in rapidly dividing tumor cells. Even in nutrient deprived states, cancer cells can gain access to nutrients, such as proteins, in the blood through the upregulation of macropinocytosis. The increased protein uptake allows for their degradation and release of amino acids including glutamine. Some RAS-transformed cancer cells have been shown to acquire needed glutamine through autophagic degradation of intracellular proteins.
In order to support energy generation the glutamine that is transported into cells, such as rapidly proliferating cells and cancer cells, is deaminated through the action of glutaminase and the resulting glutamate is further deaminated through the actions of aminotransferases or through the action of glutamate dehydrogenase. The resulting 2-oxoglutarate is fed into the TCA cycle and oxidized generating the reduced electron carriers, NADH and FADH2, which fuel oxidative phosphorylation and the synthesis of ATP.
Transcription of both the SLC1A5 and SLC38A5 genes is enhanced by the proto-oncogene c-MYC. Thus, in many cancers where overexpression of the c-MYC gene is evident there will be an associated increased access of these tumor cells to glutamine for diversion of the carbons and nitrogen into biomass.
Glutamine and NADPH Synthesis
In some tumor cells, a portion of glutamine that is converted to oxaloacetic acid via the TCA cycle, is converted to pyruvate through the activity of malic enzymes. Humans express three malic enzymes, one cytoplasmic that requires NADP+, and two mitochondrial enzymes, one that requires NADP+ and one that requires NAD+.
The NADP+-dependent cytoplasmic enzyme is called malic enzyme 1 and is encoded by the ME1 gene. The ME1 gene is located on chromosome 6q14.2 and is composed of 14 exons that encode a protein of 572 amino acids.
The NAD+-dependent mitochondrial enzyme is called malic enzyme 2 and is encoded by the ME2 gene. The ME2 gene is located on chromosome 18q21.2 and is composed of 16 exons that generate two alternatively spliced mRNAs encoding precursor proteins of 584 amino acids (isoform 1) and 479 amino acids (isoform 2).
The NADP+-dependent mitochondrial enzyme is called malic enzyme 3 and is encoded by the ME3 gene. The ME3 gene is located on chromosome 11q14.2 and is composed of 23 exons that generate five alternatively spliced mRNAs, four of which encode the same 604 amino acid protein (isoform 1).
The NADPH produced by the ME1 catalyzed reaction can be used in biomass producing reductive biosynthetic reactions such as fatty synthesis and phospholipid synthesis. Indeed, expression of ME1 is elevated in most human cancers and gene knockdown studies in cancer cells in culture results in acute growth inhibition by inducing cellular senescence or apoptosis.
Glutamine in Nucleotide Biosynthesis
Glutamine is also critical in the biosynthesis of nucleotides for RNA and DNA synthesis as is required in rapidly proliferating cells and cancer cells. Glutamine can serve as a precursor for aspartate through the concerted actions of glutaminase, yielding glutamate, followed by transfer of the nitrogen from glutamate to oxaloacetic acid (OAA), via the action of the cytoplasmic aspartate transaminase (AST; encoded by the GOT1 gene), forming aspartate. The oxaloacetic acid utilized in this reaction can also be derived from glutamine, thus indicating this pivotal role of glutamine in overall nucleotide homeostasis. The aspartate that can be derived from glutamine is required as a source of carbon for both purine and pyrimidine nucleotide synthesis.
In addition, glutamine serves as the amino donor in the formation of carbamoyl phosphate through the actions of the tri-functional enzyme carbamoyl-phosphate synthetase 2, aspartate transcarbamoylase, and dihydroorotase. This enzyme is encoded by the CAD gene. The resulting carbamoyl phosphate is the initiating precursor for the synthesis of the pyrimidine nucleotides. Thus, it should be obvious that the carbons and the nitrogen of glutamine are critical components of overall nucleotide homeostasis.
The CAD gene is located on chromosome 2p23.3 and is composed of 45 exons that generate two alternatively spliced mRNAs, one encoding a protein of 2225 amino acids (isoform 1) and the other a protein of 2162 amino acids (isoform 2).
In cancer cells, the expression of enzymes involved in glutamine metabolism varies widely and is related to factors such as tissue of origin and the overall genotype of the specific cancer. Humans express two genes encoding glutaminase enzymes, GLS encoding the GLS1 enzymes (where the kidney-type isoform is the predominant) and GLS2 encoding the GLS2 enzyme. The GLS gene is more widely expressed and is the glutaminase encoding gene believed to be of more significance in cancer cell metabolism, specifically the GAC encoded isoform.
Sirtuins, which are NAD+-dependent deacetylases, play a role in the regulation of expression of numerous genes. In cancer cells the sirtuin encoded by the SIRT3 gene has been shown to deacetylate the GLS2 gene resulting in increased expression under conditions of caloric restriction. The glutaminase C isoform (GAC) is more active than the kidney-type glutaminase (KGA) and the levels of GAC have been found to be increased in numerous cancers.
Glutamine Coupling to Glutamate Dehydrogenase
Glutamate dehydrogenase activity is also altered in cancer cells. Glutamate dehydrogenase is allosterically activated by leucine, the levels of which will be higher in cancer cells due to increased activity of various amino acid transporters. In the direction of glutamate deamination, glutamate dehydrogenase is activated by ADP. Therefore, low cellular energy states will lead to induction of 2-oxoglutarate production via the glutamate dehydrogenase reaction allowing for increased ATP synthesis from the TCA cycle-derived NADH and FADH2.
Glutaminase Expression and Inhibition in Cancer
The role of glutaminolysis in cancer has been well established. Indeed, the overexpression of several genes, such as the c-MYC proto-oncogene, in numerous cancers has been shown to result in increased expression from the GLS gene resulting in increased levels of the kidney-type glutaminase (KGA). Although the GLS encoded enzyme is normally predominately found in the mitochondria, in numerous cancers the KGA isoform can also be found in the cytosol. The c-MYC-associated increased expression of GLS is a consequence of c-MYC-mediated transcriptional repression of the microRNAs, miR-23a and miR-23b. Both the miR-23a and miR-23b miRNAs target the 3′-untranslated region of the GLS encoded mRNAs.
Given that increased expression of the GLS gene, and the consequent increased level of glutaminolysis, clearly plays a contributory role in cancer progression, it is not surprising that targeted inhibition of glutaminase enzymes holds promise as a means to inhibit the growth of certain forms of cancer. Analogs of glutamine, that target the active site of glutaminase, were the first types of drugs used to exert antitumor activity. However, these compounds were highly toxic. Compounds that target the process of oligomerization of the GLS encoded enzymes have shown promise in the treatment of certain cancers. One such compound is BPTES [bis-2-(5-phenylacetamido-1,2,4-thiadiazol-2-yl)ethyl sulfide].
Asparagine Metabolism in Cancer
Expression of the gene (ASNS) encoding asparagine synthetase in solid tumors correlates with poor prognosis. Additionally, expression of the ASNS gene has been found to be highly correlated to the metastatic potential of numerous types of cancer.
Expression of the ASNS gene is upregulated in response to deficiencies in numerous amino acids, including most essential amino acids. The mechanism of induced expression of the ASNS gene in response to amino acid deprivation involves the transcription factor, activating transcription factor 4 (ATF4).
Aside from its role as an amino acid for protein synthesis, asparagine has been shown to exchange with extracellular amino acids, specifically arginine, histidine, and serine, to regulate the activation of mTOR complex 1 (mTORC1), the synthesis of nucleotides, and to stimulate proliferation. In the context of cancer cell proliferation, glutamine-dependent asparagine synthesis plays a critical metabolic function. Cancer cells that have adapted to growth under glutamine limiting conditions do so only with adequate exogenous asparagine. Although glutamine-independent cancers can synthesize sufficient glutamine for protein and purine synthesis they cannot grow in the absence of exogenous asparagine. This indicates that in proliferating cells, such as is typical in cancer, the demand for asparagine exceeds that which can be synthesized via pathways of de novo asparagine synthesis.
Although glutamine is the most abundant amino acid in the blood and within cells it is extensively metabolized, predominantly via mitochondrial glutaminase. Because asparagine is not metabolized it is likely to be more available for exchange with extracellular amino acids contributing to cancer cell proliferation. Indeed, when asparagine synthesis is blocked by inhibition of asparagine synthetase activity there is a decrease in amino acid uptake, even when there is abundant levels of glutamine.
The role of asparagine in amino acid exchange, particularly in cancer cells, influences protein synthesis and nucleotide synthesis, both of which are required for proliferation. Asparagine exchange for other amino acids induces protein synthesis via activation of mTORC1. The role of asparagine in the regulation of nucleotide synthesis is due to exchange with extracellular serine. Asparagine is clearly an important contributor to cancer cell growth and strategies that interfere with this role are likely to be important cancer treatments.
Cystine (Cysteine Disulfide) Role in Epigenetics and Oxidative Stress
Cystine is formed from the oxidation of the —SH (sulfhydryl) of the R-groups of two cysteine molecules which generates a disulfide bond. Because this is an oxidation process the formation of cystine occurs most readily in the endoplasmic reticulum (ER), the Golgi, and the extracellular environment. Conversely, within the cytosol the actions of, primarily, glutathione and thioredoxin reduce the disulfide bond generating two molecules of cysteine.
Cystine is an important molecule for proliferating cells, normal and tumorigenic, because these cells, particularly cancer cells, experience increased oxidative stress. One major antioxidant utilized by all cells is glutathione (GSH) which is a tripeptide containing cysteine. Proliferating cells thus require access to free cysteine to deal with oxidative stress. However, because the extracellular environment is highly conducive to oxidizing cysteine to cystine the ability to transport cysteine into cells is limiting. Recent work has shown that proliferating cells and cancer cells increase the expression of the cystine transporter encoded by the SLC7A11 gene. This transporter is commonly called the xCT transporter. The Xc– transporter, a heterodimeric transporter, is composed of the SCL7A11 encoded protein and the SLC3A2 encoded protein identified as 4F2hc (4F2 cell-surface antigen heavy chain).
The increased uptake of cystine by proliferating cells provides a mechanism to increase access to cysteine. The cysteine can be used to synthesize more glutathione (GSH) allowing cells to more efficiently deal with oxidative stress. However, since the reduction of cystine to two cysteines requires NADPH, in the context of GSH activity, the increased cystine uptake can lead to depletion of NADPH which can then lead to exacerbation of oxidative stress. One mechanism to deal with this conundrum is to increase glucose diversion into the pentose phosphate pathway since this is the major NADPH generating pathway. Indeed, in cancer cells, the activation of the PKM gene and the synthesis of the PKM2 isoform of pyruvate kinase promotes glucose diversion into pathways conducive for biomass production.
Increased cystine uptake, via the increased expression of the SCL7A11 transporter, has been shown to contribute to changes in the epigenome. Cystine forms a complex with cystathionine γ-lyase (most often designated CSE) and the a-subunit of the E1 portion of the PDH complex (PDHc) which is encoded by the PDHA1 gene. The PDHA1 encoded enzyme is known to function in the nucleus in addition to its role in the PDHc in the mitochondria. Once the complex of cystine, PDH α-subunit, and cystathionine γ-lyase are transported into the nucleus, cystathionine γ-lyase oxidatively catabolizes cystine into cysteine and pyruvate. The PDH α-subunit then oxidizes pyruvate generating nuclear acetyl-CoA. This acetyl-CoA can be utilized by histone acetyltransferases (HAT) to acetylate histones. The acetylation of histones is an epigenetic process that results in open chromatin and increased transcription.
Role of Glucose Metabolism in Cancer Progression
The metabolism of glucose does much more than just provide the ATP energy required for rapid growth and proliferation of cancer cells. Glucose and metabolic intermediates derived from glucose are involved in the modulation of epigenetic control of gene expression that involves the processes of O-GlcNAcylation, acetylation, phosphorylation, and ubiquitylation.
Increased glucose uptake by cancer cells contributes to increased synthesis of UDP-N-acetylglucosamine (UDP-GlcNAc) which is the substrate for O-GlcNAcylation of numerous proteins, including histone proteins. The process of protein O-GlcNAcylation plays a critical role in the regulation numerous cellular processes that includes, but is not limited to, cell cycle progression, transcription, translation, signal transduction, protein-protein interactions, and epigenetic regulation. All of these processes contribute to the progression of cancer.
Glucose Metabolism and the Warburg Effect
It has been known for over 100 years that cancer cells metabolize glucose differently than differentiated cells. In 1924 Otto Warburg made an observation that cancer cells metabolized glucose in a manner that was distinct from the glycolytic process of cells in normal tissues. Warburg discovered that, unlike most normal tissues, cancer cells tended to “ferment” glucose into lactate even in the presence of sufficient oxygen to support mitochondrial oxidative phosphorylation. This observation became known as the Warburg Effect. Warburg assumed that this altered metabolism was the result of defects in mitochondrial function in cancer, but it was subsequently demonstrated that cancer cells did not harbor altered mitochondrial functions.
In the presence of oxygen, most differentiated cells primarily metabolize glucose to CO2 and H2O by oxidation of glycolytic pyruvate in the mitochondrial TCA cycle. Originally postulated to be the result of mitochondrial dysfunction in cancer cells, it was subsequently shown that this was not the mitigating reason for the increased production of lactate in cancer cells. It might seem counterintuitive that highly proliferative cells, such as is characteristic of cancers, would bypass oxidation of pyruvate (from glucose) in mitochondria since this is the organelle where the vast majority of the ATP from glycolysis is generated. However, it has been shown that oncogenic mutations can result in the uptake of nutrients, particularly glucose and glutamine, that meet or exceed the bioenergetic demands of cell growth and proliferation.
Proliferating cells, including cancer cells, require altered metabolism to efficiently incorporate nutrients such as glucose into biomass. The ultimate fate of glucose depends not only on the proliferative state of the cell but also on the activities of the specific glycolytic enzymes that are expressed. This is particularly true for pyruvate kinase, the terminal enzyme in glycolysis.
Pyruvate Kinase Isoforms and Cancer
In mammals, two genes encode a total of four pyruvate kinase (PK) isoforms. The PKLR gene encodes the liver (L-PK or PKL) and the erythrocyte (R-PK or PKR) isoforms of pyruvate kinase via a process of alternative promoter usage. The PKM gene, so called originally due to initial characterization in muscle tissues, encodes the PKM1 and PKM2 isoforms.
PKM1 and PKM2 are derived via alternative splicing of the PKM gene encoded mRNA. This alternative splicing results in mutual exclusion of a single conserved exon encoding 56 amino acids. In PKM1 mRNA exon 9 is included, whereas it is excluded from PKM2 mRNA. In the PKM2 mRNA exon 10 is included, whereas it is excluded from PKM1 mRNA.
Expression of PKM1 predominates in cardiac muscle, skeletal muscle, smooth muscle, brain, adrenal medulla, and mature sperm. Expression of PKM2 occurs in most proliferating cells, including in all cancer cell lines and tumors tested to date, embryonic tissues, epithelial cells of the distal tubule of the kidney, spleen, ovaries, testes, intestine, leukocytes, thymus, and adrenal cortex.
Although PKM1 and PKM2 are highly similar in amino acid sequence they have different catalytic and regulatory properties. PKM1 has high constitutive enzymatic activity and is exclusively an enzyme that utilizes the energy of phosphoenolpyruvate (PEP) to phosphorylate ADP to ATP while converting PEP to pyruvate as is typical of glycolysis.
PKM2 Structure-Function Relationships
In contrast to PKM1, PKM2 activities are more complex and, in part, depend on the oligomeric state of the enzyme. PKM2 can exist as a homodimeric or as a homotetrameric enzyme. The tetrameric form of PKM2 exhibits high catalytic activity in the conversion of phosphoenolpyruvate (PEP) to pyruvate and this is associated with ATP synthesis and catabolic metabolism of glucose. In contrast, the dimeric form of PKM2 has low catalytic activity and this low activity, in part, facilitates the diversion of glycolytic intermediates into glucose metabolic branch pathways that including glycerol synthesis and the pentose phosphate pathway. Of significance to proliferating cells, as well as cancer, is that the NADPH produced by the pentose phosphate pathway is used to reduce the level of reactive oxygen species (ROS) and is used in numerous reductive biosynthetic pathways. In addition, the ribose-5-phosphate is used for nucleotide synthesis.
The regulation of the dimeric versus tetrameric forms of PKM2 is under complex control that includes allosteric effectors and post-translational modifications. The various post-translational modifications include phosphorylation, acetylation, and oxidation. The upstream glycolytic metabolite fructose 1,6-bisphosphate (FBP) was the first characterized allosteric regulator of PKM2 which promotes the formation of the tetrameric form of the enzyme.
The amino acid serine, which can be derived from the glycolytic intermediate, 3-phosphoglycerate, is also an allosteric activator promoting the tetrameric form of PKM2.
Post-Translational Modification of PKM2
Post-translational modifications, that include phosphorylation and acetylation, promote the less active dimeric form of PKM2. In growth factor stimulated proliferative cells and in cancer cells there is increased tyrosine phosphorylation of numerous proteins including PKM2. When PKM2 is phosphorylated on tyrosine at amino acid position 105 (Y105) it releases fructose 1,6-bisphosphate (FBP) causing conversion to the less active dimeric form.
Acetylation of lysine 305 (K305) in PKM2 by the acetyltransferase KAT2B, which was originally identified as PCAF (p300/CBP-associated factor), results in decreased enzymatic activity due to the inhibition of the formation of the more active homotetrameric structure. Lysine 305 is located in the protein at a location referred to as the dimerization interface. In addition to preventing tetramerization, the presence of K305 in PKM2 promotes degradation of the protein in the lysosomes. The acetylation of K305 occurs preferentially in the presence of high glucose concentrations. The deacetylation of K305 is catalyzed by SIRT2 which prevents degradation and returns the enzyme to its active state.
Acetylation of lysine 433 (K433) by the p300/CBP acetyltransferase also inhibits the activation of PKM2 enzymatic activity by interfering with the fructose 1,6-bisphosphate (FBP)-mediated dimeric to tetrameric transition. This effect is due to the fact the K433 resides in the FBP-binding pocket of the enzyme. Deacetylation of K433 is catalyzed by SIRT6.
Another critical lysine in PKM2 that undergoes acetylation if lysine 115 (K115). When K115 is acetylated the enzymatic activity of PKM2 is significantly inhibited due to the stabilization of a closed active site conformation. Deacetylation of K115 has been shown to be carried out by both SIRT1 and SIRT2 which carry out the deacetylation if an FBP-dependent manner.
Low PKM2 activity, in conjunction with increased glucose uptake, facilitates the diversion of glucose carbons into the anabolic pathways that are derived from glycolysis. PKM2 is also converted to the less active dimeric form by direct oxidation of a cysteine residue (Cys358) as an adaptive response to increased intracellular reactive oxygen species (ROS). This inhibition does not occur in PKM1.
Nuclear-Localized PKM2 as a Transcriptional Co-Activator
PKM2 is also phosphorylated on serine residues at positions 37 (S37) and 202 (S202). Serine 37 phosphorylation of PKM2 stimulates its movement into the nucleus. Nuclear localized PKM2 functions as a transcriptional co-activator of β-catenin in the activation of the MYC gene. MYC is a potent transcription factor and elevated expression of MYC contributes to the transformed state of numerous types of cancer. In addition, several MYC target genes contribute to the increased metabolic profiles of cancer cells. These genes include the SLC2A1 gene that encodes glucose transporter 1 (GLUT1), the LDHA gene that encodes lactate dehydrogenase A, and also the PKM gene leading to increased levels of PKM2. This latter effect results in a positive feed-back loop between PKM2 and MYC.
Serine 202 phosphorylation also results in migration of PKM2 to the nucleus and this modification allows PKM2 to serve as a transcriptional co-activator of the transcription factors STAT3 (signal transducer and activator of transcription 3) and STAT5A (signal transducer and activator of transcription 5A). Activation of STAT5A results in enhanced transcription of a member of the cyclin family (cyclin D1) of cyclin-dependent kinase (CDK) regulators leading to enhanced cell cycle progression.
As indicated in the previous section, Lys433 (K433) in PKM2 is acetylated by p300/CBP. When acetylated PKM2 migrates to the nucleus where it can also serve as transcriptional coactivator of STAT3. In T-helper cells the PKM2-mediated activation of STAT3 promotes their differentiation and, as a result, the cell-mediated inflammatory processes of T-helper cells.
Nuclear PKM2 is also a transcriptional co-activator of the hypoxia-induced factor-1 (HIF-1) complex. The interaction of PKM2 with HIF-1 results in enhanced transcription of HIF-1 target genes, many of which encode metabolic enzymes, thereby enhancing the metabolic rate of cancer cells. Metabolic genes that are targets for HIF-1 include LDHA [encoding the M (muscle) form of lactate dehydrogenase), SLC2A1 (encoding GLUT1), PDK1 (encoding pyruvate dehydrogenase kinase 1), FASN (encoding fatty acid synthase), and ACACA (encoding acetyl-CoA carboxylase 1).
Within cancer cells, PKM2 interacts with succinylaminoimidazole carboxamide ribotide (SAICAR) which is an intermediate in purine nucleotide biosynthesis. This interaction stimulates the kinase activity of PKM2 as well as the nuclear localization of the enzyme. The activation of PKM2 by SAICAR occurs under glucose-limiting conditions and this activation promotes the survival of cancer cells under these conditions. More than 150 proteins, including numerous protein kinases, including members of the MAP kinase family, have been shown to be phosphorylated by PKM2-SAICAR complexes.
Nuclear-Localized PKM2 as RNA Binding Protein
Nuclear localization of PKM2, as discussed in the previous section, has been shown to function as a transcriptional coactivator. The localization of PKM2 to the nucleus has also been shown to be enhanced in numerous types of cancer. Recent evidence has found that in addition to transcriptional co-activation, nuclear localized PKM2 functions as an RNA-binding protein.
During the processing of heteronuclear pre-mRNA there are complex structures that form within individual RNA molecules as a result of nucleobase interactions. One such structure that forms in guanine (G)-rich regions of pre-mRNA is referred to as G-quadraplexes. These G-quadraplexes are found to form within RNA (and DNA) molecules possessing a motif identified as Gx–N1–7–Gx–N1–7–GxN1–7–Gx, where x is 3–6 guanine nucleotides and N corresponds to any nucleotide.
Nuclear-localized PKM2 has been shown to interact with G-quadraplex structures in pre-mRNAs which prevents the binding of repressive RNA binding proteins. The net effect of PKM2 binding to pre-mRNAs harboring G-quadraplexes is that there is a preferential enrichment of these mRNAs and, with respect to cancer, this enrichment contributes to the growth and proliferation of various cancer types.
Nuclear-Localized PKM2 as Histone Modifier
PKM2 contributes to the epigenetic regulation of gene expression following its translocation to the nucleus. The role of nuclear PKM2 in the regulation of gene expression involves both direct substrate phosphorylation and the generation of pyruvate. PKM2 has been shown to phosphorylate a Thr residue in histone H3 which leads to enhanced acetylation of Lys 9 of H3 (H3K9).
When acting as a kinase, PKM2 utilizes phosphoenolpyruvate (PEP) as the phosphate donor as opposed to ATP. The product of the reaction is a phosphorylated substrate, such as histone H3, and pyruvate. The nuclear-localized pyruvate can serve as a substrate for nuclear-localized pyruvate dehydrogenase (PDH) which generates acetyl-CoA. The resultant acetyl-CoA can then serve as a substrate for various acetyltransferases leading to altered patterns of histone acetylation and consequently, gene expression.
Phosphoglycerate Mutase in PKM2 Expressing Cells
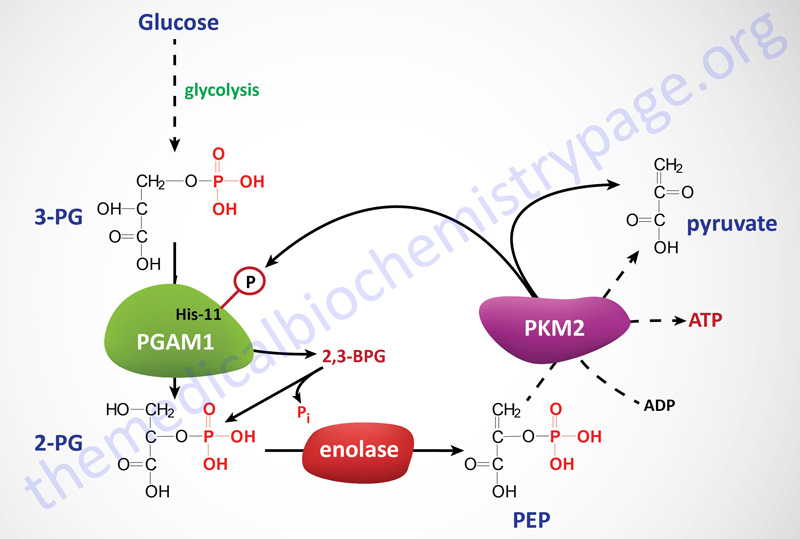
In cells in culture, the replacement of PKM2 with PKM1 (the constitutively active isoform) results in reduced lactate production and enhanced oxygen consumption. An additional observation has been made that in cells expressing PKM2 there is increased phosphorylation of an active site histidine (His11) in the upstream glycolytic enzyme phosphoglycerate mutase (PGAM1). His11 phosphorylation of PGAM1 increases its mutase activity. The phosphorylation of PGAM1 is not seen in PKM1 expressing cells.
It turns out that the phosphate donor for His11 phosphorylation of PGAM1 is phosphoenolpyruvate (PEP) which is the substrate for pyruvate kinases. Phosphate transfer from PEP to PGAM1 yields pyruvate without concomitant generation of ATP. This reaction occurs at physiological concentrations of PEP and produces pyruvate in the absence of the normal glycolytic pyruvate kinase activity. Thus, the PEP-dependent histidine phosphorylation of PGAM1 may provide an alternate glycolytic pathway that decouples ATP production from normal pyruvate kinase-mediated phosphotransfer from PEP. This alternate pathway allows for a high rate of glycolysis that is needed to support the anabolic metabolism observed in many proliferating cells. In addition to altered pyruvate kinase activity, cancer cells also have increased levels of expression of the kinases that phosphorylate the pyruvate dehydrogenase complex (PDHc), further limiting pyruvate oxidation, and increased expression of lactate dehydrogenase which drives pyruvate into lactate production.
PGAM1 is unique with respect to glycolytic enzymes because its rate of transcription is regulated by the tumor suppressor p53. In addition, increased expression of PGAM1 has been shown to immortalize primary cells, although the mechanism of this immortalization remains unknown. When PKM2 activity is down-regulated, as a consequence of growth factor-mediated tyrosine phosphorylations, PGAM1 mutase activity is enhanced due to the consequent increase in His11 phosphorylation from PEP. Thus, a positive feedback loop is activated, whereby the production of PEP increases the enzymatic activity of PGAM1.
Activation of this feedback loop between PEP and His11 modified PGAM1 may be the mechanism that promotes the redistribution of glycolytic carbons, upstream of PGAM1, into biosynthetic pathways that branch from glycolysis. In order for this alternative pathway to continue, the phosphate on His11 of PGAM1 must be removed so that it can serve as a continual acceptor of PEP phosphate. When PGAM1 converts 3-phosphoglycerate (3-PG) to 2-phosphoglycerate (2-PG) there is spontaneous hydrolysis of the phosphohistidine. In addition, it has been observed that 2,3-bisphosphoglycerate (2,3-BPG) can be formed from either 3-PG or 2-PG via phosphate transfer from His11 of PGAM1.
PKM2 Benefit for Cancer Cell Proliferation
For proliferating cells such as cancers, even though it may seem counterproductive to prevent complete oxidation of glucose solely for ATP production, the demands for carbon incorporation into biomass clearly supersedes the needs for ATP production from glucose. Also, cancer cells bypass the hormonal signals required of normal cells for nutrient uptake so there is no limit to the sources of carbon atoms for ATP production (e.g. fatty acids and amino acids).
Of metabolic significance to proliferating cells is that they must avoid ATP production in excess of demand to avoid allosteric inhibition of PFK-1 and other rate-limiting steps in glycolysis that are inhibited by a high ATP/ADP ratio. Therefore, the inhibition of PKM2 by binding to tyrosine phosphorylated proteins, following growth factor stimulation, may serve to uncouple the ability of cells to divert the carbons from nutrients (such as glucose) into biosynthetic pathways away from the production of ATP. This may, in fact, be the underlying reason why PKM2 activity towards production of pyruvate has evolved to be decreased in rapidly dividing cells.
PKM2 as an Anti-Cancer Target
Targeting PKM2 for the treatment of cancers is a distinct possibility. Recent work has demonstrated that small molecule PKM2-specific activators are functional in tumor growth models in mice. These new drugs have been shown to constitutively activate PKM2 and the activated enzyme is resistant to inhibition by tyrosine phosphorylated proteins. PKM2-specific activators reduce the incorporation of glucose into lactate and lipids. In addition, PKM2 activation results in decreased pools of nucleotide, amino acid, and lipid precursors and these effects may account for the suppression of tumorigenesis observed with these drugs.
Acetyl-CoA Roles in Cancer Metabolism
Acetyl-CoA is a central metabolic and signaling intermediate in normal cells, but particularly so in transformed cells. As an important metabolic intermediate, acetyl-CoA functions in both the cytosol and the mitochondria. As a signaling intermediate, acetyl-CoA plays a crucial role in the process of protein acetylation both in the cytosol and the nucleus. Indeed, the acetylation of histone proteins, where acetyl-CoA is the acetate donor, represents a major epigenetic mechanism controlling gene expression.
The major sources of acetyl-CoA in the mitochondria are pyruvate oxidation via the pyruvate dehydrogenase complex (PDHc), fatty acid β-oxidation, and amino acid oxidation (primarily leucine and isoleucine). Mitochondrial acetyl-CoA is also generated from acetate, via the action of the mitochondrial acetyl-CoA synthetase (encoded by the ACSS1 gene), and from the metabolism of the ketone body, β-hydroxybutyrate (BHB).
Acetyl-CoA enters the cytoplasm in the form of citrate via the tricarboxylate transport system (encoded by the SLC25A1 gene). The SLC25A1 encoded transporter is commonly referred to as the mitochondrial citrate carrier (CiC). The origin of the mitochondrial citrate is the TCA cycle where citrate synthase condenses oxaloacetate (OAA) and acetyl-CoA, with the concomitant release of coenzyme A (CoASH). In the cytoplasm, citrate and CoASH are converted to OAA and acetyl-CoA by the ATP driven ATP-citrate lyase reaction. This mechanism allows excess mitochondrial acetyl-CoA to be utilized for the synthesis of fatty acids and cholesterol. In the context of cancer cells the synthesis of membrane lipids from cytosolic acetyl-CoA is critical to proliferative expansion.
Acetate also serves as a source of cytosolic acetyl-CoA through the same reaction process by which mitochondrial acetate serves as a precursor for acetyl-CoA. Humans express cytosolic/nuclear and mitochondrial acetyl-CoA synthetases, encoded by the ACSS2 and ACSS1 genes, respectively. Normal physiological sources of acetate include bacterial fermentation in the colon which increases significantly when consuming a high fiber diet. This intestinal acetate enters the portal circulation and is taken up by the liver where it is converted to acetyl-CoA. Acetate is also generated within the nucleus of all cells via the action of histone deacetylases (HDAC). As with the other sources of acetate, this nuclear acetate must be reactivated before it can be oxidized or reused. Intracellular generation of acetate is the consequence of the ubiquitously expressed cytosolic enzyme acetyl-CoA hydrolase. The acetate can then be salvaged by reactivation to acetyl-CoA. The consumption, and subsequent metabolism, of ethanol also serves as a source of acetate.
The overall level of enzymes that are involved in the generation and utilization of acetyl-CoA are enhanced in cancer cells as a means to promote the growth of the cancer cells. The genes encoding ATP-citrate lyase (ACLY) and cytosolic acetyl-CoA synthetase (ACSS2) are transcriptionally regulated by the transcription factor, SREBP, as are many of the genes encoding enzymes of lipid metabolism. Typical of the tumor microenvironment, which includes acidic and hypoxic conditions, the transcription factor HIF-1 is activated which in turn activates SREBP (specifically the SREBP-2 isoform). The activation of SREBP results in increased expression of the ACSS2 gene contributing to increased de novo lipid synthesis. The transcription factor, NRF2 [nuclear factor erythroid 2–related factor 2 which is encoded by the NFE2L2 (NFE2 like bZIP transcription factor 2) gene], which is activated in many types of cancer, has also been shown to activate transcription of the ACSS2 gene.
Lactate Production and Cancer Cell Proliferation
The conversion of pyruvate into lactate is enhanced in the context of activated hypoxia-induced factor-1 (HIF-1) since this transcription factor activates the expression of the LDHA gene. The increased production of lactate, by cancer cells, contributes to the acidification of the tumor microenvironment which, in turn, promotes further activation of the HIF-1 pathway. Lactate accumulation also results in pyruvate accumulation in cancer cells. Pyruvate is a known inhibitor of the prolyl hydroxylases that hydroxylate the HIF1α subunit proteins. Loss of HIF1α proline hydroxylation results in increased HIF1α stability and, therefore, increased HIF-1 transcriptional activity. Thus, accumulation of lactate and pyruvate, which occurs as a result of both altered pyruvate kinase gene expression and activation of the HIF-1 pathway, further promotes activation of the HIF-1 pathway leading to a controlled and enhanced metabolic profile within cancer cells.
Lactate is the ligand for the GPCR encoded by the HCAR1 gene. This receptor was originally identified as an orphan GPCR and named GPR81. The receptor is a member of the hydroxycarboxylic acid (HCA) receptor family and as such is identified HCA1. HCA1 is expressed at high levels on adipocytes where binding of lactate results in reduced release of fatty acids from stored triglycerides.
Of significance to cancer cell proliferation is that the HCA1 receptor is also found on cancer cells and dendritic cells of the immune system. These sites of expression contribute to the role of lactate in overall cancer metabolism and immune system evasion. Due to the altered metabolism of glucose in cancer cells they produce large amounts of lactate. The lactate is transported out of the cells via transporters of the monocarboxylate transporter (MCT) family which are encoded by genes of the SLC16 family. The primary lactate efflux transporter is MCT4 (encoded by the SLC16A3 gene).
The lactate then binds to HCA1 receptors on these same cells resulting in the activation of a transcription program that contributes to immune evasion. One significant gene whose transcription is enhanced in lactate-stimulated cancer cells is CD274. The protein encoded by the CD274 gene is called programmed death ligand 1 (PD-L1). The role of PD-L1 is suppression of the adaptive immune response, thereby contributing to reduced immune system activation of T-cells. When HCA1 is activated on dendritic cells of the immune system they express reduced levels of MHC molecules as well as the interleukins, IL-6 and IL-12. IL-12 is involved in the maturation of immature T-cells and thus, reduce release of IL-12 by dendritic cells further contributes to reduced T-cell-mediated adaptive immunity.
Lactate Metabolism and Cancer Epigenetics
As outlined in the section above, glucose metabolism to lactate in cancer cells results in activation of a transcription program that results in a self-fulfilling enhanced metabolic program promoting high rates of cancer cell growth and proliferation. Recent work (2025) has also found that the enhanced lactate production in the tumor microenvironment leads to increased cancer stem cell proliferation by suppression of cancer stem cell differentiation. These effects are exerted as a result of alterations in the epigenetic profile in these cancer stem cells.
Increased levels of lactate, as is typical of most cancers, leads to increased mitochondrial pyruvate and acetyl-CoA that is independent of pyruvate transport into the mitochondria. In normal metabolism cytosolic lactate can be oxidized to pyruvate which is then transported into the mitochondria via the actions of the inner mitochondrial membrane (IMM)-localized mitochondrial pyruvate carrier (MPC). However, inhibition of the MPC does not lead to decreased mitochondrial respiration in the presence of lactate.
The mechanism by which lactate enters the mitochondria is, as yet, incompletely understood but most likely involves one of two monocarboxylate transporters, MCT1 and/or MCT4. Within the mitochondria lactate is oxidized to pyruvate by a complex referred to as the mitochondrial lactate oxidation complex, mLOC. The mLOC is composed of the monocarboxylate transporters, MCT1 and MCT4 (and possibly MCT2), the lactate dehydrogenase isoforms, LDHA and LDHB, cytochrome oxidase, and a scaffolding protein identified as basignin (previously identified as CD147).
The increased metabolism of lactate within mitochondria and the resultant increases in acetyl-CoA are associated with changes in the levels of histone acetylation, a definitive epigenetic phenomenon. The alterations in histone acetylation are coupled to increased expression of the proto-oncogene, MYC, a known regulator of stemness.
Fructose Metabolism and Cancer
Overconsumption of fructose, as is typical in Western-style diets that are enriched in highly processed foods and beverages that contain high quantities of high fructose corn syrup (HFCS) or sucrose, have been found to be highly correlated with the development of certain types of cancers, particularly those of the pancreas, colon, and liver. Many different types of cancers utilize fructose as an additional energy source thereby promoting proliferation and metastases. Although several countries have banned the use of HFCS, it is still utilized by the food industry in the US and accounts for up to 40% of the caloric intake from sweeteners.
Within the gut, intestinal bacteria metabolize fructose to acetate. The acetate is taken up by colonic epithelial cells and can be oxidized for energy production. Some of the acetate will be delivered, via the portal circulation to the liver. In normal hepatocytes the acetate contributes to fatty acid synthesis following its conversion to acetyl-CoA which is catalyzed by acyl-CoA synthetase short chain family member 2 (encoded by the ACSS2 gene) which is a cytosolic enzyme.
Fructose Metabolism and Hepatocellular Carcinoma
However, in hepatoma cells the acetate that is derived from bacterial metabolism of fructose contributes to the progression to hepatocellular carcinoma (HCC). In hepatoma cells there is increased expression of mitochondrial acyl-CoA synthetase short chain family member 1 (encoded by the ACSS1 gene) which drives the acetate carbons into the TCA cycle contributing to increased production of 2-oxoglutarate (α-ketoglutarate) which in turn enhances production of glutamate. The increased glutamate contributes to the production of nucleotides allowing for enhanced cellular proliferation.
Additional alterations in expression in hepatoma cells include the GLUL gene which encodes glutamine synthetase. The increased levels of glutamine synthetase result in increased conversion of glutamate to glutamine. The increased production of glutamine, in turn, contributes to increased synthesis of UDP-GlcNAc. UDP-GlcNAc is utilized by O-GlcNAc transferase (OGT) in the process of O-GlcNAcylation of numerous cytosolic and nuclear proteins.
Several proteins that were recently shown to be O-GlcNAcylated as a consequence of high dietary fructose are eEF-1α1 (encoded by the EEF1A1 gene), calpain small subunit 1 (encoded by the CAPNS1 gene), nuclear factor related to KappaB binding protein (encoded by the NFRKB gene), EMSY transcriptional repressor, BRCA2 interacting (encoded by the EMSY gene), and nucleoporin 214 (encoded by the NUP214 gene).
The EMSY gene was given this name because it is the name of the sister of the first author of the original publication describing the protein. The EMSY encoded protein has amino acids at positions 57 to 62 that spell “SISTER” using the single letter amino acid code.
Each of these proteins has been indicated to play a role in proliferation, in particular, the O-GlcNAcylation of the EEF1A1 encoded translation elongation factor has been shown to enhance the proliferation of HCC cells as a result of its role in protein synthesis.
Fructose Metabolism and Colorectal Carcinoma
The role of fructose metabolism in the development and progression of colorectal carcinoma (CRC) involves not only the direct effects of disturbed metabolism in colorectal epithelial cells but also the impact of altered tumor-associated macrophages. The effects of fructose metabolism on tumor-associated macrophages reflects the role of the tumor microenvironment on cancer progression.
Within the tumor microenvironment (TME) there are two distinct subsets of macrophages identified as M1-like and M2-like macrophages. M1-like macrophages are a type of activated macrophages similar to classic M1 macrophages that are activated by interferon-γ to initiate a pro-inflammatory state. M2-like macrophages are similar to classic M2 macrophages that are involved in anti-inflammatory processes. The immune system plays a critical role in repression of cancer progression and evasion of immune responses contributes to progression of most types of cancer. Enhanced activity of M1-like, or repressed activity of M2-like macrophages within the TME would, therefore, be contributory to cancer progression.
Fructose metabolism has been shown to lead to inhibition of M1-like macrophage activation, a process referred to as polarization. The inhibition of M1-like macrophage polarization results in a reduction of the inflammatory processes that would normally interfere with cancer progression.
Fructose uptake by macrophages occurs via the action of the GLUT5 transporter and the level of GLUT5 expression has been shown to directly correlate with the progression of colorectal cancer. Upon uptake by M1-like macrophages fructose acts as a signaling molecule that promotes the interaction of the hexokinase 2 (HK2) isoform of hexokinase with inositol-1,4,5-trisphosphate, IP3 (also designated Ins-1,4,5-P3) receptor in the membrane of the endoplasmic reticulum, ER. Specifically, the interaction is between HK2 and the ITPR3 gene encoded receptor. The interaction of HK2 with the IP3 receptor results in increased Ca2+ release from the ER which contributes to the reduced pro-inflammatory capacity of M1-like macrophages. The inhibition of inflammation by the released Ca2+ primarily from decreased NFκB expression and increased cleavage of IL-1β.
Fructose Metabolism and Leukemia
Acute myeloid leukemia (AML) progression has been shown to be affected by excess fructose consumption. The level of expression of GLUT5, the major fructose transporter, is elevated in AML, compared to normal myeloid progenitor cells. In addition, elevated levels of GLUT5 expression are associated with poor prognosis and negative outcomes in patients with AML. Like many other types of cancer, AML metabolize fructose via HK2-mediated phosphorylation as opposed to via ketohexokinase (KHK)-mediated phosphorylation.
In AML cells, increased fructose leads to increased synthesis of serine. This is significant given that the serine synthesis pathway contributes to the generation of 2-oxoglutarate (α-ketoglutarate) from glutamate via the actions of phosphoserine aminotransferase 1 (PSAT1). The 2-oxoglutarate can be metabolized via the TCA cycle as well as serving as a critical cofactor for numerous dioxygenases that regulate DNA and histone methylation status.
The metabolism of fructose into the serine synthesis pathway, and subsequently the TCA cycle, ultimately results in the synthesis of precursors for nucleotide synthesis as well as the antioxidant protein, glutathione.
Hypoxia-Induced Factors and Metabolism in Cancer
As described in the previous section, altered metabolism of glucose is a hallmark of all types of cancer. The diversion of glucose carbons into biomass in cancer cells necessitates an increased delivery of glucose into these cells and an increase in the rate of anaerobic glycolysis to lactate. This is accomplished by an increase in the expression of genes encoding glucose transporters and glycolytic enzymes. These transcriptional changes can be observed in over 70% of human cancers and is driven in part through activation of the hypoxia induced factor 1 (HIF-1) pathway and by increased expression of various proto-oncogenes and decreased expression of various tumor suppressors.
Details of the hypoxia induced factor pathway are covered in the Hypoxia and Metabolism page.
Pentose Phosphate Pathway Genes and Cancer
6-Phosphogluconate Dehydrogenase
The third enzyme of the pentose phosphate pathway, 6-phosphogluconate dehydrogenase (6PGD), is encoded by the phosphogluconate dehydrogenase gene (symbol: PGD). The PGD gene is located on chromosome 1p36.33 and is composed of 13 exons that generate several alternatively spliced mRNAs that encode three different sized isoforms of 6-phosphogluconate dehydrogenase. Enhanced expression of the PGD gene has been reported in numerous different human cancers such as colorectal carcinomas, breast and ovarian cancers, cervical cancers, hepatocellular carcinomas, thyroid cancer, lung cancers, and leukemias.
Altered expression of the PGD gene and activity of the encoded 6PGD enzyme is mediated via both post-transcriptional and post-translational mechanisms. These alterations are the result of the activities of YTH N6-methyladenosine RNA binding protein 2 (encoded by the YTHDF2 gene), nuclear factor erythroid 2-related factor 2 (NRF2; encoded by the NFE2L2 gene), and epidermal growth factor receptor (EGFR) signaling. In addition, 6PGD activity is modified by direct protein-protein interactions with malic enzyme 1 (ME1).
Methylation of eukaryotic mRNA is a relatively recently identified modification that involves a family of methylating enzymes (termed writers), methylated mRNA-binding proteins (termed readers), and mRNA demethylases (termed erasers). The details of mRNA methylation are discussed in the RNA: Transcription and Processing page.
The methylation of mRNA has the potential to affect most of the post-transcriptional steps in the processes of regulated gene expression. Indeed, evidence has demonstrated that nuclear mRNA methylation readers are involved in the control of mRNA stability and splicing, and micro-RNA (miRNA) processing, whereas cytoplasmic readers are known to be involved in the regulation of mRNA translation.
Formation of N6-methyladenosine (m6A or 6mA) is one of the major mRNA methylation processes used to regulate gene expression. The YTHDF2 encoded protein is a m6A reader protein and as a result of its binding, it affects mRNA stability as well as mRNA translational potential. YTHDF2 has been shown to be an important promoter of 6PGD mRNA translation by directly binding to 6mA modification sites in the mRNA. The consequences of YTHDF2 activation of 6PGD translation, with respect to lung cancer, are promotion of cancer growth and an increased potential for metastasis.
Overexpression of the epidermal growth factor receptor (EGFR) gene has been identified in up to 50% of human cancers. Recent evidence has shown that the activation of the EGFR signal transduction cascade leads to tyrosine phosphorylation of 6PGD by FYN, a SRC family tyrosine kinase. Tyrosine phosphorylation of 6PGD results in upregulated binding of NADP+ and a consequent increase in the production of NADPH and ribose-5-phosphate. NADPH is critical for biomass production in cancer cells as well as being important in the detoxification of reactive oxygen species, ROS (see the Pentose Phosphate Pathway page). The increased ribose-5-phosphate enhances nucleotide biosynthesis which subsequently enhances DNA synthesis.
Acetylation of lysine residues in metabolic enzymes is associated with altered catalytic activity. Acetylation of 6PGD has been identified in numerous human cancer cells. Acetylation of 6PGD at lysine 76 (K76) and lysine 294 (K294) promotes the formation of active dimers of 6PGD. Two metabolic enzymes, acetyl-CoA acetyltransferase 2 (encoded by the ACAT2 gene) and dihydrolipoamide S-acetyltransferase (encoded by the DLAT gene), have been shown to promote the formation of K76 and K294 in 6PGD. ACAT2 is involved in cholesterol biosynthesis and in the utilization of ketone bodies by the brain, cardiac muscle, and several other tissues. Dihydrolipoamide S-acetyltransferase is a subunit of the pyruvate dehydrogenase complex and as such is involved in the production of acetyl-CoA which can then be utilized as the substrate for lysine acetyltransferases (KAT), the enzymes that acetylate proteins. Inhibition of the deacetylation of 6PGD has been shown to enhance tumor progression most likely as a result of the enhanced metabolic advantage of increased 6PGD activity.
The malic enzyme encoded by the ME1 gene is an NADP+-dependent enzyme that functions as a major contributor to the transfer of acetyl-CoA from the mitochondria to the cytosol where it can serve as a substrate for fatty acid and cholesterol biosynthesis as well as cytosolic protein acetylation. Increased expression of the ME1 gene, in several cancers, has been shown to be associated with increased activity of 6PGD and overall elevated activity of the pentose phosphate pathway. Increased levels of ME1 activity are associated with hetero-oligomerization with 6PGD which enhances the activity of 6PGD. ME1-mediated activation of 6PGD results in enhanced NADP+ binding and NADPH production. Like the consequences for tumor promotion from 6PGD acetylation, interaction of 6PGD with ME1 leads to enhanced production of NADPH and ribose-5-phosphate, in turn resulting in enhanced biomass production and DNA synthesis in cancer cells.
NRF2 (nuclear factor erythroid 2-related factor 2) is a master transcriptional regulator whose overexpression has been shown to be responsible for poor prognosis in numerous cancers. NRF2 transcriptionally activates the PGD gene resulting in increased levels of 6PGD. The PGD gene contains promoter elements termed antioxidant response elements (ARE) to which NRF2 binds. All human cancer cells express the PKM2 isoform of pyruvate kinase (for details on PKM2 function see the Glucose Metabolism and the Warburg Effect section above and go to the Glycolysis and the Regulation of Blood Glucose page) which, in addition to its metabolic function, acts as a transcriptional co-activator. One of the transcriptional activities of PKM2 is to activate the transcription factor, STAT3 (signal transducer and activator of transcription 3). STAT3 and NRF2 have been shown to interact and this interaction is associated with enhanced PGD expression. The interaction between metabolic enzymes and the transcriptional machinery is likely to be critical in the development of cancer.
Malic Enzyme and Enhanced Mitochondrial Biogenesis in Cancer
Humans express three malic enzymes, one cytoplasmic that requires NADP+ and two mitochondrial enzymes, one that requires NADP+ and one that requires NAD+.
The NADP+-dependent cytoplasmic enzyme is called malic enzyme 1 and is encoded by the ME1 gene. The highest level of ME1 expression is in adipose tissue attesting to the major role of the ME1 enzyme in fatty acid synthesis. The NADPH produced by the ME1 catalyzed reaction can be used in biomass producing reductive biosynthetic reactions such as fatty synthesis, cholesterol synthesis, and phospholipid synthesis.
The NAD+-dependent mitochondrial enzyme is called malic enzyme 2 and is encoded by the ME2 gene. The NADP+-dependent mitochondrial enzyme is called malic enzyme 3 and is encoded by the ME3 gene.
The role of the mitochondrial malic enzymes is principally to provide the cell with an alternate source of pyruvate under conditions where glycolytic flux in reduced. In these circumstances, the pyruvate generated by the actions of ME2 and/or ME3 comes from fumarate precursors such as glutamine. However, recent evidence has demonstrated that ME2 activity is critically involved in mitochondrial biogenesis and this ME2 function can contribute to the rapid generation of ATP required for cancer cell proliferation. Indeed, under experimental conditions where the level of ME2 is reduced there is a concomitant decrease in the level of mitochondrial biogenesis and respiration. The effects of ME2 on mitochondrial biogenesis are not shared by the other mitochondrial malic enzyme, ME3.
The role of ME2 in cancer cell proliferation through increased mitochondrial biogenesis requires its interaction with fumarate. Fumarate functions as an allosteric activator of ME2 activity by stimulating dimerization of the enzyme. When fumarate induced ME2 dimers are formed there is increased production of mitochondrial nucleotides that are required for mitochondrial DNA (mtDNA) replication. Fumarate-induced ME2 dimerization results in the interaction of these dimers with two enzymes of mitochondrial nucleotide biosynthesis, deoxyuridine triphosphatase (DUT) and dCTP pyrophosphatase 1 (DCTPP1). DUT converts dUTP to dUMP and enables dTTP production. DCTPP1 converts dCTP to dCMP and inorganic pyrophosphate.
In addition to the role of fumarate-induced ME2 dimerization in mtDNA replication, there is a function in mitochondrial protein synthesis. ME2 dimers interact with mitochondrial ribosome recycling factor (MRRF) as well as mitochondrial ribosomal protein L45 (MRPL45). The function of MRRF is to induce dissociation of the mitochondrial ribosomes (mitoribosomes) from the mRNA at the termination of translation.. MRPL45 anchors the mitoribosomes to the inner mitochondrial membrane.
The ability of ME2 to contribute to mitochondrial biogenesis is not only regulated by its interaction with fumarate but also by its state of methylation, specifically methylation of arginine 67 (R67). The arginine methyltransferase responsible for the R67 methylation is encoded by the PRMT1 gene. When ME2 is methylated by PRMT1 its ability to dimerize is reduced and there is a concomitant decrease in the level of mtDNA replication and mitochondrial protein synthesis. The clinical significance of the PRMT1-mediated regulation of ME2 dimerization is evident in numerous leukemias where the level of PRMT1 expression is reduced and ME2 is hypomethylated. In these cancer cells fumarate-induced dimerization and mitochondrial biogenesis is enhanced promoting their proliferation.
TCA Cycle Activity in Cancer Versus Normal Cells
Evidence has demonstrated that the level of TCA cycle activity is lower in cells from solid tumors when compared to cells from the surrounding normal tissue. This appears paradoxical since tumor cells proliferate rapidly and the TCA cycle the metabolic pathway with the greatest potential to support the synthesis of ATP. One mechanism that can account for low TCA cycle activity is increased use of glycolysis to generate ATP. Indeed, glucose flux is higher in tumors than in corresponding healthy tissues. This is to be expected given the well documented and studied Warburg effect. Nonetheless, it has been found that solid tumors produce ATP more slowly when compared with the corresponding healthy tissue. These observations imply that that tumors are not, in fact, hypermetabolic.
Tumor cells can generate less ATP that normal cells and still be proliferative because of the fact that the cells have altered biochemical and physiological functions relative to the otherwise healthy normal cells of the same tissue. Many proteins that are required for the normal functions of normal cells are likely to be lost and unnecessary in tumor cells thus allowing for conservation of energy consumption and this alone could account for a reduced need for ATP synthesis.
When the pattern of gene expression has been examined in tumor cells, and compared with that of normal cells, it has been found that numerous genes, for example those encoding proteins of ATP costly metabolic pathways, are expressed at lower levels than in normal cells. Data such as this suggests that solid tumors selectively downregulate ATP-expensive functions that allows for a sparing of ATP that can then be directed to support proliferation.
TCA Cycle Alterations in Cancer Metabolism
At least three of the genes that encode TCA cycle enzymes (or homologues of TCA cycle enzymes), or protein subunits of enzyme complexes of the TCA cycle, have been shown to harbor mutations in certain types of familial cancer syndromes. These genes include all four of the genes encoding subunits of succinate dehydrogenase, the fumarate hydratase (fumarase) gene, and two of the isocitrate dehydrogenase family member genes (IDH1 and IDH2).
Isocitrate Dehydrogenase Mutations
Both IDH1 and IDH2 enzymes utilize NADP+ as their cofactor and generate NADPH via oxidative decarboxylation of isocitrate outside the context of the TCA cycle. The IDH1 enzyme is localized to the cytosol while the IDH2 enzyme is localized to the mitochondria. The primary functions of IDH1 and IDH2 are to serve as producers of NADPH to enhance the cellular responses to oxidative stress and the generation of reactive oxygen species, ROS. The principal sources of NADPH are from the glucose-6-phosphate dehydrogenase (G6PD) reaction in the pentose phosphate pathway and via malic enzyme which is involved in the mobilization of mitochondrial acetyl-CoA to the cytosol. The significance of IDH1 and IDH2 produced NADPH can be made clear by pointing out that cells with low level IDH1 and IDH2 expression are more sensitive to oxidative stress than cells with higher levels of expression of these two enzymes.
Clinical significance is associated with the IDH1 and IDH2 genes as there has been a close association made between mutations in these two genes and the development of certain forms of cancer, specifically gliomas of the brain. All of the mutations in IDH1 that are associated with gliomagenesis are missense mutations that alter the normal Arg residue at position 132 to a histidine (R132H). The mutations in IDH2 are also single amino acid missense mutations at the comparable Arg residue at position 172 and are predominantly Lys substitutions (R172K). In some cases of IDH2 mutations another Arg residues is mutated to Glu (R140Q). The characterized mutations in IDH1 and IDH2 that are associated with the early onset of gliomagenesis result in altered activities of the encoded proteins.
Although associated with gliomagenesis, gliomas that contain mutant IDH1 or IDH2 genes are associated with a better prognosis than gliomas with wild-type IDH1 or IDH2.
In addition to gliomas, the same IDH1 missense mutation is found in acute myelogenous leukemia, cholangiocarcinoma, cartilaginous tumors, prostate cancer, papillary breast carcinoma, acute lymphoblastic leukemia, angioimmunoblastic T-cell lymphoma, and primary myelofibrosis.
2-Hydroxyglutarate (2-HG) is an Oncometabolite
Normally IDH1 (cytosolic) and IDH2 (mitochondrial) convert isocitrate to 2-oxoglutarate (α-ketoglutarate) while also generating NADPH. The R132H mutation in IDH1 and the R172K mutation in IDH2 result in a loss of isocitrate binding while at the same time altering the catalytic activity of the enzymes such that they reduce 2-oxoglutarate (α-ketoglutarate), their normal product, to 2-hydroxyglutarate (2-HG). Indeed, in gliomas with IDH1 mutations the levels of 2-HG are elevated relative to normal cells.
When IDH1 and IDH2 reduce 2-oxoglutarate to 2-HG there is a simultaneous oxidation of NADPH to NAPD+. The reductions in NADPH levels will lead to cells being more susceptible to the effects of oxidative stress, primarily as a result of decreased levels of reduced glutathione.
2-Hydroxyglutarate exists in two stereoisomeric configurations, D-2-hydroxyglutarate (D-2-HG) and L-2-hydroxyglutarate (L-2-HG). The normal pathways for the synthesis of 2-HG utilize 2-oxoglutarate (α-ketoglutarate) or gamma(γ)-hydroxybutyrate (GHB; also identified as 4-hydroxybutyrate) as the substrate.
GHB is a normal catabolic byproduct of the inhibitory neurotransmitter, γ-aminobutyrate, GABA. GABA is transported into the mitochondria where it is metabolized to succinic semialdehyde by GABA transaminase [encoded by the ABAT (4-aminobutyrate aminotransferase) gene]. Succinic semialdehyde can be converted to succinate and to GHB by succinic semialdehyde dehydrogenase (SSADH). The SSADH enzyme is encoded by the ALDH5A1 (aldehyde dehydrogenase 5 family member A1) gene. The metabolism of GABA to succinate allows the succinate to be converted to 2-oxoglutarate which is then converted to glutamate which can then be transported out of the mitochondria and utilized for the synthesis of GABA. These reactions constitute what is referred to as the GABA shunt.
Under normal conditions, mitochondrial malate dehydrogenase (MDH2) converts 2-oxoglutarate to L-2-HG. Cytosolic malate dehydrogenase (MDH1) converts 2-oxoglutarate to D-2-HG. Within the cytosol the LDHA encoded lactate dehydrogenase can also convert 2-oxoglutarate to L-2-HG. The rate-limiting enzyme of serine biosynthesis, phosphoglycerate dehydrogenase (encoded by the PHGDH gene), can also convert cytosolic 2-oxoglutarate to D-2-HG and to L-2-HG.
Alcohol dehydrogenase iron containing 1 (encoded by the ADHFE1 gene) converts GHB to 2-HG. The ADHFE1 encoded enzyme is also known as hydroxyacid-oxoacid transhydrogenase (HOT).
The catabolism of 2-HG is catalyzed by stereospecific dehydrogenases localized to the mitochondria, D-2-hydroxyglutarate dehydrogenase (encoded by the D2HGDH gene) and L-2-hydroxyglutarate dehydrogenase (encoded by the L2HGDH gene). Both dehydrogenases are capable of catalyzing the reverse direct and so can contribute to production of D-2-HG (D2HGDH reaction) or L-2-HG (L2HGDH reaction). Mutations in either the D2HGDH gene or the L2HGDH gene, or both genes, are the cause of an organic aciduria referred to as 2-hydroxyglutaric aciduria. Gain-of-function mutations in the IDH2 gene also result a form of 2-hydroxyglutaric aciduria. This disorder is termed D-2-hydroxyglutaric aciduria, type 2 (D2HGA2).
Mutations in IDH2 are much less common than mutations in IDH1 and the appearance of the mutations in these two genes are mutually exclusive meaning that a glioma with mutated IDH1 does not also harbor a mutant IDH2 gene and vice versa. Mutant forms of IDH2 are capable of generating 2-HG from 2-oxoglutarate (α-ketoglutarate) just as is the case for mutant forms of IDH1.
Increases in 2-HG levels, coupled with decreases in 2-oxoglutarate levels, results in competitive inhibition of several important 2-oxoglutarate and Fe2+-dependent dioxygenases (2OG-oxygenases) such as DNA demethylases and histone demethylases that are involved in regulating chromatin structure and regulating gene expression. Both D-2-HG and L-2-HG are capable of inhibition of 2OG-oxygenases. Due to its effects as a competitive inhibitor of these 2OG-oxygenases, 2-HG is referred to as an oncometabolite.
In addition, inhibition of prolyl hydroxylase domain (PDH) containing enzymes, such as those that are involved in the regulation of the hypoxia-inducible factor (HIF) signaling pathways, also results from increased 2-HG levels.
Studies of multiple different types of cancer have demonstrated the key roles that 2-HG plays in the tumor cells themselves as well as in the tumor microenvironment. 2-HG has been shown to stimulate angiogenesis (new vessels) in solid tumors and also to suppress the antitumor activity of T-cells. The latter effect results in immune system evasion by tumor cells allowing them to grown and proliferate.
When 2-HG is taken up by T-cells it leads to inhibition of mitochondrial ATP synthesis via inhibition of the ATP synthase subunit, ATP synthase F1 subunit beta (ATP5B) which is encoded by the ATP5F1B gene. Decreased ATP production results in the activation of AMPK and a reduction in Ca2+ signaling. Reduced Ca2+ signaling impairs the activity of the transcription factor NFAT (nuclear factor of activated T cells) which in turn reduces the expression of NFAT target genes, many of which are responsible for the anti-tumor functions of T-cell.
Additional effects of 2-HG in T-cells that reduce their ability to respond appropriately include inhibition of ornithine decarboxylase (ODC), the rate -limiting enzyme of polyamine biosynthesis, and reduction in phosphorylation of phospholipase C-gamma (PLC-γ). Reduced levels of polyamines results in impaired DNA replication which impairs T-cell proliferation. Reduced levels of PLC-γ phosphorylation contributes to the impairment in nuclear translocation of NFAT.
In addition to the effects of 2-HG at the level of increased tumor cells growth via enhanced angiogenesis as well as immune evasion, 2-HG inhibits several enzymes involved in DNA damage repair. Many of these enzymes are members of the 2OG-oxygenase family. Reduced DNA damage repair will lead to increased mutations as tumor cells proliferate which can contribute to the acquisition of new mutations that may be beneficial for cancer proliferation.
Two of the DNA repair enzymes that have been shown to be inhibited by 2-HG are ALKBH2 and ALKBH3 (members of the AlkB homolog family). ALKBH2 and ALKBH3 function to demethylate N1-methyladenosine (1mA) and N3-methylcytosine (3mC) residues in DNA. The ALKBH2 protein is the primary enzyme responsible for demethylation repair of alkylated DNA. The ALKBH3 enzyme is also involved the dealkylation of RNA.
Several lysine demethylases of the KDM family, that are also members of the 2OG-oxygenase family, have also been found to be inhibited by 2-HG and this effect contributes to faulty DNA double-strand break (DSB) repair. These demethylases include KDM4A and KDM4B. The inhibition of KDM4B results in altered histone H3 lysine 9 (H3K9) methylation at sites of DNA breakage where the trimethylated form of H3K9 (H3K9me3) is normally found.
Additional mitochondrial enzymes whose activities are inhibited by 2-HG include succinate dehydrogenase and cytochrome c oxidase (COX). Succinate dehydrogenase (SDH) is a complex constituting one of the reactions of the TCA cycle as well as being complex II of oxidative phosphorylation. Loss of SDH activity impairs overall mitochondrial function leading to mitochondrial stress and the recruitment of the anti-apoptotic protein, BCL-2, to the mitochondrial membrane preventing the release of cytochrome c which is the normal activation process for mitochondrially-induced apoptosis. The inhibition of cytochrome c oxidase, which is complex IV of oxidative phosphorylation, also impairs mitochondrial activity resulting in mitochondrial stress.
Succinate Dehydrogenase Mutations
The succinate dehydrogenase (SDH) complex of the TCA cycle (also known as complex II of oxidative phosphorylation) is composed of four subunits encoded by four distinct genes, SDHA, SDHB, SDHC, and SDHD. Mutations in all four of these genes are associated with several familial cancer syndromes. In addition to the four functional components of the SDH complex, mutations in the SDH assembly factor gene, SDHAF2, are also found in certain forms of cancers. Although mutations in the SDH subunit assembly factor gene, SDHAF1, have not been associated with cancer syndromes, they are associated with mitochondrial deficiency syndromes. The SDH complex gene associated cancers include paragangliomas, pheochromocytomas, gastrointestinal stromal tumors (GIST), and an aggressive form of renal cancer identified as SDH-RCC (SDH-deficient renal cell carcinoma).
Mutations in the genes encoding the four proteins comprising the SDH complex result in a pathophysiological state defined as pseudo-hypoxia. This pathophysiologic state leads to a subsequent increase in angiogenesis, via activation of the HIF-1 hypoxia response pathway, resulting in the ability of tumors to acquire nutrients encouraging their aberrant growth. Activation of the HIF-1 pathway in SDH-deficient tumors is most likely the result of accumulating levels of succinate which is then transported into the cytosol via the action of the mitochondrial dicarboxylate transporter.
Accumulation of cytosolic succinate inhibits the 2-oxoglutarate and Fe2+-dependent dioxygenases [prolyl hydroxylase domain (PDH) enzymes] responsible for the hydroxylation of the α-subunit (HIF-1α) of HIF-1. For this reason succinate is considered an oncometabolite as is 2-hydroxyglutarate.
The hydroxylation of HIF-1α normally occurs continuously under normoxic conditions which stimulates ubiquitylation and proteasome degradation of HIF-1α, thereby restricting the transcription factor function of HIF-1. The loss of HIF-1α hydroxylation results stabilization of the protein and consequent activation of the HIF-1 pathway in the absence of hypoxia. Activation of HIF-1 in the absence of hypoxia leads to increased angiogenesis within the SDH-deficient tumor environment promoting the growth of the tumor cells. Because loss of function of any one of the four subunits of the SDH complex results in the progression to cancer, all four genes encoding proteins of the complex (SDHA, SDHB, SDHC, and SDHD) have been classified as tumor suppressors.
Gastrointestinal stromal tumors (GIST) are the most common mesenchymal tumor of the gastrointestinal tract. Until very recently the vast majority of GIST were associated with mutations resulting in the constitutive activation of the KIT gene encoded tyrosine kinase (75% to 80%). Additional activating mutations, associated with GIST, although not as common as the KIT mutations, are those in the PDGF receptor A (PDGFRA: 5% to 8%) gene. Less than 10 years ago a new class of GIST was identified that is due to mutations in several of the genes encoding protein components of the SDH complex. The initially characterized SDH-deficient GIST were found to be associated with mutations in the SDHB, SDHC, and SDHD genes. More recently SDHA deficient GIST have been characterized. The SDH-deficient GIST are wild type for both the KIT and PDGFRA genes. The SDH-deficient tumors occur exclusively in the stomach and commonly metastasize to lymph nodes. These SDH-deficient tumors are also characterized by having a unique epithelioid morphology often with a plexiform and multinodular growth pattern and many of the tumors also overexpress the insulin-like growth factor 1 receptor (IGFR1) gene. Since their initial characterization, the SDH-deficient GISTs have been shown to account for up to 7.5% of these forms of cancer in adult patients and represent the vast majority of these types of tumors that are found in childhood.
Mutations in all four SDH complex genes, as well as the SDHAF2 gene, are associated with additional forms of cancer. These additional forms are defined as the hereditary paraganglioma and pheochromocytoma syndrome (HPGL/PCC). Paragangliomas (PGL) are rare tumors derived from the paraganglia. The paraganglia are neuroendocrine tissues that are symmetrically distributed along the paravertebral axis from the base of the skull and the neck to the pelvis. In the adult the major paraganglionic organs are the carotid body and the adrenal medulla. The carotid body represents a small cluster of chemoreceptors and associated supporting cells that runs along the bottom of the bifurcation of the carotid artery. The primary function of the carotid body is to detect changes in the partial pressures of O2 and CO2 within the arterial blood, but is also involved in the sensation of pH and temperature.
Chromaffin cells of the adrenal medulla secrete the catecholamines, epinephrine and norepinephrine, in response to stress-mediated activation of preganglionic sympathetic neurons. Paragangliomas are primarily found in the head and neck region, particularly in the carotid bodies. Paragangliomas arising from chromaffin cells of the adrenal medulla are defined as pheochromocytomas and they characteristically hypersecrete catecholamines. The paraganglioma-pheochromocytoma syndrome (HPGL/PCC) was originally identified as being caused by mutations in the SDHB, SDHC, and SDHD genes and later to be associated with SDHA and SDHAF2 gene mutations.
Fumarate Hydratase Mutations
The fumarate hydratase gene (symbol: FH) is located on chromosome 1q43 and is composed of 10 exons that encode two distinct mRNAs as a result of alternative transcriptional initiation directed by a broad promoter region. It was originally thought the the two forms of human fumarate hydratase resulted from a single FH mRNA as a result of alternative translational start site utilization as is the case for the rat fumarate hydratase forms.
One form of the enzyme is localized to the cytosol while the TCA cycle enzyme is localized to the mitochondria. The mitochondrial version of fumarate hydratase (a 510 amino acid protein) is extended at the N-terminus, relative to the cytosolic form of the enzyme. The N-terminal extension contains a mitochondrial targeting sequence. The cytosolic fumarate hydratase is 43 amino acids shorter than the mitochondrial version and this shorter FH protein lacks the mitochondrial targeting sequence. The cytosolic form of FH is involved in a process of cellular protection from DNA double strand breaks as a part of the DNA damage response pathway. In response to DNA damage the cytosolic FH migrates into the nucleus where its activity is required to inhibit histone demethylases, the effect of which is enhanced DNA repair.
Mutations in the fumarate hydratase (FH: also called fumarase) gene have been shown to be associated with hereditary leiomyomatosis and renal cell carcinoma (HLRCC). Leiomyoma is a form of smooth muscle tumor that can develop in any organ but is most commonly found in the uterus, esophagus, and small intestine. Uterine fibroids are a common form of leiomyoma of the uterine smooth muscle that are benign. HLRCC is an autosomal dominant hereditary cancer syndrome that is characterized by a predisposition to the development of cutaneous and uterine leiomyomas and a very aggressive form of papillary kidney cancer. HLRCC-associated renal tumors readily metastasize to both regional and distant lymph nodes making this form of renal cancer significantly more aggressive than other forms of genetically defined renal cancer.
Mutations resulting in loss of function of fumarate hydratase are the primary genetic alterations associated with HLRCC. The FH mutations found in HLRCC are missense, frameshift, partial deletions, or complete deletions. Because loss of function of the FH gene results in the progression to cancer, the gene has been classified as a tumor suppressor.
Similar to the alterations that occur in the metabolic programs in SDH mutation containing cancers, FH mutation cancer cells exhibit a substantial metabolic reorganization. Fumarate hydratase mutant tumor cells satisfy their need for high rates of ATP production by developing high rates of glucose uptake and higher rates of glucose oxidation via glycolysis. This form of metabolic reprogramming in cancer cells (as well as that occurring in normal rapidly proliferating cells) is referred to as the Warburg effect. The reduced entry of glycolytic intermediates into the TCA cycle, due to FH deficiency, results in diversion of glucose to lactate as well as into other metabolic pathways of biomass production.
All of these shifts in metabolism are not unique to FH-mutant tumors but are a common occurrence in most tumors. As indicated in the Hypoxia and Metabolism page and in the section above on SDH-mutant tumors, the common changes in metabolic profiles in cancer cells is, in part, the result of the activation of the hypoxia induced factor 1 (HIF-1) transcriptional program.
When fumarate accumulates in the mitochondria of FH-mutant cells the metabolite is transported to the cytosol by the dicarboxylate transporter. Within the cytosol the accumulating fumarate, similar to the effect of accumulating succinate, results in the inhibition of the family of 2-oxoglutarate and Fe2+-dependent dioxygenases (2OG-oxygenases). For this reason, fumarate, like 2-hydroxyglutarate and succinate, is considered an oncometabolite.
The prolyl hydroxylase domain (PHD) members of this family of enzymes are responsible for the hydroxylation of the α-subunit (HIF1α) of the HIF-1 complex. The hydroxylation of HIF1α presents binding sites for the VHL ubiquitin ligase which then ubiquitylates HIF1α resulting in its proteasome degradation. Inhibition of the PDH enzymes by fumarate results in loss of HIF1α turnover which then allows the HIF-1 complex to migrate to the nucleus where a program of genes are activated that are normally responsible for cell survival in response to hypoxia. One of the major responses to HIF-1 transcriptional activity is increased angiogenesis allowing tumor cells higher rates of access to biomolecules for growth. Additional metabolic changes in FH-deficient tumors have been shown to be the result of reduced activation and reduced expression of the master metabolic regulatory kinase, AMPK. The loss of AMPK activity results in increased rates of lipid and protein synthesis in the tumor cells.
Fumarate is a normal byproduct of the urea cycle and of the purine nucleotide cycle. When FH is deficient, resulting in the accumulation of fumarate in the cytosol, the normal equilibrium of these two processes is disturbed.
The accumulation of cytosolic fumarate alters the argininosuccinate lyase (ASL) catalyzed reaction of the urea cycle causing it to proceed in the reverse direction resulting in the accumulation of argininosuccinate. Under these conditions cells become dependent on arginine for survival.
The increased fumarate also alters the equilibrium of the adenylosuccinate lyase (ADSL) reaction of the purine nucleotide cycle causing it to proceed in the reverse direction resulting in increased levels of succinylaminoimidazole carboxamide riboside (SAICAr) and adenylosuccinate. SAICAr is the dephosphorylated form of SAICAR (succinylaminoimidazole carboxamide ribotide), an intermediate in de novo purine nucleotide biosynthesis. Increased adenylosuccinate results in increased production of the toxic metabolite succinyladenosine (S-Ado). The result of altered ADSL activity causes cells to become dependent on purine salvage pathways since the de novo pathway is blocked.
Accumulation of fumarate also results in increased post-translational modification of cysteine residues in proteins via a spontaneous reaction referred to as a “Michael addition”. The spontaneous addition of fumarate to cysteine residues in proteins, as well as the cysteine in glutathione (GSH), is referred to as succination, not to be confused with protein lysine succinylation which involves succinyl-CoA. During the addition, the double bond of fumarate is reduced forming succinate which is why the process is termed succination. The resulting residue in proteins and GSH is termed 2-(succino)cysteine (2SC).
Critical proteins that have been found to undergo succination, thus altering their activities, in FH deficient cancers are nuclear factor erythroid 2-related factor 2 (NRF2), phosphatase and tensin homolog (PTEN), and SWI/SNF-related, matrix-associated, actin-dependent regulator of chromatin subfamily C member 1 (SMARCC1). NRF2 is a transcription factor, PTEN is a phosphatidylinositol-3-phosphate phosphatase, and SMARCC1 is a core component of what is identified as the SWI/SNF chromatin remodeling complex. SWI/SNF refers to switching of mating type/sucrose non-fermenting, a chromatin remodeling complex originally identified in yeast. NRF2 is a transcription factor and PTEN is a phosphatidylinositol-3-phosphate phosphatase.
Metabolic enzymes have also been found to be subject to succination under conditions of excess cytosolic fumarate. Both glyceraldehyde-3-phosphate dehydrogenase (GAPDH) of glycolysis and glucose-6-phosphate dehydrogenase (G6PDH) of the pentose phosphate pathway (PPP) are subject to succination. The effect of this post-translational modification is increased glycolytic flux and increase PPP activity. Both of these effects are beneficial to rapidly proliferating cells, such as cancer cells.
Recent (2024) evidence has discovered that the urea cycle enzyme, argininosuccinate synthetase, encoded by the ASS1 gene, plays a role in the tumor suppressor p53-mediated responses to DNA damage. The loss of expression of the ASS1 gene has been correlated to increased DNA damage in colorectal carcinoma cells as well as in fibroblasts in patients with ASS1 deficiency (known as citrullinemia type 1, CTLN1). When DNA damage is sensed and p53 is activated to migrate to the nucleus, there is a concomitant increase in both argininosuccinate synthetase and argininosuccinate lyase localization to the nucleus. Within the nucleus these two enzymes increase the level of fumarate which in turn enhances the succination of the SMARCC1 protein. The consequences of the succination of SMARCC1 is a decrease in the level of expression of p53 target genes and the cessation of cell cycle progression.
The importance of argininosuccinate synthetase to the suppression of cancer progression is evidenced by the fact that ASS1 promoter hypermethylation, and thus transcriptional silencing, has been shown to occur in numerous types of cancer.
The Role of the TCA Cycle in Immune Modulation
The TCA cycle plays a central metabolic role in all cells, excluding red blood cells, and is of particular significance in cells of the immune system. Within activated macrophages there are distinct changes in TCA cycle activity that lead to the accumulation of TCA cycle intermediates, particularly fumarate and itaconate. These TCA cycle changes serve to regulate metabolic processes, immune system functions, cancer, and other diseases.
Fumarate Links Metabolism, Immune System Function, and Cancer
The major source of fumarate is the TCA cycle but in addition fumarate is generated in the context of the urea cycle, the biosynthesis of the purine nucleotides, and in the catabolism of the amino acids phenylalanine and tyrosine. Within the context of the urea cycle fumarate is generated by the actions of argininosuccinate lyase in the conversion of argininosuccinate to arginine. Within the context of purine nucleotide biosynthesis, fumarate is generated by the actions of adenylosuccinate lyase in the conversion of succinylaminoimidazole carboxamide ribotide (SAICAR) to 5-aminoimidazole-4-carboxamide ribotide (AICAR) and in the conversion of adenylosuccinate to AMP. Within the context of phenylalanine and tyrosine catabolism, fumarate is generated in the final reaction catalyzed by fumarylacetoacetate hydrolase which converts 4-fumarylacetoacetate to fumarate and acetoacetate.
As indicated in the Fumarate Hydratase Mutations section above, accumulation of fumarate in the cytosol leads to inhibition of 2-oxoglutarate and Fe2+-dependent dioxygenases and to the post-translational modification of numerous proteins. Fumarate is involved in these functions whether there are pre-existing mutations in the FH gene or not.
The ability of fumarate to non-enzymatically react with the sulfhydryl of cysteine residues in proteins, in a reaction referred to as “Michael addition”, generates a 2-(succino)cysteine (2SC) residue. This fumarate-mediated post-translational modification is referred to as succination.
The process of fumarate-mediated succination has been shown to occur on numerous proteins, resulting in modification of the activity of the modified proteins. The first protein shown to be modified by succination was glyceraldehyde-3-phosphate dehydrogenase (GAPDH). The succination of GAPDH leads to inhibition of its enzymatic activity. Although succination of GAPDH results in inhibition of its enzymatic activity, not all succination reactions result in inhibition.
Subsequent to the identification of GAPDH succination, numerous additional proteins, many being metabolic enzymes and anti-oxidant proteins, have been identified as undergoing succination. Other metabolic enzymes that are succinated include glucose-6-phosphate dehydrogenase and alcohol dehydrogenase. Anti-oxidant proteins that are succinated include KEAP1 (Kelch-like ECH-associated protein 1) and glutathione (GSH).
Numerous signaling proteins have been shown to be succinated, including PTEN (phosphatase and tensin homolog). PTEN is a phosphoinositide phosphatase, specifically a phosphatidylinositol-3-phosphate phosphatase. Succination of PTEN inhibits its activity which results in increased signaling via the PI3K pathway.
Succinate Modulation of Immune Functions
As discussed in Succinate Dehydrogenase Mutations section above, increased levels of succinate in the cytosol result in the inhibition of the PHD enzymes that hydroxylate, and thereby destabilize, the HIF-1α subunit of the HIF-1 transcription factor complex. When HIF-1α is not hydroxylated, and is thus not targeted for degradation by ubiquitylation, it migrates to the nucleus leading to an increase in the transcription of HIF-1 target genes. In response to inflammatory processes, macrophages increase the rate of glycolysis and this change is, in part, due to the stabilization of HIF-1α as a consequence of succinate.
Succinate also enhances immune functions via the activation of the succinate receptor encoded by the SUCNR1 gene. Upon binding to its receptor, succinate initiates a signaling cascade that includes the activation of PLCβ. Activation of PLCβ results in the hydrolysis of membrane phosphatidylinositol-4,5-bisphosphate (PIP2; PtdIns-4,5-P2) generating inositol-1,4,5-trisphosphate (IP3; Ins-1,4,5-P3) diacylglycerol (DAG). The generation of IP3 results in release of endoplasmic reticulum (ER) stored Ca2+ and the Ca2+, along with DAG, leads to activation of PKC.
The activation of PKC results in phosphorylation of downstream kinases leading to activation of the MAPK signaling pathway. In the context of immune responses, the MAPK pathway results ultimately, in the enhanced transcription of pro-inflammatory genes. Activated PKC also leads to enhanced transcriptional activity of the critical immune modulatory transcription factor, NFκB.
Itaconate Synthesis and Immune System Modulation
One metabolite that accumulates in activated macrophages, as a consequence of alterations in the TCA cycle, is itaconate. Itaconate is a dicarboxylic acid that is produced from the TCA cycle intermediate, cis-aconitate, by the enzyme aconitate decarboxylase 1 which is encoded by the ACOD1 gene. The ACOD1 gene was originally identified as immunoresponsive gene 1 (IRG1). In addition to intracellular effects, itaconate is secreted from activated macrophages, particularly within the tumor microenvironment, where it exerts numerous additional effects. Itaconate is a critical metabolite that contributes to the negative feedback suppression of inflammatory cytokines.
The ACOD1 gene is located on chromosome 13q22.3 and is composed of 5 exons that encode a protein of 481 amino acids. Expression of the ACOD1 gene is near exclusive to macrophages and myeloid cells.
Itaconate produced by macrophages has antimicrobial activity which is crucial in the killing of phagocytosed bacteria. In vacuoles of macrophages where phagocytosed bacteria are killed, itaconate inhibits the bacterial enzyme, isocitrate lyase (ICL), a bacterial enzyme required for the glyoxylate shunt that functions during host infection by bacteria. The itaconate metabolite, itaconyl-CoA, inhibits bacterial methylmalonyl-CoA mutase (MCM) which in turn blocks propionyl-CoA–dependent bacterial growth. Itaconyl-CoA has also been shown to be a competitive inhibitor of mammalian methylmalonyl-CoA mutase and of the erythroid-specific form of δ-aminolevulinic (5-aminolevulinic) acid synthase encoded by the ALAS2 gene.
When macrophages are stimulated, for example by ligand interaction with the toll-like receptor 4 (TLR4), a signal transduction cascade is activated leading to enhanced expression of the ACOD1 gene. In addition to activation of ACOD1, the TLR4 signal transduction cascade results in enhanced production of interferon-alpha and -beta (INFα and INFβ). These interferons activate interferon receptors whose signal transduction cascades contribute to the activation of ACOD1 expression.
The increased levels of aconitate decarboxylase 1 result in diversion of cis-aconitate away from the TCA cycle via its metabolism to itaconate. Within the mitochondria itaconate inhibits the succinate dehydrogenase (SDH) complex which results in reduced flow of carbons through the TCA cycle as well as inhibition of oxidative phosphorylation since TCA cycle SDH is also complex II of the electron transport chain.
The inhibition of SDH reduces the generation of reactive oxygen species (ROS) at complex I. Decreased ROS production in the mitochondria allows for prolyl hydroxylase domain (PHD) enzymes to hydroxylate HIF1α. The prolyl hydroxylases that hydroxylate HIF α-subunits (HIF1α and HIF2α) are all 2-oxoglutarate (α-ketoglutarate) and Fe2+-dependent enzymes (2OG-oxygenases). Hydroxylation of HIF α-subunits renders the proteins susceptible to proteasomal degradation. The reduced levels of active HIF1α result in reduced transcription of genes encoding pro-inflammatory cytokines such as IL-1β. The net effect of itaconate at the level of SDH is anti-inflammatory.
Itaconate is transported to the cytosol where several enzymes of glucose metabolism are inhibited including aldolase A (encoded by the ALDOA gene), glyceraldehyde-3-phosphate dehydrogenase (encoded by the GAPDH gene), lactate dehydrogenase (encoded by the LDHA gene), enolase (encoded by the ENO1 gene), and the PKM2 isoform of pyruvate kinase (encoded by the PKM gene). The mechanism of itaconate-mediated inhibition of these enzymes is via alkylation of cysteine residues as well as by acylation of lysine residues. These forms of post-translational modification are collectively referred to as itaconylation (also referred to as itaconation).
Cytosolic itaconate has been shown to inhibit the NLRP3 inflammasome. NLRP3 refers to Nucleotide-binding oligomerization domain and Leucine-rich repeat-containing Receptor Protein 3. The NLRP3 inflammasome is a complex consists of a sensor (NLRP3 protein), an adaptor (apoptosis-associated speck-like protein, ASC), and an effector (caspase-1). Normally, one function of the NLRP3 inflammasome is to convert proinflammatory IL-1β (pro-IL-1β) to its active inflammatory state, IL-1β. The activity of the NLRP3 protein of the inflammasome is inhibited as a result of direct itaconation. Thus, inhibition of the NLRP3 inflammasome by itaconate represents an anti-inflammatory function of itaconate.
The NLRP3 inflammasome is also inhibited as a result of itaconate complexing with homocysteine forming homocysitaconate. Homocysteine is generated in the context of the methionine cycle which is the pathway by which methionine is utilized in the synthesis of S-adenosylmethionine (SAM or AdoMet) as well for diversion into cysteine biosynthesis.
During the process of protein synthesis, methionyl tRNA synthetase (MARS) catalyzes the conversion of homocysteine to homocysteine thiolactone. Homocysteine thiolactone is reactive towards lysine residues in a process termed N-homocysteinylation. The NLRP3 protein is one substrate for N-homocysteinylation, the consequences of which are enhanced inflammatory responses. The effect of homocysitaconate in this process is inhibition of the the MARS enzyme.
Thus, increased levels of itaconate repress inflammatory process mediated by the NLRP3 inflammasome by direct itaconation as well as through the inhibitory effects of homocystitaconate on the activity of the NLRP3 inflammasome.
Additional anti-inflammatory effects exerted by itaconate include activation of the basic region leucine zipper (bZIP) transcription factor identified as nuclear factor erythroid erythroid 2 (NEF2)-related factor 2 (NRF2). The human NRF2 protein is encoded by the NFE2L2 gene.
The transcriptional activity of NRF2 is regulated post-translationally by Kelch-like ECH-associated protein 1 (KEAP1) which mediates the degradation of NRF2. The activity of KEAP1 is inhibited by itaconylation. The itaconylation of KEAP1 allows NRF2 to activate genes whose encoded proteins serve antioxidant functions. The reduction in ROS production, as a result of NRF2 activity, contributes to the destabilization of HIF1α, furthering the anti-inflammatory activity of itaconate.
Two important targets of NRF2 are the HMOX1 (heme oxygenase 1) and NQO1 [NAD(P)H quinone oxidoreductase 1] genes. NRF2 also actively represses the transcription of pro-inflammatory genes in macrophages. Heme oxygenase 1 catalyzes the degradation of heme to free iron, biliverdin, and carbon monoxide (CO) thereby removing free heme which is pro-oxidant. The function of NQO1 is to catalyze the NADPH-dependent reduction of quinones to hydroquinones, thereby preventing the formation of highly reactive semiquinones and associated reactive oxygen species (ROS).
In addition to the effects of itaconate on macrophage functions, it is transported out of macrophages via the action of the ABCG2 encoded transporter. When exported from macrophages, itaconate limits the extent of anti-bacterial immunity.
Anti-Tumor Effects of Itaconate
When macrophage produced itaconate is transported to the tumor microenvironment, via the ABCG2 transporter, it is taken up by tumor cells. Within tumor cells, itaconate exerts anti-tumor effects predominantly as a result of the inhibition of the pentose phosphate pathway (PPP) enzyme glucose-6-phosphate dehydrogenase, G6PDH (encoded by the G6PD gene). Inhibition of G6PDH by itaconate occurs through direct binding of itaconate to the enzyme. As a consequence of the inhibition of G6PDH, tumor cells are less able to produce the NADPH necessary to handle the increased oxidative stress of the tumor microenvironment.
In addition, when produced by tumor cells, as opposed to macrophages or tumor infiltrating macrophages, itaconate enhances the presentation of tumor antigens which, in turn, enhances tumor cell recognition by T cells. In addition, the inhibition of G6PDH in tumor macrophages results in their conversion to anti-tumor macrophages.
Tumor Promoting Effects of Itaconate
Itaconate that is excreted from activated macrophages is transported into cells via the SLC13A3 encoded Na+-dicarboxylate cotransporter. In the context of tumor cells, the uptake of itaconate has been shown to result in the itaconylation of PD-L1 (programmed death ligand 1). The itaconylation of PD-L1 stabilizes the protein by preventing the protein from being ubiquitylated. Enhanced PD-L1 activity in tumor cells promotes immune evasion, thereby enhancing tumor cell proliferation.
Metabolism of Itaconate
Metabolism of itaconate generates acetyl-CoA and pyruvate. Succinyl coenzyme A synthetase converts itaconate to itaconyl-CoA. 3-Methylglutaconyl-CoA hydratase (encoded by the AUH gene) then converts itaconyl-CoA to citramalyl-CoA. Finally, citramalyl-CoA lyase (encoded by the CLYBL gene, also known as citrate lyase subunit beta-like ) converts citramlyl-CoA to acetyl-CoA and pyruvate.