

Last Updated: October 28, 2025
Primary Structure of Proteins
The primary structure of peptides and proteins refers to the linear number and order of the amino acids present. The convention for the designation of the order of amino acids is that the N-terminal end (i.e. the end bearing the residue with the free α-amino group) is to the left (and the number 1 amino acid) and the C-terminal end (i.e. the end with the residue containing a free α-carboxyl group) is to the right.
Secondary Structure in Proteins
The ordered array of amino acids in a protein confer regular conformational forms upon that protein. These conformations constitute the secondary structures of a protein. In general proteins fold into two broad classes of structure termed, globular proteins or fibrous proteins. Globular proteins are compactly folded and coiled, whereas, fibrous proteins are more filamentous or elongated. It is the partial double-bond character of the peptide bond that defines the conformations a polypeptide chain may assume. Within a single protein different regions of the polypeptide chain may assume different conformations determined by the primary sequence of the amino acids.
The α-Helix
The α-helix is a common secondary structure encountered in proteins of the globular class. The formation of the α-helix is spontaneous and is stabilized by H-bonding between amide nitrogens and carbonyl carbons of peptide bonds spaced four residues apart. This orientation of H-bonding produces a helical coiling of the peptide backbone such that the R-groups lie on the exterior of the helix and perpendicular to its axis.
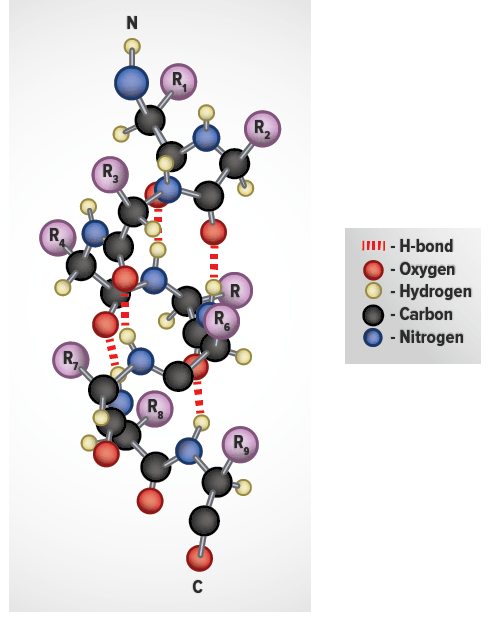
Not all amino acids favor the formation of the (α-helix due to steric constraints of the R-groups. Amino acids such as A, D, E, I, L and M favor the formation of α-helices, whereas, G and P favor disruption of the helix. This is particularly true for P since it is a pyrrolidine based imino acid (HN=) whose structure significantly restricts movement about the peptide bond in which it is present, thereby, interfering with extension of the helix. The disruption of the helix is important as it introduces additional folding of the polypeptide backbone to allow the formation of globular proteins.
β-Sheets
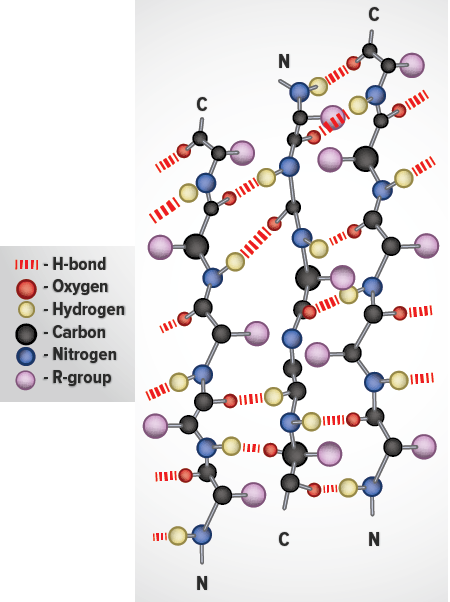
Whereas an α-helix is composed of a single linear array of helically disposed amino acids, β-sheets are composed of 2 or more different regions of stretches of at least 5-10 amino acids. The folding and alignment of stretches of the polypeptide backbone aside one another to form β-sheets is stabilized by H-bonding between amide nitrogens and carbonyl carbons. However, the H-bonding residues are present in adjacently opposed stretches of the polypeptide backbone as opposed to a linearly contiguous region of the backbone in the α-helix. β-sheets are said to be pleated. This is due to positioning of the α-carbons of the peptide bond which alternates above and below the plane of the sheet. β-sheets are either parallel or antiparallel. In parallel sheets adjacent peptide chains proceed in the same direction (i.e. the direction of N-terminal to C-terminal ends is the same), whereas, in antiparallel sheets adjacent chains are aligned in opposite directions. β-sheets can be depicted in ball and stick format or as ribbons in certain protein formats.
Super-Secondary Structure
Some proteins contain an ordered organization of secondary structures that form distinct functional domains or structural motifs. Examples include the helix-turn-helix domain of bacterial proteins that regulate transcription and the leucine zipper, helix-loop-helix and zinc finger domains of eukaryotic transcriptional regulators. These domains are termed super-secondary structures.
Tertiary Structure of Proteins
Tertiary structure refers to the complete three-dimensional structure of the polypeptide units of a given protein. Included in this description is the spatial relationship of different secondary structures to one another within a polypeptide chain and how these secondary structures themselves fold into the three-dimensional form of the protein. Secondary structures of proteins often constitute distinct domains. Therefore, tertiary structure also describes the relationship of different domains to one another within a protein. The interactions of different domains is governed by several forces: These include hydrogen bonding, hydrophobic interactions, electrostatic interactions and van der Waals forces.
Forces Controlling Protein Structure
Hydrogen Bonding:
Polypeptides contain numerous proton donors and acceptors both in their backbone and in the R-groups of the amino acids. The environment in which proteins are found also contains the ample H-bond donors and acceptors of the water molecule. H-bonding, therefore, occurs not only within and between polypeptide chains but with the surrounding aqueous medium.
Hydrophobic Forces:
Proteins are composed of amino acids that contain either hydrophilic or hydrophobic R-groups. It is the nature of the interaction of the different R-groups with the aqueous environment that plays the major role in shaping protein structure. The spontaneous folded state of globular proteins is a reflection of a balance between the opposing energetics of H-bonding between hydrophilic R-groups and the aqueous environment and the repulsion from the aqueous environment by the hydrophobic R-groups. The hydrophobicity of certain amino acid R-groups tends to drive them away from the exterior of proteins and into the interior. This driving force restricts the available conformations into which a protein may fold.
Electrostatic Forces:
Electrostatic forces are mainly of three types; charge-charge, charge-dipole and dipole-dipole. Typical charge-charge interactions that favor protein folding are those between oppositely charged R-groups such as K or R and D or E. A substantial component of the energy involved in protein folding is charge-dipole interactions. This refers to the interaction of ionized R-groups of amino acids with the dipole of the water molecule. The slight dipole moment that exist in the polar R-groups of amino acid also influences their interaction with water. It is, therefore, understandable that the majority of the amino acids found on the exterior surfaces of globular proteins contain charged or polar R-groups.
van der Waals Forces:
There are both attractive and repulsive van der Waals forces that control protein folding. Attractive van der Waals forces involve the interactions among induced dipoles that arise from fluctuations in the charge densities that occur between adjacent uncharged non-bonded atoms. Repulsive van der Waals forces involve the interactions that occur when uncharged non-bonded atoms come very close together but do not induce dipoles. The repulsion is the result of the electron-electron repulsion that occurs as two clouds of electrons begin to overlap.
Although van der Waals forces are extremely weak, relative to other forces governing conformation, it is the huge number of such interactions that occur in large protein molecules that make them significant to the folding of proteins.
Quaternary Structure
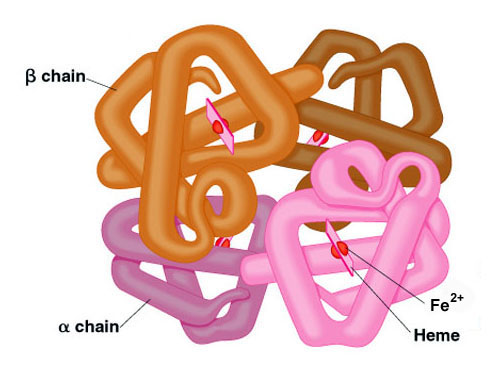
Many proteins contain 2 or more different polypeptide chains that are held in association by the same non-covalent forces that stabilize the tertiary structures of proteins. Proteins with multiple polypeptide chains are oligomeric proteins. The structure formed by monomer-monomer interaction in an oligomeric protein is known as quaternary structure.
Oligomeric proteins can be composed of multiple identical polypeptide chains or multiple distinct polypeptide chains. Proteins with identical subunits are termed homo-oligomers. Proteins containing several distinct polypeptide chains are termed hetero-oligomers.
Hemoglobin, the oxygen carrying protein of the blood, contains two α and two β subunits arranged with a quaternary structure in the form, α2β2. Hemoglobin is, therefore, a hetero-oligomeric protein.
Complex Protein Structures
Proteins also are found to be covalently conjugated with carbohydrates. These modifications occur following the synthesis (translation) of proteins and are, therefore, termed post-translational modifications. These forms of modification impart specialized functions upon the resultant proteins. Proteins covalently associated with carbohydrates are termed glycoproteins. Glycoproteins are of two classes, N-linked and O-linked, referring to the site of covalent attachment of the sugar moieties. N-linked sugars are attached to the amide nitrogen of the R-group of asparagine; O-linked sugars are attached to the hydroxyl groups of either serine or threonine and occasionally to the hydroxyl group of the modified amino acid, hydroxylysine.
There are extremely important glycoproteins found on the surface of erythrocytes. It is the variability in the composition of the carbohydrate portions of many glycoproteins and glycolipids of erythrocytes that determines blood group specificities. There are at least 100 blood group determinants, most of which are due to carbohydrate differences. The most common blood groups, A, B, and O, are specified by the activity of specific gene products whose activities are to incorporate distinct sugar groups onto RBC membrane glycosphingolipids as well as secreted glycoproteins.
Structural complexes involving protein associated with lipid via noncovalent interactions are termed lipoproteins. The distinct roles of lipoproteins are described on the linked page. Their major function in the body is to aid in the transport of triglycerides, phospholipids, and cholesterol.
Clinical Significances
This discussion is not intended to be a complete review of all disorders that result from defects in protein structure and function.
The substitution of a hydrophobic amino acid (V) for an acidic amino acid (E) in the β-chain of hemoglobin results in sickle cell anemia (HbS). This change of a single amino acid alters the structure of hemoglobin molecules in such a way that the deoxygenated proteins polymerize and precipitate within the erythrocyte, leading to their characteristic sickle shape.
Collagens are the most abundant proteins in the body. Alterations in collagen structure arising from abnormal collagen genes or abnormal processing of collagen proteins results in numerous diseases, including Larsen syndrome, osteogenesis imperfecta and Ehlers-Danlos syndrome.
Ehlers-Danlos syndrome is actually the name associated with at least ten distinct disorders that are biochemically and clinically distinct yet all manifest structural weakness in connective tissue as a result of defective collagen structure. Osteogenesis imperfecta also encompasses more than one disorder. There are at least 15 biochemically distinguishable yet clinically related maladies that have been identified as osteogenesis imperfecta, OI. All forms of OI are characterized by multiple fractures and resultant bone deformities. The four major forms of OI (types I-IV) are each due to mutations in type I collagen genes. Marfan syndrome manifests itself as a disorder of the connective tissue and was originally believed to be the result of abnormal collagens. However, it was subsequently determined that Marfan syndrome results from mutations in the extracellular protein, fibrillin 1, which is an integral constituent of the non-collagenous microfibrils of the extracellular matrix.
Several forms of familial hypercholesterolemia are the result of genetic defects in the gene encoding the low-density lipoprotein receptor (LDLR). These defects result in the synthesis of abnormal LDL receptors that are incapable of binding to LDLs, or that bind LDLs but the receptor/LDL complexes are not properly internalized and degraded. The outcome is an elevation in serum cholesterol levels and increased propensity toward the development of atherosclerosis.
A number of proteins can contribute to cellular transformation and carcinogenesis when their basic structure is disrupted by mutations in their genes. These genes are termed proto-oncogenes. For some of these proteins, all that is required to convert them to the oncogenic form is a single amino acid substitution. The cellular gene, RAS, is observed to sustain single amino acid substitutions at positions 12 or 61 with high frequency in colon carcinomas. Mutations in the RAS gene are some of the most frequently observed genetic alterations in colorectal carcinomas.
Analysis of Protein Structure and Composition
The most current techniques for determination of the amino acid sequence of a protein involve the use of mass spectrometry (MS). MS involves the analysis of mass-to-charge ratios of ionized protein fragments to deduce the sequence of amino acids. The most commonly used MS techniques are tandem mass spectrometry (MS/MS), electrospray ionization (ESI), and matrix-assisted laser desorption/ionization (MALDI). With the MALDI technique the ions generated from a protein, or fragments of proteins, are analyzed by time-of-flight mass analysis. This technique is referred to as MALDI-TOF.
Amino-Terminal Sequence Determination
Prior to sequencing peptides it is necessary to eliminate disulfide bonds within peptides and between peptides. Several different chemical reactions can be used in order to permit separation of peptide strands and prevent protein conformations that are dependent upon disulfide bonds. The most common treatments are to use either 2-mercaptoethanol or dithiothreitol (DTT). Both of these chemicals reduce disulfide bonds. To prevent reformation of the disulfide bonds the peptides are treated with iodoacetic acid in order to alkylate the free sulfhydryls.
There are three major chemical techniques for sequencing peptides and proteins from the N-terminus. These are the Sanger, Dansyl chloride and Edman techniques.
Sanger’s Reagent: This sequencing technique utilizes the compound, 2,4-dinitrofluorobenzene (DNF) which reacts with the N-terminal residue under alkaline conditions. The derivatized amino acid can be hydrolyzed and will be labeled with a dinitrobenzene group that imparts a yellow color to the amino acid. Separation of the modified amino acids (DNP-derivative) by electrophoresis and comparison with the migration of DNP-derivative standards allows for the identification of the N-terminal amino acid.
Dansyl chloride: Like DNF, dansyl chloride reacts with the N-terminal residue under alkaline conditions. Analysis of the modified amino acids is carried out similarly to the Sanger method except that the dansylated amino acids are detected by fluorescence. This imparts a higher sensitivity into this technique over that of the Sanger method.
Edman degradation: The utility of the Edman degradation technique is that it allows for additional amino acid sequence to be obtained from the N-terminus inward. Using this method it is possible to obtain the entire sequence of peptides. This method utilizes phenylisothiocyanate to react with the N-terminal residue under alkaline conditions. The resultant phenylthiocarbamyl derivatized amino acid is hydrolyzed in anhydrous acid. The hydrolysis reaction results in a rearrangement of the released N-terminal residue to a phenylthiohydantoin derivative. As in the Sanger and Dansyl chloride methods, the N-terminal residue is tagged with an identifiable marker, however, the added advantage of the Edman process is that the remainder of the peptide is intact. The entire sequence of reactions can be repeated over and over to obtain the sequences of the peptide. This process has subsequently been automated to allow rapid and efficient sequencing of even extremely small quantities of peptide.
Protease Digestion
Due to the limitations of the Edman degradation technique, peptides longer than around 50 residues can not be sequenced completely. The ability to obtain peptides of this length, from proteins of greater length, is facilitated by the use of enzymes, endopeptidases, that cleave at specific sites within the primary sequence of proteins. The resultant smaller peptides can be chromatographically separated and subjected to Edman degradation sequencing reactions.
Table of the Specificities of Several Endoproteases
| Enzyme | Source | Specificity | Additional Points |
| Trypsin | Bovine pancreas | peptide bond C-terminal to R, K, but not if next to P | highly specific for positively charged residues |
| Chymotrypsin | Bovine pancreas | peptide bond C-terminal to F, Y, W but not if next to P | prefers bulky hydrophobic residues, cleaves slowly at N, H, M, L |
| Elastase | Bovine pancreas | peptide bond C-terminal to A, G, S, V, but not if next to P | |
| Thermolysin | Bacillus thermoproteolyticus | peptide bond N-terminal to I, M, F, W, Y, V, but not if next to P | prefers small neutral residues, can cleave at A, D, H, T |
| Pepsin | Bovine gastric mucosa | peptide bond N-terminal to L, F, W, Y, but not when next to P | exhibits little specificity, requires low pH |
| Endopeptidase V8 | Staphylococcus aureus | peptide bond C-terminal to E |
Carboxy-Terminal Sequence Determination
No reliable chemical techniques exist for sequencing the C-terminal amino acid of peptides. However, there are enzymes, exopeptidases, that have been identified that cleave peptides at the C-terminal residue which can then be analyzed chromatographically and compared to standard amino acids. This class of exopeptidases are called, carboxypeptidases.
Table of the Specificities of Several Exopeptidases
| Enzyme | Source | Specificity |
| Carboxypeptidase A | Bovine pancreas | will not cleave when C-terminal residue = R, K or P or if P resides next to terminal residue |
| Carboxypeptidase B | Bovine pancreas | cleaves when C-terminal residue = R or K; not when P resides next to terminal reside |
| Carboxypeptidase C | Citrus leaves | all free C-terminal residues, pH optimum = 3.5 |
| Carboxypeptidase Y | Yeast | all free C-terminal residues, slowly at G residues |
Chemical Digestion of Proteins
The most commonly utilized chemical reagent that cleaves peptide bonds by recognition of specific amino acid residues is cyanogen bromide (CNBr). This reagent causes specific cleavage at the C-terminal side of M residues. The number of peptide fragments that result from CNBr cleavage is equivalent to one more than the number of M residues in a protein.
The most reliable chemical technique for C-terminal residue identification is hydrazinolysis. A peptide is treated with hydrazine, NH2–NH2, at high temperature (90°C) for an extended length of time (20-100hr). This treatment cleaves all of the peptide bonds yielding amino-acyl hydrazides of all the amino acids excluding the C-terminal residue which can be identified chromatographically compared to amino acid standards. Due to the high percentage of hydrazine induced side reactions this technique is only used on carboxypeptidase resistant peptides.
Size Exclusion Chromatography
This chromatographic technique is based upon the use of a porous gel in the form of insoluble beads placed into a column. As a solution of proteins is passed through the column, small proteins can penetrate into the pores of the beads and, therefore, are retarded in their rate of travel through the column. The larger proteins a protein is the less likely it will enter the pores. Different beads with different pore sizes can be used depending upon the desired protein size separation profile.
Ion Exchange Chromatography
Each individual protein exhibits a distinct overall net charge at a given pH. Some proteins will be negatively charged and some will be positively charged at the same pH. This property of proteins is the basis for ion exchange chromatography. Fine cellulose resins are used that are either negatively (cation exchanger) or positively (anion exchanger) charged. Proteins of opposite charge to the resin are retained as a solution of proteins is passed through the column. The bound proteins are then eluted by passing a solution of ions bearing a charge opposite to that of the column. By utilizing a gradient of increasing ionic strength, proteins with increasing affinity for the resin are progressively eluted.
Affinity Chromatography
Proteins have high affinities for their substrates or co-factors or prosthetic groups or receptors or antibodies raised against them. This affinity can be exploited in the purification of proteins. A column of beads bearing the high affinity compound can be prepared and a solution of protein passed through the column. The bound proteins are then eluted by passing a solution of unbound soluble high affinity compound through the column.
High Performance Liquid Chromatography (HPLC)
In column chromatography the smaller and more tightly packed a resin is the greater the separation capability of the column. In gravity flow columns the limitation column packing is the time it takes to pass the solution of proteins through the column. HPLC utilizes tightly packed fine diameter resins to impart increased resolution and overcomes the flow limitations by pumping the solution of proteins through the column under high pressure. Like standard column chromatography, HPLC columns can be used for size exclusion or charge separation. An additional separation technique commonly used with HPLC is to utilize hydrophobic resins to retard the movement of nonpolar proteins. The proteins are then eluted from the column with a gradient of increasing concentration of an organic solvent. This latter form of HPLC is termed reversed-phase HPLC.
Electrophoresis of Proteins
Proteins also can be characterized according to size and charge by separation in an electric current (electrophoresis) within solid sieving gels made from polymerized and cross-linked acrylamide. The most commonly used technique is termed SDS polyacrylamide gel electrophoresis (SDS-PAGE). The gel is a thin slab of acrylamide polymerized between two glass plates. This technique utilizes a negatively charged detergent (sodium dodecyl sulfate) to denature and solubilize proteins. SDS denatured proteins have a uniform negative charge such that all proteins will migrate through the gel in the electric field based solely upon size. The larger the protein the more slowly it will move through the matrix of the polyacrylamide. Following electrophoresis the migration distance of unknown proteins relative to known standard proteins is assessed by various staining or radiographic detection techniques.
The use of polyacrylamide gel electrophoresis also can be used to determine the isoelectric charge of proteins (pI). This technique is termed isoelectric focusing. Isoelectric focusing utilizes a thin tube of polyacrylamide made in the presence of a mixture of small positively and negatively charged molecules termed ampholytes. The ampholytes have a range of pIs that establish a pH gradient along the gel when current is applied. Proteins will, therefore, cease migration in the gel when they reach the point where the ampholytes have established a pH equal to the proteins pI.
Centrifugation of Proteins
Proteins will sediment through a solution in a centrifugal field dependent upon their mass. Analytical centrifugation measures the rate that proteins sediment through various density solutions. The most common solution utilized is a linear gradient of sucrose (generally from 5–20%). Proteins are layered atop the gradient in an ultracentrifuge tube then subjected to centrifugal fields in excess of 100,000 x g. The sizes of unknown proteins can then be determined by comparing their migration distance in the gradient with those of known standard proteins.