
Last Updated: July 13, 2026
Introduction to Cori Disease
Glycogen storage disease type 3 (GSD3) is also known as Cori disease, Forbes disease, and limit dextrinosis. Cori disease is inherited as an autosomal recessive disorder. The symptoms associated with Cori disease were first described in 1952 by Barbara Illingworth and Gerti T. Cori and was studied clinically by Gilbert B. Forbes hence the associated names for this disorder.
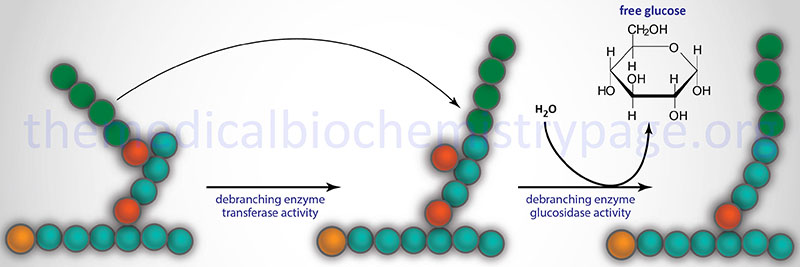
The disorder is associated with the accumulation of an abnormally structured glycogen having very short outer chains. This structure is similar to what would be seen in a phosphorylase-limit dextrin. The disorder was predicted, and later shown, to be the result of a deficiency of amylo-α-1,6-glucosidase, 4-α-glucanotransferase, also known more simply as the glycogen debranching enzyme (GDE).
Molecular Biology of Cori Disease
The amylo-α-1,6-glucosidase, 4-α-glucanotransferase enzyme is encoded by the AGL gene. The AGL gene is located on chromosome 1p21.2 spanning 85 kb and is composed of 37 exons that generate five alternatively spliced mRNAs. Four of these mRNAs encode the same 1532 amino acid protein identified as AGL isoform 1. AGL isoform 3 is 1516 amino acids. The AGL encoded protein has a predicted molecular mass of 173 kDa.
The AGL encoded enzyme has two distinct catalytic activities, amylo-α-1,6-glucosidase and 4-α-glucanotransferase. The two activities encompass distinct catalytic sites on the enzyme and can function independent of each other. However, both activities, as well as the binding of glycogen, are required for complete function. The AGL isoform 1 represents the major form of the enzyme and it results from mRNAs that begin transcription at exon 1 and initiate translation in exon 3. There are at least 3 muscle-specific isoforms which begin transcription at exon 2. There are two promoters for the AGL gene which direct the tissue-specific expression pattern of the various isoforms of AGL mRNA.
Clinical Features of Cori Disease
Deficiency in glycogen debranching activity causes hepatomegaly, ketotic hypoglycemia, hyperlipidemia, variable skeletal myopathy, cardiomyopathy and results in short stature. Patients with both liver and muscle involvement have GSD3a and those with only liver involvement (~15% of GSD3 patients) are classified as GSD3b. Because hepatomegaly, hypoglycemia, hyperlipidemia, and growth retardation are common symptoms in both von Gierke disease (GSD1) and Cori disease (GSD3), it is difficult to initially determine from which disease an infant is suffering.
Definitive diagnosis is only made by examining the structure of the glycogen in patients as well as assaying for the level of activity of the debranching enzyme. This diagnosis is best accomplished from liver and/or muscle biopsy. The hepatic symptoms of Cori disease usually resolve after puberty. However, liver failure due to cirrhosis may occur.
Therapeutic Intervention in Cori Disease Patients
During periods of hypoglycemia, Cori disease is treated with frequent high carbohydrate meals with cornstarch supplements. A high protein diet is also an effective treatment as this drives gluconeogenesis. There is currently no effective treatment for the progressive cardiomyopathy.