

Last Updated: July 6, 2026
Brief Overview of Human Nervous System
The human nervous system consists of two main parts, the central nervous system (CNS) and the peripheral nervous system (PNS). The CNS contains the brain and spinal cord. The PNS comprises the nerve fibers that connect the CNS to every other part of the body. The biochemistry of nerve transmission relates to the synthesis and function of the various neurotransmitters that control nerve processes in the CNS and the PNS.
The PNS includes the motor neurons that are responsible for mediating voluntary movement. The PNS also includes the autonomic nervous system which encompasses the sympathetic nervous system, the parasympathetic nervous system, and the enteric nervous system. The sympathetic and parasympathetic nervous systems are tasked with the regulation of all involuntary activities. The enteric nervous system is unique in that it represents a semi-independent part of the nervous system whose function is to control processes specific to the gastrointestinal system. The nervous systems of the body are composed of two primary types of cell: the neurons that carry the chemical signals of nerve transmission, and the glial cells that serve to support and protect the neurons.
Two important concepts relate to the functioning of the nervous system. These terms are efferent and afferent. Efferent connections in the nervous system refer to those that send signals from the CNS to the effector cells of the body such as muscles and glands. Efferent nerves are, therefore, also referred to as motor neurons. Afferent connections refer to those that send signals from sense organs to the CNS. For this reason these nerves are commonly referred to as sensory neurons.
Another important cellular structure in nervous systems are the ganglia. The term ganglion refers to a bundle (mass) of nerve cell bodies. In the context of the nervous system, ganglia are composed of soma (cell bodies) and dendritic structures. The dendritic trees of most ganglia are interconnected to other dendritic trees resulting in the formation of a plexus. In the human nervous system there are two main groups of ganglia. The dorsal root ganglia, which is also referred to as the spinal ganglia, contains the cell bodies of the sensory nerves. The autonomic ganglia contain the cell bodies of the nerves of the autonomic nervous system. Nerves that project from the CNS to autonomic ganglia are referred to as preganglionic nerves (or fibers). Conversely, nerves projecting from ganglia to effector organs are referred to as postganglionic nerves (or fibers). Generally the term ganglion relates to the peripheral nervous system. However, the term basal ganglia (also basal nuclei) is used commonly to describe the neuroanatomical region of the brain that connects the hypothalamus, cerebral cortex, and the brainstem.
Neurons
Neurons are the highly specialized cells of all nervous systems (e.g. CNS and PNS) that are tasked with transmitting signals from one location to another. These cells accomplish this role through specialized membrane-to-membrane junctions called synapses. Most neuron possess an axon which is a long protrusion from the body (soma) of the neuron to the synapse. Axons can extend to distant parts of the body and make thousands of synaptic contacts such as is the case with the CNS neurons of the spinal cord. Axons frequently travel through the body in bundles called nerves. The synapses are termed pre-synaptic and post-synaptic. The pre-synaptic synapse will release secretory granule contents in response to the propagation of an electrochemical signal (action potential) down its axon. The released substance (termed a neurotransmitter) will then, most likely, bind to a specific receptor on the membrane of the post-synaptic synapse, thereby, propagating the initial action potential to the next neuron. The human nervous system is composed of hundreds of different types of neurons. These include sensory neurons that transmute physical stimuli such as light and sound into neural signals, and motor neurons that are responsible for converting neural signals into activation of muscles or glands.
Glial Cells
Glial cells (named from the Greek for “glue”) are the specialized non-neuronal cells of the nervous system that provide protection, support and nutrition for neurons. Astrocytes represent specialized glial cells tasked with maintaining neural metabolic functions, regulating neurotransmitter, and help maintain normal neuronal function.
As the Greek name glue infers, glial cells hold neurons in place and provide guidance cues which directs axons of the neurons to their appropriate target cell(s). Glial cells, in particular astrocytes, are responsible for the maintenance of neural homeostasis, for the formation of myelin, and they play a participatory role in signal transmission in the nervous system. Glial cells provide an electrical insulation (myelin) for neurons which allows for rapid transmission of action potentials and also prevents the abnormal propagation of nerve impulses to inappropriate neurons. The glial cells that produce the myelin sheath are called oligodendrocytes in the CNS and Schwann cells in the PNS.
Microglial cells are another specialized cell type in the nervous system. Microglia are immune cells in the brain and spinal cord that respond to infections by producing cytokines. Microglia also destroy pathogens and remove dead neurons, protein aggregates, and other debris that might otherwise compromise normal functions in the brain.
Autonomic Nervous System
Sympathetic Nervous System
The sympathetic nervous system (SNS) is predominantly responsible for excitatory action potentials with the goal of inducing the “fight-or-flight” responses of the body under conditions of stress. In general, activation of the SNS results in contraction, for example, vasoconstriction. Although stress is a major trigger of the SNS, it is constantly active at a basal level to maintain homeostasis. The activation of the neurons of the SNS occurs as a result of signals arising in the region of the brain stem called the nucleus of the solitary tract (NTS, for the Latin term nucleus tractus solitarii). The NTS receives a wide range of sensory inputs from both systemic and central baroreceptors and chemoreceptors.
The neurons of the SNS emanate from the medulla, specifically the rostral ventrolateral medulla (RVLM), and travel down the spinal cord where they synapse with short preganglionic neurons within the sympathetic ganglia. The ganglia of the SNS are the nerve cell bodies that lie on either side of the spinal cord. Preganglionic sympathetic fibers are those that exit the spinal cord synapses within these ganglia. The preganglionic neurotransmitter is acetylcholine, ACh. ACh released from the sympathetic preganglionic neuron binds to nicotinic ACh receptors (nAChR) on the postganglionic neuron. ACh binding depolarizes the cell body of the postganglionic neuron generating an action potential that travels to the target organ to elicit a response. The neurotransmitter released from sympathetic postganglionic neurons is norepinephrine which binds to its receptor expressed in the target cell.
The target organ receptors responsive to signals from the SNS are those of the adrenergic family, specifically α1, α2, β1, and β2 (see below). Although the primary neurotransmitter released from sympathetic postganglionic neurons is norepinephrine, there are two important exceptions. These exceptions are the postganglionic neurons that innervate chromaffin cells of the adrenal medulla and those that innervate the sweat glands. When the postganglionic neurons that innervate sweat glands are activated, they release ACh (not epinephrine) which binds to muscarinic ACh receptors (mAChR: specifically the M1 and M3 receptors) on the target cell. Adrenal medullary chromaffin cells are functionally analogous to sympathetic postganglionic neurons and when stimulated by ACh from a sympathetic preganglionic neuron these cells release epinephrine and norepinephrine into the circulation. The receptors triggering the release of adrenal epinephrine and norepinephrine are nicotinic (nAChR).
Parasympathetic Nervous System
The parasympathetic nervous system is predominantly responsible for inhibitory action potentials resulting in relaxation, for example, vasodilation. The parasympathetic nervous system is responsible for stimulation of “rest-and-digest” and “feed-and-breed” activities that occur when the body is at rest. These responses include, but are not limited to, sexual arousal, salivation, lacrimation (tears), urination, digestion, and defecation.
Within the head the parasympathetic nervous system includes cranial nerves III, VII, and IX, while cranial nerve X (comprising the vagus nerves) exits the brain stem to innervate the organs of the body. Like the SNS, the activation of the vagus nerves of the parasympathetic nervous system occurs as a result of signals arising in the NTS. There are three nuclei within the medulla that send out vagal nerves of the parasympathetic nervous system. These nuclei are the dorsal motor nucleus, the solitary nucleus, and the nucleus ambiguus (located in the reticular formation in the medulla oblongata).
Parasympathetic neural outputs to the heart arise primarily within the nucleus ambiguus. The ganglia of the parasympathetic nervous system are also referred to as terminal ganglia as they lie close to, or within, the organs that they innervate. The exceptions to this are the parasympathetic ganglia of the head and neck. Parasympathetic ganglia are those that are found within the target organ. Preganglionic parasympathetic fibers associated with the vagal nerve all exit the brain stem, they do not travel down the spinal chord except for the pelvic splanchnic nerves which exit the spinal cord in the S2-S4 region.
The parasympathetic preganglionic nerves enter their target organs where they form synapses with postganglionic neurons. Like the sympathetic ganglia, the neurotransmitter of parasympathetic preganglionic nerves is ACh. When released from these nerves the ACh binds to nicotinic ACh receptors (nAChR) on the postganglionic nerve. However, unlike sympathetic postganglionic nerves, activation of the parasympathetic postganglionic nerves results in the release of ACh. When released from the parasympathetic postganglionic neuron, the ACh binds to muscarinic ACh receptors (mAChR) in the target cells, primarily the M2 and M3 receptors.
Autonomic Control of Cardiovascular System
Within the cardiovascular system the norepinephrine released from sympathetic postganglionic neurons binds to β1 (and to a lesser extent β2) adrenergic receptors expressed on cardiac myocytes of the heart within the sinoatrial (SA) node (primary cardiac pacemaker cells), the atrioventricular (AV) node, the ventricles, and the Purkinje fibers of the cardiac conduction system.
The acetylcholine (ACh) released from parasympathetic postganglionic neurons binds to the muscarinic acetylcholine type 2 (M2) receptors in the SA node (from the right vagus nerve), the AV node (from the left vagus nerve), and atrial cells.
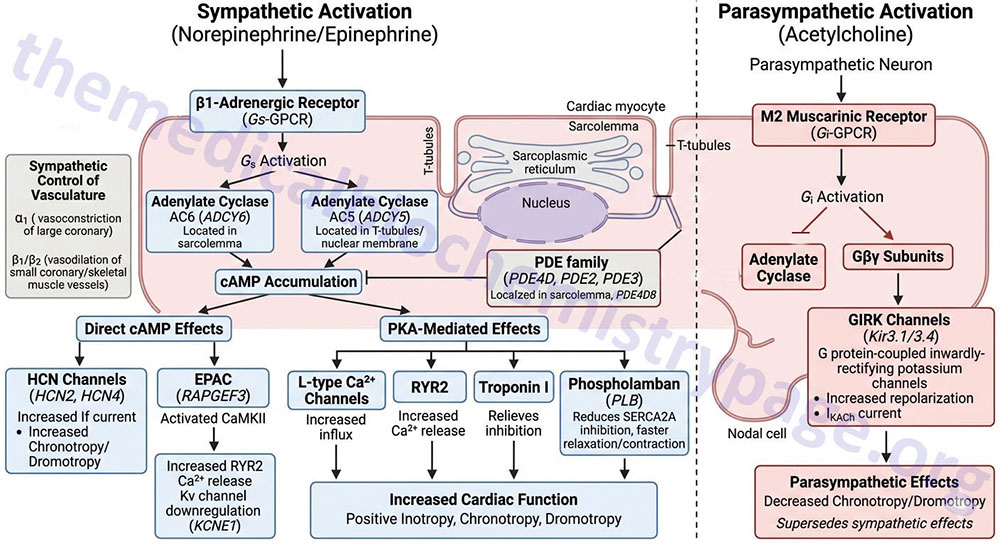
Activation of the β1 receptor in the heart results in increased force of contraction (inotropy), increased heart rate (chronotropy), and increased cardiac conductance (dromotropy). These effects are exerted as a result of the increased levels of cAMP and the activation of PKA that result from β1 receptor activation of Gs-type G-proteins.
Activation of the β1 receptor associated Gs-type G-proteins results in the consequent activation of adenylate cyclases. The major adenylate cyclases activated by β-adrenergic receptors in the heart are encoded by the ADCY5 and ADCY6 genes. The ADCY5 encoded enzyme, identified as AC5, is localized to the nuclear membrane and to specialized domains of the membranes of the T-tubule system of the cardiac myocyte. AC5 activity is regulated by both β1– and β2-adrenergic receptors in cardiac myocytes. The ADCY6 encoded enzyme, identified as AC6, is localized to the sarcolemma (muscle cell plasma membrane) of the cardiac myocyte outside the T-tubule system. AC6 activity is regulated exclusively by β1-adrenergic receptors.
The extent to which cAMP can exert its effects, either directly or through the activation of PKA, is controlled by the receptor-mediated activation of the various adenylate cyclases as well as by the activities of various enzymes of the phosphodiesterase (PDE) family. Multiple PDE isoforms are expressed within various tissues of the heart, however, within the cardiac myocyte the predominate PDE are members of the PDE4D family, specifically the PDE4D5 and PDE4D8 isoforms. However, it should be noted that members of the PDE2 and PDE3 families are also expressed within cardiac myocytes. The PDE4D8 isoform is localized to protein complexes at the sarcolemma (muscle cell plasma membrane) that includes the β1-adrenergic receptor.
Numerous responses, exerted by β-adrenergic receptor activation, are the result of cAMP itself. Direct effects of cAMP within cardiac myocytes of the SA node, AV node, and the Purkinje fibers contribute to positive chronotropic and dromotropic effects elicited as a result of β1-adrenergic receptor activation. The effects of direct cAMP action include binding to, and enhancing of the activation of, the hyperpolarization-activated cyclic nucleotide-gated channel 2 and 4 (HCN2 and HCN4) that are responsible for the pacemaker current (If or IKf) in cardiac nodal cells. The HCN channels are often referred to as funny current channels. The resulting cAMP-mediated activation increases the pacemaker current leading to positive chronotropic and dromotropic activities of the cardiac myocytes.
Another important protein activated by direct interaction with cAMP is RAP guanine nucleotide exchange factor 3 (encoded by the RAPGEF3 gene). The RAPGEF3 encoded protein was originally, and is commonly, referred to as exchange protein directly activated by cAMP, EPAC (also known as EPAC1). The activation of EPAC by cAMP results in the activation of Ca2+/calmodulin-dependent kinase II (CaMKII or CaMK2).
When activated, CaMKII phosphorylates the calcium release channel, ryanodine receptor 2, RYR2, resulting in increased release of Ca2+ stored within the sarcoplasmic reticulum, SR. Increased free intracellular Ca2+ plays a major role in the contractile activity of muscle cells through, among other effects, the activation of the calmodulin subunit of myosin light chain kinases. Thus, the stimulated release of Ca2+ from the SR contributes to the increased inotropic and chronotropic effects of β1-adrenergic receptor activation.
Activation of EPAC also results in the downregulation of the regulatory subunit of voltage-gated potassium channels which are commonly identified as Kv channels. The regulatory subunit of the Kv channels is encoded by the KCNE1 gene and is commonly referred to as the β-subunit. The specific cardiac Kv family channels that are regulated by the KCNE1 encoded protein are Kv2.1 channels encoded by the KCNB1 gene and Kv1.9 channels encoded by the KCNQ1 gene. The KCNQ1 encoded potassium channels are required for the repolarization phase of cardiac myocyte action potentials. The KCNQ1 (Kv1.9) channels constitute the cardiac K+ current identified as IKs where the I refers to inward current and the Ks refers to slow acting outwardly rectifying K+ transport.
Within the cardiac myocyte PKA is localized to specific subcellular locations through its anchoring to proteins of the A-kinase anchoring protein (AKAP) family. Upon its activation via cAMP, PKA mediates its effects on cardiac contractility and chronotropy via the phosphorylation of numerous proteins controlling these processes. PKA phosphorylates L-type Ca2+ channels in the plasma membrane (sarcolemma) and RYR2 in the sarcoplasmic reticulum (SR).
The PKA-mediated phosphorylation of plasma membrane L-type Ca2+ channels results in increased influx of Ca2+ and the phosphorylation of the RYR2 channel results in increase release of Ca2+ stored in the SR. Both of these events lead to increased Ca2+ availability for activation of the calmodulin subunit of myosin light-chain kinase, MLCK. It is important to note that the effects of PKA in the striated muscle cells of the heart are distinct from the effects of PKA in smooth muscle cells. Whereas, in striated cardiomyocytes the effect of PKA is increased contractile activity, in smooth muscle the phosphorylation of the plasma membrane calcium channels leads to inhibition and PKA phosphorylates the smooth muscle MLCK isoform rendering it less active. The net effect in smooth muscle is relaxation, not contraction.
The released Ca2+ also interacts with troponin C (TnC) resulting a conformational change to the troponin complex (troponin C, I, and T) that moves the attached tropomyosin away from the myosin binding sites on actin. This conformational change abolishes the inhibitory action of the TnI protein of the complex. In addition, the conformational change permits nearby myosin heads to interact with myosin binding sites, and contractile activity ensues. PKA also phosphorylates troponin I preventing it from inhibiting the interactions of actin and myosin.
Another important target of PKA is the transmembrane protein identified as phospholamban (PLB). Phospholamban (encoded by the PLN gene) interacts with SR membrane-associated Ca2+ reuptake channels identified as sarco/endoplasmic reticulum Ca2+-ATPases (SERCA: encoded by ATP2A family genes). The cardiac myocyte SERCA is identified as SERCA2A and is encoded by the ATP2A2 gene. Phospholamban exists in monomeric and homopentameric conformations where the monomeric form interacts with SERCA to regulate Ca2+ transport. Phosphorylation of PLB causes it to dissociate from SERCA and form homopentamers. The normal function of monomeric PLB is to inhibit the reuptake of Ca2+ into the SR via the action of SERCA2A transporters. Phosphorylation of PLB by PKA reduces the inhibitory action of PLN on SERCA2A promoting Ca2+ reuptake. Diastolic relaxation of cardiac myocytes requires Ca2+ reuptake by the SR, thus, the inhibition of PLB allows for an increased rate of myocyte contraction and relaxation resulting in an overall increased force of contraction.
Within the large coronary arteries (predominantly the left and right coronary arteries, LCA and RCA, respectively) sympathetic postganglionic nerve release of norepinephrine results in activation of the α1 (and to a lessor extent α2) adrenergic receptors in the smooth muscle cells resulting in vasoconstriction. This vasoconstriction of the large coronary arteries, which are branches of the aorta, allow a greater volume of blood to remain in the aorta for delivery of oxygen- and nutrient-rich blood to the tissues during sympathetic activation such as is typified by the fight-or-flight response.
However, the heart muscle cannot be starved of blood and this is prevented by the presence of high concentrations of β1-adrenergic receptors on the smooth muscle cells of the small coronary arteries. The activation of these β1 receptors results in vasodilation of the small coronary arteries. Within the periphery, the smooth muscle cells of the vessels in skeletal muscle also possess β-adrenergic receptors, predominantly the β2 class. Stimulation of these β2 receptors also results in vasodilation. This effect allows the skeletal muscle vasculature to receive larger volumes of blood during the fight-or-flight response. The primary activator of the β2 adrenergic receptors in skeletal muscle vasculature is the epinephrine released from the adrenal medulla in response to sympathetic activation.
The cardiac muscarinic receptor that binds the ACh released from parasympathetic postganglionic nerves is the M2 type receptor. Each of the muscarinic ACh receptors is a GPCR and the M2 receptors are coupled to a Gi-type G-protein. Activation of the M2 receptor results in decreased levels of cAMP leading to reduced direct effects of cAMP and reduced activation of PKA, thereby reducing all of the processes, detailed above, that are activated in response to the release of norepinephrine from postganglionic sympathetic neurons.
Activation of the M2 receptor also results in the activation of inwardly rectifying K+ channels resulting in rapid repolarization of cardiac myocytes leading to termination of an action potential. The particular class of K+ channels that are responsive to G-proteins are activated by the βγ subunits of the G-protein. These K+ channels are commonly referred to as G protein-coupled inwardly-rectifying potassium channels (GIRK). The GIRK are members of the KCNJ subfamily of voltage-gated K+ channels. The specific cardiac GIRK channels are heterodimers formed from the proteins encoded by the KCNJ3 and KCNJ5 genes. These proteins are commonly identified as Kir3.1 and Kir3.4. This cardiac nodal cell potassium channel constitutes the IKACh current that is responsible for phase 4 of nodal cell action potentials.
The net effect of parasympathetic activation of nodal M2 receptors is decreased heart rate (chronotropy) and decreased cardiac conductance (dromotropy). The effects of the parasympathetic nervous system on the heart supersede the effects of the sympathetic nervous system such that even in the face of sympathetic stimulation, parasympathetic stimulation can depress cardiac activity.
Within the vasculature ACh binds to the M3 receptor on endothelial cells leading to increased NO production resulting in vasodilation. However, this ACh is not derived from parasympathetic nerves but directly from the circulation. Parasympathetic postganglionic ACh does stimulate M3 receptor-mediated NO production but this is only seen in the external genitalia.
Introduction to Neurotransmitters
Neurotransmitters are endogenous substances that act as chemical messengers by transmitting signals from a neuron to a target cell across a synapse. Prior to their release into the synaptic cleft, neurotransmitters are stored in secretory vesicles (called synaptic vesicles) near the plasma membrane of the axon terminal. The release of the neurotransmitter occurs most often in response to the arrival of an action potential at the synapse. When released, the neurotransmitter crosses the synaptic gap and binds to specific receptors in the membrane of the post-synaptic neuron or cell.
Neurotransmitters are generally classified into two main categories related to their overall activity, excitatory or inhibitory. Excitatory neurotransmitters exert excitatory effects on the neuron, thereby, increasing the likelihood that the neuron will fire an action potential. Major excitatory neurotransmitters include glutamate, epinephrine and norepinephrine. Inhibitory neurotransmitters exert inhibitory effects on the neuron, thereby, decreasing the likelihood that the neuron will fire an action potential. Major inhibitory neurotransmitters include GABA, glycine, and serotonin. Some neurotransmitters, can exert both excitatory and inhibitory effects depending upon the type of receptors that are present.
In addition to excitation or inhibition, neurotransmitters can be broadly categorized into two groups defined as small molecule neurotransmitters or peptide neurotransmitters. Many peptides that exhibit neurotransmitter activity also possess hormonal activity since some cells that produce the peptide secrete it into the blood where it then can act on distant cells. Small molecule neurotransmitters include (but are not limited to) acetylcholine, GABA, amino acid neurotransmitters, ATP and nitric oxide (NO). The peptide neurotransmitters include more than 50 different peptides. Many of the gut-derived and hypothalamic neurotransmitter peptides are discussed in detail in the Gut-Brain Interrelationships page. Several peptide neurotransmitters are all derived from the same precursor protein, pro-opiomelanocortin (POMC), as discussed in the Peptide Hormones page.
Many neurotransmitters can also be divided into two broad categories dependent upon whether the receptor activated by the binding of transmitter is a metabotropic or an ionotropic receptor. Metabotropic receptors activate signal transduction upon transmitter binding similar to many peptide hormone receptors which involves a second messenger. Metabotropic receptors are members of the G-protein coupled receptor (GPCR) family. Ionotropic receptors ligand-gated ion channels. Some neurotransmitters, for example glutamate and acetylcholine, bind to multiple receptors some of which are metabotropic and some of which are ionotropic.
Nerve Cell Action Potentials and Synaptic Transmission
The transmission of an efferent signal from the CNS to a target tissue, or an afferent signal from a peripheral tissue back to the CNS occurs as a result of the propagation of action potentials along a nerve cell. Nerve cells are excitable cells and they can respond to various stimuli such as electrical, chemical, or mechanical. When the excitation event is propagated along the nerve cell membrane it is referred to as a nerve impulse or more often as an action potential. When a nerve cell terminates on another it does so at a specialized structure called a synapse. Synaptic transmission refers to the propagation of nerve impulses (action potentials) from one nerve cell to another. The synapse is a junction at which the axon of the presynaptic neuron terminates at some location upon the postsynaptic neuron. The end of a presynaptic axon, where it is juxtaposed to the postsynaptic neuron, is enlarged and forms a structure known as the terminal button (pronounced “boo-tawn”). An axon can make contact anywhere along the second neuron: on the dendrites (an axodendritic synapse), the cell body (an axosomatic synapse) or the axons (an axoaxonal synapse).
Action potentials are the result of membrane depolarization which is brought about by a change in the distribution of ions across the membrane. Differences in ion concentrations on either side of a membrane result in a electrical charge differential across the membrane which is referred to as an electrochemical potential. Changes in ion concentrations on either side of a membrane result in depolarization of the membrane. Once a portion of a membrane is depolarized, the ion gradients need to be returned to the “resting” state, a process referred to as repolarization. The movement of ions across the membrane is the function of proteins and protein complexes termed ion channels. Because nerve transmission involves changes in voltage (charge) across the plasma membrane, these ion channels respond to the voltage changes and are, therefore, referred to as voltage-gated ion channels.
The resting membrane potential of a neuron is maintained by the differential distribution of K+ and Na+ ions. The concentration of intracellular K+ is much higher than the extracellular concentration. This situation is just the opposite for Na+, which is at a much higher concentration outside the cell than inside. This differential is maintained through the action plasma membrane transporters of the Na+,K+-ATPase family. The initiation and propagation of an action potential is the result of the opening and closing of voltage-gated K+ channels and voltage-gated Na+ channels. In the rested stated both types of voltage-gated channels are closed. In response to a depolarizing signal (an excitation signal) the fast acting voltage-gated Na+ channels open allowing an influx of Na+ ions into the cell. The influx of Na+ causes more voltage-gated Na+ channels to open propagating the depolarization event. The Na+ channels ultimately close (within milliseconds) to an inactivated state, meaning they cannot be re-opened prior to the membrane returning to its initial rested state.
The opening of voltage-gated K+ channels occurs much slower than for the Na+ channels and they are not fully open until the Na+ channels have re-closed. The opening of the K+ channels allows K+ to exit the cell which brings the net charge inside the cell back to the rested state potential. The opening of the K+ channels, following closure of the Na+ channels, represents the repolarization stage and brings the action potential to an end.
Nerve impulses are transmitted from one neuron to another, or from a neuron to a target tissue cell, at synapses by the release of neurotransmitters. As discussed in detail throughout this page, neurotransmitters can be small chemicals, such as amino acids or amino acid derivatives, or they can be lipids, such as the endocannabinoid, anandamide. As a nerve impulse, or action potential, reaches the end of a presynaptic axon, molecules of neurotransmitter are released into the synaptic space. The release of neurotransmitter involves the processes of exocytosis. When an action potential reaches the presynaptic terminal the membrane depolarization results in the opening of voltage-gated Ca2+ channels. The influx of Ca2+ ions induces the membranes of neurotransmitter secretory vesicles to fuse with the plasma membrane allowing the contents to be released into the synaptic cleft.
Neuromuscular Transmission
In order to move a skeletal muscle cell, an action potential must be initiated from a peripheral motor neuron. Cardiac muscle (myocardial) cells on the other hand, can initiate their own electrical activity in the absence of an autonomic nerve-mediated action potential. With respect to skeletal muscle, nerve transmission occurs when an axon of a post-ganglionic nerve terminates on a skeletal muscle fiber, at specialized structures called the neuromuscular junction. An action potential occurring at this site is known as neuromuscular transmission. At a neuromuscular junction, the axon subdivides and branches into numerous structures, referred to as terminal buttons (pronounced “boo-tawns”) or end bulbs, that can then innervate numerous skeletal muscle fibers. The result is that many muscle fibers can be innervated by a single neuron instead of each fiber having to be dependent upon an individual neuron for contractile activation.
The skeletal muscle fibers that are innervated by branches from the same neuron constitute a motor unit. Large muscles in the body (e.g. the gastrocnemius) contain numerous motor units. This arrangement of the motor units in a particular muscle allows for activation of only a specific part of a muscle at any given time. This represents a form of spatial control over muscle fiber contraction within a muscle, a feature not associated with cardiac muscle excitation as discussed below.
The terminal buttons (end bulbs) of the motor neurons reside within depressions formed in the skeletal muscle plasma membrane (sarcolemma). At these locations the skeletal muscle membrane is thickened and is referred to as the motor end plate. The space between the terminal buttons (end bulbs) and the motor end plate is similar to the synaptic cleft that exists where the pre-synaptic and post-synaptic membranes of neurons are in close proximity. The particular neurotransmitter in use at the neuromuscular junction is acetylcholine, ACh.
When an action potential reaches the pre-synaptic membrane of a motor neuron the permeability of the membrane changes. This change in permeability allows Ca2+ to enter the nerve endings triggering exocytosis of ACh. The released ACh then binds to nicotinic ACh receptors (nAChR) that are concentrated in the motor end plate membrane. Once released from the motor neuron, the level of active ACh is controlled by its catabolism through the action of acetylcholinesterase. As discussed below, nAChR are members of the ionotropic receptor superfamily (ion channel receptors). Activation of nicotinic ACh receptors in the motor end plate results in an increase in Na+ and K+ conductance through the nAChR channel.
The resulting influx of Na+ into the skeletal muscle cell produces a depolarizing potential. As a result of this depolarization, action potentials are conducted in both directions, away from the motor end plate, along the muscle fiber. These action potentials are the result of the initial membrane depolarization and propagated across the surface membrane via the opening of voltage-gated Na+ channels. The action potential is then propagated down the T-tubule system which directly interacts with the sarcoplasmic reticulum, SR. Activation of the SR leads to the release, into the sarcoplasm (cytoplasm of muscle cells), of stored Ca2+ through the opening of Ca2+ release channels. The SR calcium release channels are also known as the ryanodine receptor (RYR) due to the fact that they were originally identified by their high affinity for the plant alkaloid ryanodine. The end result of the ACh-initiated propagating action potential is muscle contraction.
A particularly devastating disease that results from defects in the overall processes of neuromuscular nerve transmission is myasthenia gravis, MG. MG is a very serious disorder that is often times fatal. The characteristic features of the disease are weakened skeletal muscles that tire with very little exertion. MG is an auto-immune disease associated with antibodies to the nAChR of the neuromuscular junction. Binding of the antibodies to the receptor results in receptor destruction as well as receptor cross-linking. In most patients with MG there is a 70%–90% reduction in motor end plate nicotinic receptor number. Two major forms of MG exist, one in which the extraocular muscles are the ones primarily affected and in the other form there is a generalized skeletal muscle involvement. In the latter form of MG, the muscles of the diaphragm become affected resulting in respiratory failure which contributes to the mortality of MG. Treatment of MG involves numerous approaches including the use of acetylcholinesterase inhibitors. The use of these types of drugs allows for enhanced levels of ACh at the motor end plate during repeated muscle stimulation.
Neurotransmitter Receptors
Once the molecules of neurotransmitter are released from a cell as the result of the firing of an action potential, they bind to specific receptors on the surface of the postsynaptic cell. In all cases in which these receptors have been cloned and characterized in detail, it has been shown that there are numerous subtypes of receptor for any given neurotransmitter. As well as being present on the surfaces of postsynaptic neurons, neurotransmitter receptors are found on presynaptic neurons. In general, presynaptic neuron receptors act to inhibit further release of neurotransmitter.
The vast majority of neurotransmitter receptors belong to a class of proteins known as the G-protein coupled receptors, GPCR. The GPCR are also called serpentine receptors because they exhibit a characteristic transmembrane structure: that is, they span the cell membrane, not once but seven times. The link between neurotransmitters and intracellular signaling is carried out by association, either with the receptor-associated G-protein, with protein kinases, or by the receptor itself in the form of a ligand-gated ion channel (for example, the nicotinic acetylcholine receptors). The receptors that are of the GPCR family are referred to as metabotropic receptors, whereas, the ligand-gated ion channel receptors are referred to as ionotropic receptors.
One additional characteristic of neurotransmitter receptors is that they are subject to ligand-induced desensitization. Receptor desensitization refers to the phenomenon whereby upon prolonged exposure to ligand results in uncoupling of the receptor from its signaling cascade. A common means of receptor desensitization involves receptor phosphorylation by receptor-specific kinases. Following phosphorylation of the receptor there is increased affinity for inhibitory molecules that uncouple the interaction of receptor with its associated G-protein. One major class of these desensitizing inhibitors are the arrestins. Arrestins were first identified in studies of β-adrenergic receptor desensitization and so were called β-arrestins.
Table of Common Neurotransmitters
This Table is a non-inclusive listing of the neurotransmitters
| Transmitter Molecule | Transmitter Class | Derived from | Receptors / Activities / Comments |
| Acetylcholine | other | Choline | functions in both the CNS and the PNS; receptors are cholinergic; 2 receptor classes: muscarinic (metabotropic) and nicotinic (ionotropic); within the periphery ACh is the major transmitter of the autonomic nervous system where it activates muscles; within the brain its major effects are inhibitory or anti-excitatory; its actions in cardiac tissue are also inhibitory |
| Adenosine | other | ATP | is an inhibitory neurotransmitter within the CNS, suppresses arousal thus promoting sleep; within the periphery adenosine exerts anti-inflammatory actions, stimulates vasodilation through vascular smooth muscle, induces bronchospasm in the lungs; within the heart it affects the cardiac conduction system; adenosine binds to a family of adenosine receptors (members of GPCR family) identified as A1 (coupled to Gi/o), A2A (coupled to Gs), A2B (coupled to Gs or Gq-dependent upon tissue), and A3 (coupled to Gi or Gq-dependent upon tissue); activation of A1 receptors expressed in cardiac pacemaker cells of sinoatrial node leads to reduced heart rate (chronotropy); activation of A2A receptors in coronary vascular smooth muscle cells induces vasodilation via the actions of the associated Gq; transport of adenosine in and out of cells is the function of the equilibrative nucleoside transporters (ENT) which are encoded by the SLC29 family of genes |
| Anandamide | other | phospholipids via at least two pathways | an endocannabinoid, binds to the cannabinoid receptors (CB1 and CB2) with highest affinity for CB1; CB1 is most abundant receptor in the CNS; classic response to CB1 activation is stimulation of food intake; exerts peripheral effects on overall energy homeostasis |
| Aspartate | amino acid | stimulates the NMDA receptor but not as strongly as glutamate | |
| ATP | other | as a neurotransmitter ATP is released from sympathetic, sensory and enteric nerves; ATP binds to metabotropic G-protein coupled receptors (GPCR) of the P2Y family of purinergic/pyrimidinergic receptors of which there are ten in humans: P2Y1, 2, 4, 6, 8, 10–14; P2Y12 is primarily expressed on the surface of platelets where it serves as a major ATP-mediating receptor of blood coagulation ATP also binds to the ionotropic family of purinergic receptors (P2X) which consists of seven members (P2X1-P2X7); these receptors modulate synaptic transmission throughout the autonomic nervous systems of the CNS and PNS; in the periphery the P2X receptors activate contractile activity of various muscle types | |
| Dopamine synthesis pathway | monoamine | tyrosine | within the CNS dopamine plays a major role in reward-motivated behavior such as feeding and drug-seeking behaviors; also involved in motor control; in the periphery dopamine regulates the release of several hormones such as insulin from the pancreas and norepinephrine from blood vessels; functions by binding to a family of dopaminergic receptors (GPCRs) |
| Epinephrine synthesis pathway | monoamine | tyrosine | catecholamine neurotransmitter and hormone; binds to both α- and β-adrenergic receptors (GPCRs); produced in the adrenal medulla and neurons in the CNS and PNS; primary hormone of the fight-or-flight response of the sympathetic nervous system; is a major regulator of metabolic processes in numerous tissues; regulates heart rate, induces vasoconstriction and bronchodilation |
| GABA | amino acid | Glutamate | major inhibitory neurotransmitter in the CNS; also exerts effects in the periphery; binds to two classes of receptor termed GABAA (ionotropic) and GABAB (metabotropic) |
| Glutamate | amino acid | most abundant excitatory neurotransmitter in the CNS; glutamate binds to the metabotropic glutamate receptors (mGluRs) of which there are eight (mGluR1–mGluR8) divided into three families; glutamate also binds to several ionotropic receptors including the N-methyl-D-aspartate (NMDA) receptor (NMDAR), the kainate receptors (KAR), and the α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) receptor (AMPAR) | |
| Glycine | amino acid | inhibitory neurotransmitter in the CNS primarily within the brainstem, spinal cord, and retina; binds to glycine receptors (GlyR) which are ionotropic; there are two separate subunit proteins of each GlyR (α and β) that combine in various ways to generate a pentameric structure; there are four α-subunit genes (α1–4) and one β-subunit gene; the primary adult form of GlyR is composed of three α1 subunits and two β subunits; is also a required co-agonist with glutamate on NMDA receptors and in this capacity exerts an excitatory effect | |
| Histamine | diamine | histidine | produced by mast cells, basophils, enterochromaffin-like cells (ECL) of the stomach, and hypothalamus; within the gut histamine stimulates gastric parietal cells to secrete acid; released from mast cells when allergens bind to IgE-antibody complexes; there are four histamine receptors (H1–H4) all of which are GPCRs |
| Nitric oxide, NO | gas | arginine | endothelial cells, phagocytic cells, CNS, gastrointestinal tract; binds to and activates soluble guanylate cyclase, oxidizes iron-containing proteins, nitrosylates protein sulfhydryl groups |
| Norepinephrine synthesis pathway | monoamine | tyrosine | catecholamine neurotransmitter and hormone; binds to both α- and β-adrenergic receptors (GPCRs); produced in CNS and PNS by sympathetic nerves; major neurotransmitter function is in regulation of cardiac inotropic (force) and chronotropic (rate) activities; functions along with epinephrine in the fight-or-flight response; involved in adaptive thermogenesis in brown adipose tissue (BAT) |
| Serotonin 5-hydroxytryptamine (5-HT) synthesis pathway | monoamine | tryptophan | most abundantly expressed in enterochromaffin cells of the gut where it regulates motility, also found in the CNS and platelets; released from activated platelets where it stimulates further activation propagating role of platelet aggregation in coagulation; in the CNS 5-HT regulates mood, appetite, sleep, memory and learning; selective serotonin re-uptake inhibitors (SSRIs) used in the treatment of depression |
Glutamate: Major Excitatory Neurotransmitter
Within the CNS glutamate is the main excitatory neurotransmitter. Neurons that respond to glutamate are referred to as glutamatergic neurons. Postsynaptic glutamatergic neurons possess four distinct classes of ionotropic receptors that bind glutamate released from presynaptic neurons. Three of these ionotropic receptor classes have been identified on the basis of their binding affinities for certain substrates and are thus, referred to as the kainate, 2-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA), and N-methyl-D-aspartate (NMDA) receptors. The fourth class of ionotropic glutamate receptor is termed the delta (δ) receptors. Each of these classes of glutamate receptor form ligand-gated ion channels, thus the derivation of the term ionotropic. There are also multiple subtypes of each of these classes of ionotropic glutamate receptor subunits.
Within the CNS glutamatergic neurons are responsible for the mediation of many vital processes such as the encoding of information, the formation and retrieval of memories, spatial recognition and the maintenance of consciousness. Excessive excitation of glutamate receptors has been associated with the pathophysiology of hypoxic injury, hypoglycemia, stroke and epilepsy.
AMPA Receptors
The AMPA receptor subunits are referred to as GluA1 (GluR1) through GluA4 (GluR4) and each is encoded by separate genes. Functional AMPA receptors consist of heterotetramers that are formed from dimers of GluA2 and dimers of either GluA1, GluA3, or GluA4. The GluA2 subunit of the receptor is responsible for regulating the permeability of the channel to calcium ions. The GluA2 mRNA is subject to RNA editing which alters the function of the calcium permeability character of the subunit. For details on the editing of the GluA2 mRNA go to the RNA: Transcription and Processing page. The AMPA receptors are found on most excitatory postsynaptic neurons where they mediate fast excitation. Indeed, AMPA receptors are responsible for the bulk of fast excitatory synaptic transmission throughout the CNS. The concept of fast synaptic transmission relates to the fact that the ion channel opens and closes quickly in response to ligand (e.g. glutamate) binding. The ion permeability of the AMPA receptors is controlled by the GluA2 subunit. AMPA receptors have low permeability to calcium ions even in the ligand-activated state and this is to prevent excitotoxicity in these neurons.
NMDA Receptors
The NMDA receptor is generated from two separate subunit families. These subunit families are identified as GluN1 (also called NMDAR1) and GluN2. There are four GluN2 subunits (GluN2A–GluN2D; also NMDAR2A–NMDAR2D). The four different GluN2 subunits are encoded by distinct genes. Although there is a single gene encoding the GluN1 subunit, multiple isoforms of this subunit are generated through alternative splicing events. The functional NMDA receptor is composed of a heterotetramer with all forms containing the GluN1 subunit and one of the different GluN2 subunits.
Unlike the other ionotropic glutamate receptors, the NMDA receptors are activated by simultaneous binding of glutamate and glycine. Glycine serves as a co-agonist and both amino acid neurotransmitters must bind in order for the receptor to be activated. Glycine binds to the GluN1 subunit while glutamate binds to the GluN2 subunit.
Glutamate binding to NMDA receptors results in calcium influx into the postsynaptic cells leading to the activation of a number of signaling cascades. These signaling cascades can include activation of Ca2+/calmodulin-dependent kinase II (CaMKII) leading to phosphorylation of the GluA2 AMPA receptor subunit. This latter effect results in long-term potentiation (LTP).
NMDA receptor activation also triggers PKC-dependent insertion of AMPA receptors into the synaptic membrane during LTP as well as activation of the kinases phosphatidylinositol-3-kinase (PI3K), protein kinase B/AK strain transforming (PKB/AKT), and glycogen synthase kinase 3 (GSK3), each of which modulates LTP. PKB (protein kinase B)/AKT (AK strain transforming) was originally identified as the tumor inducing gene in the AKT8 retrovirus found in the AKR strain of mice. Humans express three genes in the AKT family identified as AKT1 (PKBα), AKT2 (PKBβ), and AKT3 (PKBγ).
Kainate Receptors
The kainate receptor subunits are known as GluK1 through GluK5 (formerly GluR5, GluR6, GluR7, KA1, and KA2). The GluK1–GluK3 subunits can form hetero- and homomeric receptor complexes. In addition, alternative splicing of the GluK1 and GluK2 mRNAs results in at least five distinct subtypes (GluK1a–GluK1c, GluK2a, GluK2b). Less is known about the physiological significance of the kainate receptors.
One major role of the kainate receptors is in the regulation of synaptic plasticity. Another important function of the kainate receptors is in the regulation of the release of the inhibitory neurotransmitter GABA. This function of the kainate receptors is due to their presence on presynaptic GABAergic neurons.
Delta Receptors
The delta (δ) glutamate receptors were identified as ionotropic glutamate receptors based upon amino acid sequence similarity to the other more well-characterized ionotropic glutamate receptors. However, these proteins do not form glutamate-gated functional ion channels either alone or in combination with any of the other ionotropic glutamate receptor proteins. Indeed, these proteins do not bind glutamate or any other excitatory amino acid receptor ligands.
The GluD1 receptor (encoded by the GRID1 gene) is prominently expressed in inner ear hair cells and neurons of the hippocampus. The presentation of GluD1 in the inner ear indicates that it has a role in hearing.
The GluD2 receptor (encoded by the GRID2 gene) is expressed exclusively in the Purkinje cells of the cerebellum. GluD2 function is critical for the development of neuronal circuits and functions that includes long-term depression (LTD), learning and memory.
LTD refers to activity-dependent reduction in the signaling at neuronal synapses following a particular stimulus such as ligand-mediated receptor activation. LTD is the opposite long-term potentiation (LTP) which is a process where there are activity-dependent increases in synaptic activity.
Metabotropic Glutamate Receptors
Glutamate can also bind to another class of receptor termed the metabotropic glutamate receptors (mGluR; where the small m refers to metabotropic). There are eight known metabotropic glutamate receptors identified as mGluR1–mGluR8. Unlike the ionotropic receptors, the mGluR are members of the G-protein coupled receptor (GPCR) family. The mGluR can be divided into three distinct subclasses based upon sequence similarities and receptor associated G-protein.
Group I mGluR include mGluR1 and mGluR5, both of which are coupled to Gq type G-proteins and upon activation trigger increased production of DAG and IP3. Group II is composed of mGluR2 and mGluR3. Group III is composed of mGluR4, mGluR6, mGluR7, and mGluR8. Both group II and III mGluR activate an associated Gi type G-protein resulting in decreased production of cAMP.
The mGluR are primarily expressed on neurons and glial cells in close proximity to the synaptic cleft. Within the CNS, mGluR modulate the neurotransmitter effects of glutamate as well as a variety of other neurotransmitters. In addition to the CNS, mGluR have a widespread distribution in the periphery. Given their wide pattern of expression, diverse roles for mGluR have been suggested. Some of these processes include control of hormone production in the adrenal gland and pancreas, regulation of mineralization in the developing cartilage, modulation of cytokine production by lymphocytes, directing the state of differentiation in embryonic stem cells, and modulation of secretory functions within the gastrointestinal tract.
Table of Glutamate Receptors
| Receptor Name (other names) | Gene Symbol | Type / Class | Functions / Comments |
| mGluR1 | GRM1 | metabotropic, group I family | GPCR coupled to Gq-type G-protein; primarily a post-synaptic receptor; involved in nerve transduction events related to long-term potentiation (LTP) and long-term depression (LTD); increases NMDA receptor activity |
| mGluR2 | GRM2 | metabotropic, group II family | GPCR coupled to Gi-type G-protein; primarily a presynaptic receptor; involved in synaptic plasticity by exerting transient suppression of synaptic transmission occurring in response to receptor activation, induces persistent long-term depression (LTD), and mediates inhibition of long-term potentiation (LTP) |
| mGluR3 | GRM3 | metabotropic, group II family | GPCR coupled to Gi-type G-protein; primarily a presynaptic receptor; polymorphisms in GRM3 gene associated with psychosis and schizophrenia; modulates expression of glutamate transporters; affects NMDA receptor activity |
| mGluR4 | GRM4 | metabotropic, group III family | GPCR coupled to Gi-type G-protein; primarily a presynaptic receptor; depresses excitatory transmission by preventing glutamate release |
| mGluR5 | GRM5 | metabotropic, group I family | GPCR coupled to Gq-type G-protein; primarily a postsynaptic receptor; critical receptor involved in inhibitory learning processes such as drug-related self-administration learning; reduced signaling from this receptor can reverse fragile X phenotypes |
| mGluR6 | GRM6 | metabotropic, group III family | GPCR coupled to Gi-type G-protein; primarily a presynaptic receptor; involved in a photoreceptor-independent form of light adaptation within the retina; found on the photoreceptor–On bipolar cell synapse |
| mGluR7 | GRM7 | metabotropic, group III family | GPCR coupled to Gi-type G-protein; most widely distributed pre-synaptic mGluR; found at a wide range of synapses postulated to be critical for both normal CNS function and several human disorders; is a key regulator in shaping synaptic responses at glutamatergic synapses as well as in regulating critical aspects of inhibitory GABAergic transmission |
| mGluR8 | GRM8 | metabotropic, group III family | GPCR coupled to Gi-type G-protein; primarily a pre-synaptic receptor; involved in anxiety by depressing excitatory synaptic transmission in the bed nucleus of the stria terminalis (BNST) |
| GluA1 (GluR1) | GRIA1 | ionotropic: AMPA | responsible for the bulk of fast excitatory synaptic transmission throughout the CNS |
| GluA2 (GluR2) | GRIA2 | ionotropic, AMPA | controls the Ca2+ permeability of the AMPA receptor channels; RNA editing controls the permeability by altering a single amino acid (the Q/R site) in the second transmembrane domain (TMII) of the protein, if unedited the Q residue allows Ca2+ permeability whereas the edited amino acid (R) does not; almost all the CNS GluA2 is edited |
| GluA3 (GluR3) | GRIA3 | ionotropic, AMPA | responsible for the bulk of fast excitatory synaptic transmission throughout the CNS |
| GluA4 (GluR4) | GRIA4 | ionotropic, AMPA | responsible for the bulk of fast excitatory synaptic transmission throughout the CNS |
| GluK1 (GluR5) | GRIK1 | ionotropic, Kainate | three splice variants |
| GluK2 (GluR6) | GRIK2 | ionotropic, Kainate | two splice variants |
| GluK3 (GluR7) | GRIK3 | ionotropic, Kainate | |
| GluK4 (KA1) | GRIK4 | ionotropic, Kainate | expressed almost exclusively in the hippocampus |
| GluK5 (KA2) | GRIK5 | ionotropic, Kainate | protein retained within the ER unless assembled into a complex with either GluK1, GluK2, or GluK3 |
| GluN1 (NR1, NMDAR1) | GRIN1 | ionotropic, NMDA | functional NMDA receptors requires simultaneous binding of both glutamate and glycine; GluN1 provides the glycine-binding site as does the GluN3 subunits; receptors function as modulators of synaptic response and are involved in co-incidence detection (bidirectional current flow at a synapse) |
| GluN2A (NR2A, NMDAR2A) | GRIN2A | ionotropic, NMDA | functional NMDA receptors requires simultaneous binding of both glutamate and glycine; the GluN2 subunits provide the glutamate-binding sites; receptors function as modulators of synaptic response and are involved in co-incidence detection (bidirectional current flow at a synapse) |
| GluN2B (NR2B, NMDAR2B) | GRIN2B | ionotropic, NMDA | functional NMDA receptors requires simultaneous binding of both glutamate and glycine; the GluN2 subunits provide the glutamate-binding sites; receptors function as modulators of synaptic response and are involved in co-incidence detection (bidirectional current flow at a synapse) |
| GluN2C (NR2C, NMDAR2C) | GRIN2C | ionotropic, NMDA | functional NMDA receptors requires simultaneous binding of both glutamate and glycine; the GluN2 subunits provide the glutamate-binding sites; receptors function as modulators of synaptic response and are involved in co-incidence detection (bidirectional current flow at a synapse) |
| GluN2D (NR2D, NMDAR2D) | GRIN2D | ionotropic, NMDA | functional NMDA receptors requires simultaneous binding of both glutamate and glycine; the GluN2 subunits provide the glutamate-binding sites; receptors function as modulators of synaptic response and are involved in co-incidence detection (bidirectional current flow at a synapse) |
| GluN3A (NR3A, NMDAR3A) | GRIN3A | ionotropic, NMDA | functional NMDA receptors requires simultaneous binding of both glutamate and glycine; GluN3 subunits provide the glycine-binding sites as does the GluN1 subunit; receptors function as modulators of synaptic response and are involved in co-incidence detection (bidirectional current flow at a synapse) |
| GluN3B (NR3B, NMDAR3B) | GRIN3B | ionotropic, NMDA | functional NMDA receptors requires simultaneous binding of both glutamate and glycine; GluN3 subunits provide the glycine-binding sites as does the GluN1 subunit; receptors function as modulators of synaptic response and are involved in co-incidence detection (bidirectional current flow at a synapse) |
The Glutamate-Glutamine Cycle in the Brain
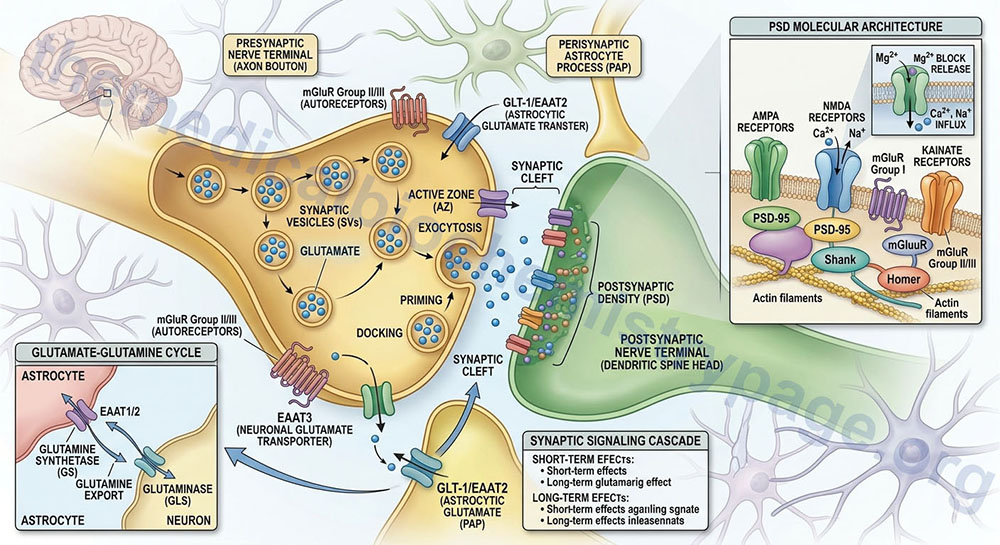
Within the CNS there is an interaction between the cerebral blood flow, neurons, and the protective astrocytes that regulates the metabolism of glutamate, glutamine, and ammonia. This process is referred to as the glutamate-glutamine cycle and it is a critical metabolic process central to overall brain glutamate metabolism. Using presynaptic neurons as the starting point, the cycle begins with the release of glutamate from presynaptic secretory vesicles in response to the propagation of a nerve impulse along the axon. The release of glutamate is a Ca2+-dependent process that involves fusion of glutamate containing presynaptic vesicles with the neuronal membrane.
Following release of the glutamate into the synapse it must be rapidly removed to prevent over excitation of the postsynaptic neurons. Synaptic glutamate is removed by three distinct process. It can be taken up into the postsynaptic cell, it can undergo reuptake into the presynaptic cell from which it was released or it can be taken up by a third non-neuronal cell, namely astrocytes. Postsynaptic neurons remove little glutamate from the synapse and although there is active reuptake into presynaptic neurons the latter process is less important than transport into astrocytes.
The membrane potential of astrocytes is much lower than that of neuronal membranes and this favors the uptake of glutamate by the astrocyte. Glutamate uptake by astrocytes is mediated by Na+-independent and Na+-dependent systems. The Na+-dependent systems have high affinity for glutamate and are the predominant glutamate uptake mechanism in the central nervous system. There are two distinct astrocytic Na+-dependent glutamate transporters identified as EAAT1 (for Excitatory Amino Acid Transporter 1; also called GLAST) and EAAT2 (also called GLT-1).
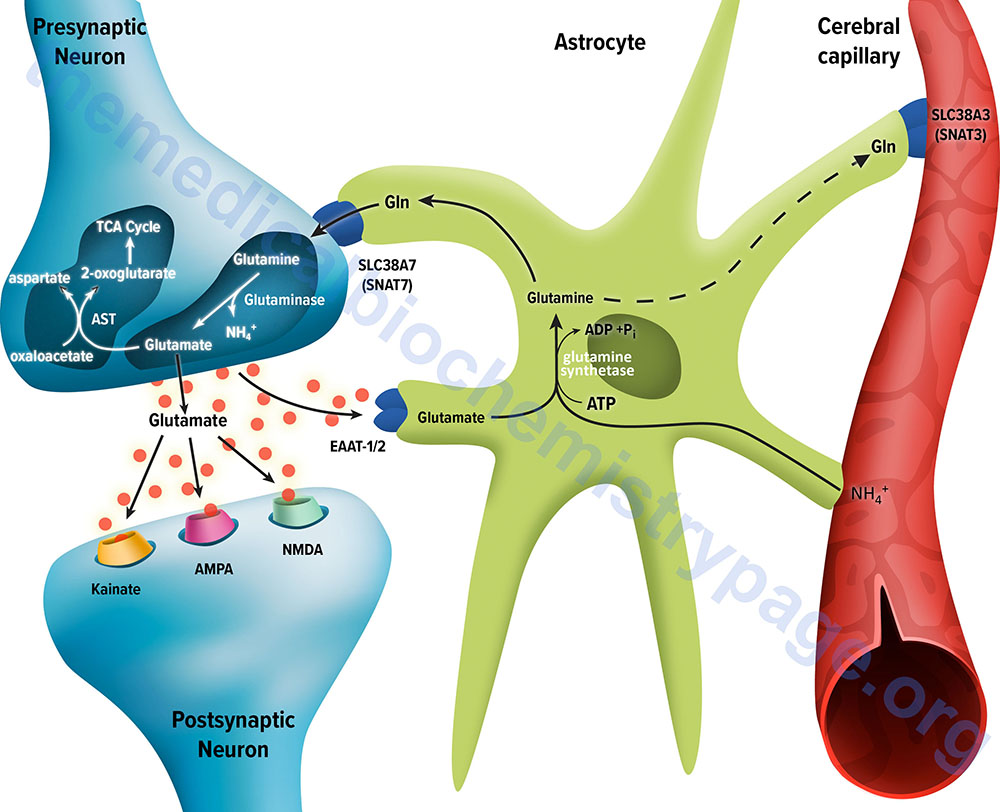
Following uptake of glutamate, astrocytes have the ability to dispose of the amino acid via export into the blood though capillaries that contact the foot processes of the astrocytes. The problem with glutamate disposal via this mechanism is that it would eventually result in a net loss of carbon and nitrogen from the CNS. In fact, the outcome of astrocytic glutamate uptake is its conversion to glutamine. Glutamine thus serves as a “reservoir” for glutamate but in the form of a non-neuroactive compound. Release of glutamine from astrocytes allows neurons to derive glutamate from this parent compound.
Astrocytes readily convert glutamate to glutamine via the glutamine synthetase catalyzed reaction as this microsomal enzyme is abundant in these cells. Indeed, histochemical data demonstrate that the glia are essentially the only cells of the CNS that carry out the glutamine synthetase reaction. The ammonia that is used to generate glutamine is derived from either the blood or from metabolic processes occurring in the brain.
Like the uptake of glutamate by astrocytes, neuronal glutamine uptake proceeds via both Na+-dependent and Na+-independent mechanisms. The major glutamine transporter in both excitatory and inhibitory neurons is the system N neutral amino acid transporter SNAT7 which is encoded by the SLC38A7 gene. The predominant metabolic fate of the glutamine taken up by neurons is hydrolysis to glutamate and ammonia via the action of the mitochondrial form of glutaminase encoded by the GLS2 gene. This form of glutaminase is referred to as phosphate-dependent glutaminase (PAG). The inorganic phosphate (Pi) necessary for this reaction is primarily derived from the hydrolysis of ATP and its function is to lower the KM of the enzyme for glutamine. During depolarization there is a sudden increase in energy consumption. The hydrolysis of ATP to ADP and Pi thus favors the concomitant hydrolysis of glutamine to glutamate via the resulting increased Pi.
Because there is a need to replenish the ATP lost during neuronal depolarization, metabolic reactions that generate ATP must increase. It has been found that not all neuronal glutamate derived from glutamine is utilized to replenish the neurotransmitter pool. A portion of the glutamate can be oxidized within the nerve cells following transamination. The principal transamination reaction involves aspartate aminotransferase (AST) and yields α-ketoglutarate (2-oxoglutarate) which is a substrate in the TCA cycle. Glutamine, therefore, is not simply a precursor to neuronal glutamate but a potential fuel, which, like glucose, supports neuronal energy requirements.
Glutamate, released as a neurotransmitter, is taken up by astrocytes, converted to glutamine, released back to neurons where it is then converted back to glutamate represents the complete glutamate-glutamine cycle. The significance of this cycle to brain glutamate handling is that it promotes several critical processes of CNS function. Glutamate is rapidly removed from the synapse by astrocytic uptake thereby preventing over-excitation of the postsynaptic neuron. Within the astrocyte glutamate is converted to glutamine which is, in effect, a non-neuroactive compound that can be transported back to the neurons. The uptake of glutamine by neurons provides a mechanism for the regeneration of glutamate which is augmented by the generation of Pi as a result of ATP consumption during depolarization. Since the neurons also need to regenerate the lost ATP, the glutamate can serve as a carbon skeleton for oxidation in the TCA cycle.
Lastly, but significantly, the incorporation of ammonia into glutamate in the astrocyte serves as a mechanism to buffer brain ammonia.
Glycine
Glycine, as an amino acid found in proteins, is critical to the functions of several different classes of protein, particularly those of the extracellular matrix. However, glycine as a free amino acid also functions as a highly important neurotransmitter within the central nervous system, CNS. Glycine and GABA are the major inhibitory neurotransmitters in the CNS, whereas, glutamate is the major excitatory neurotransmitter. In conjunction with glutamate, glycine can also function in an excitatory capacity as a co-agonist acting on the NMDA subtype of glutamate receptors (see section above).
The receptors to which glycine binds were originally identified by their sensitivity to the alkaloid strychnine. Strychnine-sensitive glycine receptors (GlyR) mediate the synaptic inhibition exerted in response to glycine binding. Glycinergic synapses mediate fast inhibitory neurotransmission within the spinal cord, brainstem, and caudal brain. The effects of glycine exert control over a variety of motor and sensory functions, including vision and audition.
The GlyR are members of the ionotropic family of ligand-gated ion channels. The binding of glycine leads to the opening of the GlyR integral anion channel, and the resulting influx of Cl– ions hyperpolarizes the postsynaptic cell, thereby inhibiting neuronal firing.
Glycine Transporters
Cellular uptake of glycine, particularly within neurons in the central nervous system (CNS), is regulated by the presence of specific glycine transporters identified as GlyT. There are two subtypes of GlyT identified as GlyT1 and GlyT2. Both glycine transporters are members of the solute carrier family of membrane transporters. The GlyT1 protein is encoded by the SLC6A9 gene and the GlyT2 protein is encoded by the SLC6A5 gene. The tissue distribution and function of the two glycine transporters are distinct. GlyT1 is predominantly expressed in glutamatergic neurons where it functions in the regulation of glycine levels in the vicinity of the NMDA-type glutamate receptors. GlyT2 is predominantly expressed in glycinergic neurons where it functions to regulate inhibitory glycinergic neurotransmission by decreasing synaptic glycine concentrations after presynaptic release. A form of inherited hyperekplexia of presynaptic origin (HKPX3) results from mutations in the SLC6A5 (GlyT2) gene.
Impaired glutamatergic neurotransmission via the NMDA receptors has been associated with the symptoms of schizophrenia and the associated cognitive deficit. Pharmacologic inhibitors of GlyT1 have some utility to improve impaired NMDA receptor function in psychosis by increasing synaptic glycine concentrations. These transport inhibitors function by increasing extrasynaptic glycine concentrations via inhibition of its neuronal or glial reuptake processes. When used in combination with other antipsychotic medications, GlyT1 inhibitors have been shown to be capable of restoring disturbed glutamatergic-GABAergic-dopaminergic balance in psychosis.
Glycine Ionotropic Receptors
The primary receptors to which glycine binds (GlyR) are ionotropic receptors that are members of the group I ligand-gated ion channel (LGIC) class of receptors. The LGIC receptors are members of the Cys loop receptor family that also includes the nicotinic acetylcholine receptors (nAChR), the serotonin type 3 receptor (5-HT3), and the GABAA receptors (GABAAR). The GlyR are composed of three different proteins, two of which constitute the actual receptor and a third protein that serves a scaffolding function.
The receptor subunits are referred to as GlyRα and GlyRβ. These subunits are tightly bound to a cytosolic scaffolding protein identified as gephyrin that is encoded by the GPHN gene. Gephyrin is tightly bound to the GlyRβ subunit. In addition to its role in GlyR function, gephryin functions to regulate the activity of the GABAA receptor and it is required for molybdenum cofactor biosynthesis. Functional GlyR are heteropentameric proteins similar to the organization of the nAChR found in skeletal muscle. The typical subunit composition of the heteropentameric GlyR is (GlyRα)2(GlyRβ)3.
Humans express four GlyR genes encoding α subunits (GLRA1–GLRA4) and a single GlyR gene encoding the β subunit (GLRB). All GlyRα subunits display high amino acid sequence identity and form functional homomeric glycine-gated channels. The GlyRα subunits possess critical determinants of ligand binding.
The GLRA1 gene is located on chromosome 5q32 and is composed of 10 exons that generate three alternatively spliced mRNAs. Glycine receptors that contain the GlyRα1 subunit represent the predominant form of the α-subunit in adult glycine receptors. Several mutations in the GLRA1 gene have been shown to be associated with the startle disease known as hereditary hyperekplexia type 1, HKPX1. The hallmark symptoms of HKPX1 are an exaggerated startle response to auditory or tactile stimuli and, particularly in neonates, transient muscle rigidity referred to as “stiff baby syndrome”.
The GLRA2 gene is located on the X chromosome (Xp22.2) and is composed of 13 exons that generate four alternatively spliced mRNAs. Two of the splice variant GLRA2 mRNAs encode the same protein, thus, the four variant mRNAs generate three different GLRA2 proteins.
The GLRA3 gene is located on chromosome 4q34.1 and is composed of 13 exons that generate two alternatively spliced mRNAs. In addition to alternative splicing, the GLRA3 mRNA is subject to editing that results in the substitution of a Pro residue for a Leu residue at amino acid 185 in the extracellular domain. This version of the GlyRα3 protein confers an increased agonist affinity to GlyRα3-containing glycine receptors. The GlyRα3-containing GlyRs are involved in the pathways of nociception (pain sensation) within the spinal cord. Specific spinal cord neurons (in laminae I and II) mediate pain sensation in response to the inflammatory mediator, prostaglandin E2 (PGE2). When PGE2 binds to its receptor in these neurons (the EP2 receptor), PKA is activated which then phosphorylates the GlyRα3 protein in the glycine receptor resulting in down-regulation of glycine stimulated inhibitory circuits in these neurons. The analgesic effects of cannabinoids and endocannabinoids involves the modulation of GlyRα3-containing glycine receptors. Thus, it is postulated that GlyRα3 represents a potentially useful target for the pharmacologic intervention in chronic pain syndromes.
The GLRA4 gene is located on the X chromosome (Xq22.2) on the other arm relative to the position of the GLRA2 gene. The GLRA4 gene is composed of 9 exons that generate two alternatively spliced mRNAs.
The GlyRβ gene is located on chromosome 4q31.3 and is composed of 12 exons that generate three alternatively spliced mRNAs that encode two distinct GlyRβ isoforms. Unlike the GlyRα subunits which can form a functional glycine-gated ion channel, the GlyRβ protein cannot form a functional glycine receptor on its own. The role of the GlyRβ subunit is to regulate agonist binding and intracellular trafficking and synaptic clustering of post-synaptic GlyR. Mutations in the GLRB gene are associated with another form of hyperekplexia identified as HKPX2.
Glycine Metabotropic Receptors
Recent studies (2023) identified that glycine binds to and modulates the signaling of a receptor identified as GPR158. GPR158 is an atypical, class C orphan GPCR whose levels of expression are highest in the brain within the hippocampus and the prefrontal cortex. This finding demonstrated that glycine initiates effects through metabotropic receptors (mGlyR) in addition to the classically identified ionotropic glycine receptors (GlyR).
GPR158 is a Gs-type GPCR that also has been shown to bind the non-protein amino acid, taurine. Within cortical neurons GPR158 enhances the production of cAMP that in turn modulates the activity of ion channels, kinases, and neurotrophic factors that are known to be involved in neuronal excitability and synaptic transmission. This effect of GPR158 signaling has lead to its implication in affective states and cognition. Indeed, in patients with Major Depressive Disorder (MDD), the level of GPR158 is upregulated in the prefrontal cortex.
The effect of glycine binding to GPR158 is decreased production of cAMP indicating that glycine functions as an antagonist of GPR158 signaling. This effect of glycine on GPR158 signaling is potentiated by the association of the GTPase activating protein (GAP) identified as RGS7 (regulatory of G-protein signaling 7) and the atypical heterotrimeric G-protein subunit identified as G-protein beta 5 (Gβ5: encoded by the GNB5 gene). Gβ5 is classified as an atypical G-protein subunit because because it binds directly to the GAP, in this case directly to RGS7. The complex of RG77 and Gβ5 accelerates the inactivation of the G-protein.
GABA (γ-Aminobutyric acid)
Several amino acids have distinct excitatory or inhibitory effects upon the nervous system. The amino acid derivative, γ-aminobutyrate (GABA; also called 4-aminobutyrate) is a major inhibitor of presynaptic transmission in the CNS, and also in the retina. Neurons that secrete GABA are termed GABAergic. GABA cannot cross the blood-brain-barrier and as such must be synthesized within neurons in the CNS. The synthesis of GABA in the brain occurs via a metabolic pathway referred to as the GABA shunt.
GABA Shunt
Glucose is the principal precursor for GABA production via its conversion to 2-oxoglutarate (α-ketoglutarate) in the TCA cycle. Within the context of the GABA shunt, GABA is synthesized in the cytosol via the reaction described in the next section.
GABA is transported into the mitochondria and converted to succinic semialdehyde through the action of 4-aminobutyrate aminotransferase (commonly called GABA transaminase). GABA transaminase uses pyruvate as the amino group acceptor converting pyruvate to alanine. Succinic semialdehyde is then oxidized to succinate by the NAD+-dependent enzyme, succinic semialdehyde dehydrogenase.
The succinate can then enter the TCA cycle and be converted to 2-oxoglutarate which can, in turn be converted to glutamate through the action of the mitochondrial glutamate dehydrogenase encoded by the GLUD1 gene. The glutamate is transported out of the mitochondria and can again serve as the substrate for GABA synthesis.
GABA transaminase is encoded by the ABAT gene which is located on chromosome 16p13.2 and is composed of 23 exons that generate 20 alternatively spliced mRNAs that collectively encode 11 different protein isoforms.
Succinic semialdehyde dehydrogenase is encoded by the ALDH5A1 (aldehyde dehydrogenase 5 family member A1) gene which is located on chromosome 6p22.3 and is composed of 11 exons that generate three alternatively spliced mRNAs, each of which encode a distinct protein isoforms.
GABA Synthesis by GABAergic Neurons
Glutamate decarboxylase (GAD) catalyzes the decarboxylation of glutamic acid to form GABA. There are two GAD genes in humans identified as GAD1 and GAD2. The major GAD isoforms produced by these two genes are identified as GAD67 (GAD1 gene) and GAD65 (GAD2 gene) which is reflective of their molecular weights. Both the GAD1 and GAD2 genes are expressed in the brain and GAD2 expression also occurs in the pancreas but at significantly lower levels than in the brain.
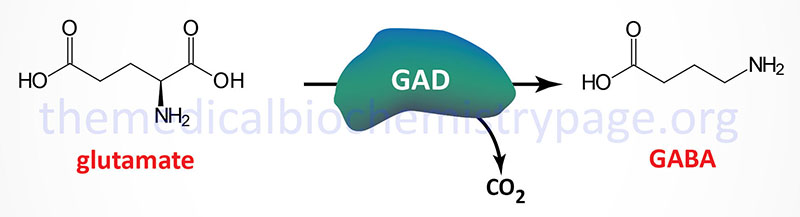
The GAD1 gene is located on chromosome 2q31.1 and is composed of 21 exons that generates two alternatively spliced mRNAs. The major GAD1 encoded mRNA generates a protein of 591 amino acids (67 kDa: GAD67) while the minor mRNA generates a protein of 224 amino acids (25 kDa: GAD25).
The GAD2 gene is located on chromosome 10p12.1 and is composed of 16 exons that generate two alternatively spliced mRNAs, both of which encode the same protein of 585 amino acid (65 kDa: GAD65).
The activity of GAD requires pyridoxal phosphate (PLP) as a cofactor. PLP is generated from the B6 vitamins (pyridoxine, pyridoxal, and pyridoxamine) through the action of pyridoxal kinase. Pyridoxal kinase itself requires zinc for activation. A deficiency in zinc or defects in pyridoxal kinase can lead to seizure disorders, particularly in seizure-prone pre-eclamptic (hypertensive condition in late pregnancy) patients.
The presence of anti-GAD antibodies (both anti-GAD65 and anti-GAD67) is a strong predictor of the future development of type 1 diabetes in high-risk populations.
GABA Synthesis in Astrocytes
In addition to neurons, the specialized glial cells called astrocytes are also capable of synthesizing GABA. Unlike GABAergic neurons that can only utilize glutamate as the substrate for GABA synthesis, astrocytes can synthesize GABA from the polyamine, putrescine. A major brain location for astrocyte GABA synthesis is the hippocampus and experiments in mice have demonstrated that increased hippocampal putrescine levels are associated with increased levels of GABA.
Astrocyte GABA synthesis from putrescine can occur via two distinct pathways. One pathway is initiated by the activity of the enzyme identified as diamine oxidase, DAO. In humans, DAO is encoded by the AOC1 (amin oxidase copper containing 1) gene. This pathway is described and diagrammed in the Agmatine Synthesis and Catabolism section below.
The second pathway by which astrocytes divert putrescine into GABA synthesis involves a monoamine oxidase (MAO) pathway. In this pathway putrescine is converted to N1-acetylputrescine via the enzyme identified as spermidine/spermine N1-acetyltransferase 1, encoded by the SAT1 gene. The SAT1 encoded enzyme is commonly referred to as putrescine acetyltransferase, PAT. N1-acetylputrescine is then oxidized by monoamine oxidase B (MAO-B; encoded by the MAOB gene) forming N1-acetyl-γ-aminobutyraldehyde (4-acetamidobutanal; also referred to as N-acetyl-GABA aldehyde). Reduction of N-acetyl-GABA aldehyde by the ALDH1A1 encoded enzyme yields N1-acetyl-GABA. Lastly, N1-acetyl-GABA is deacetylated to GABA by SIRT2.
GABA Receptors
GABA exerts its effects by binding to two distinct receptor subtypes. The GABA-A (GABAA) receptors are members of the ionotropic receptors, specifically the Cys-loop subfamily of ligand-gated ion channels that includes the nicotinic ACh receptors (nAChR), glycine receptors (GlyR), and the 5-HT3 (serotonin) receptors. The GABA-B (GABAB) receptors belong to the class C family of metabotropic G-protein coupled receptors (GPCR). The GABA-A receptors are members of the ionotropic receptor family and are chloride channels that, in response to GABA binding, increase chloride influx into the GABAergic neuron. The GABA-B receptors are coupled to a G-protein that activates an associated potassium channel that when activated by GABA leads to potassium efflux from the cell. The anxiolytic drugs of the benzodiazepine family exert their soothing effects by potentiating the responses of GABA-A receptors to GABA binding.
GABA-A Receptors
Functional GABA-A receptors are generated by the combination of a wide array of different subunits. A total of 19 GABA-A receptor subunit genes have been identified in humans that code for α (alpha), β (beta), γ (gamma), δ (delta), ε (epsilon), π (pi), θ (theta), and ρ (rho). The overall diversity of GABA-A receptors is further increased as several of these genes undergo alternative splicing. The complexity of the diverse array of molecular compositions of the GABA-A receptors has important functional and clinical consequences as they determine the properties and pharmacological modulations of a given receptor complex. In addition, zinc ions are known to regulate GABA-A receptor activity via inhibition of the receptor through an allosteric mechanism that is critically dependent on the receptor subunit composition. The GABRG3 (γ3 subunit gene) encoded protein is critical to this zinc-mediated regulation. Although the minimal requirement to produce a functional GABA-gated ion channel is the inclusion of both α and β subunits, the most common type in the brain is a heteropentameric complex composed of two α subunits, two β subunits, and a γ subunit (α2β2γ). The GABA-A receptors bind two molecules of GABA and in the heteropentameric receptors this binding site is created by the interface between the α and β subunits.
The GABA-Aρ subunits do not form heteromeric complexes with other GABA-A receptor subunits but only form homomeric receptor complexes. The GABA-Aρ receptors were formerly referred to as the GABA-C receptors.
The anxiolytic/sedative effects of the barbiturates and benzodiazepines are exerted via their binding to subunits of the GABA-A receptors. Benzodiazepines bind to a site on the GABA-A receptor created by the association of the gamma (γ) subunit and one of the alpha (α) subunits. There are two distinct subtypes of benzodiazepine receptors termed BZ1 (BZ1) and BZ2 (BZ2). The BZ1 receptor is formed by the interaction of γ and α1 subunits, whereas the BZ2 receptors is formed by the interaction of the γ and α2, α3 or α5 subunits. The receptor for the barbiturates is the beta (β) subunit of the GABA-A receptor. When benzodiazepines bind to the GABA-A receptor they potentiate the actions of GABA and require the presence of GABA in order for activation of the ion channel. Barbiturates can induce GABA-A channel opening in the absence of GABA when administered at high dose and as a result they can be lethal due to the level of CNS suppression. The potential for lethal toxicity of a benzodiazepine requires an extremely large dose. This difference in toxicity between barbiturates and benzodiazepines is the major reason barbiturates are not often used clinically any longer.
Under physiological conditions the binding of GABA to any of the GABA-A receptors leads to membrane hyperpolarization and a reduction of action potential firing. However, studies have also demonstrated the GABA-A activation can result in membrane reversal potential that is close to, or even at a more depolarized potential than the resting membrane potential at a synapse. This results in a membrane depolarization referred to as shunting inhibition. Shunting inhibition is also called divisive inhibition and defines a form of post-synaptic potential inhibition. The term shunting is used because the synaptic conductance short-circuits currents that are generated at adjacent excitatory synapses. If a shunting inhibitory synapse is activated, the amplitude of subsequent excitatory postsynaptic potentials (EPSPs) is reduced.
The major effect of GABA-A receptor activation is reduced dendritic excitatory glutamatergic responses as a consequence of a local increase in conductance across the plasma membrane. In addition to shunting inhibition, the polarity of GABA-A receptor-mediated responses can change during different physiological or pathological conditions. For example, GABA triggers excitation during the day and inhibition during the night within neural circuits of the suprachiasmatic nucleus. Also, the repeated activation of GABA-A receptors can lead to a switch from a hyperpolarizing to depolarizing direction and can, thus, enhance cell firing. The activation of GABA-A receptors results in both phasic inhibitory postsynaptic currents (IPSCs) and tonic currents. The GABA-A-induced tonic current result from GABA acting on extrasynaptic receptors composed of a different subunit composition and therefore, different pharmacological activity compared with the synaptic receptors.
Table of GABA-A Receptor Subunits
| Receptor Subunit | Gene Symbol | Functions / Comments |
| GABA-A alpha 1 (α1) | GABRA1 | GABRA1 protein is phosphorylated in a glycolysis-dependent reaction involving a kinase activity associated with the enzyme glyceraldehyde 3-phosphate dehydrogenase (GAPDH), GAPDH-mediated phosphorylation maintains functionality of the protein; this process implicates a link between regional cerebral glucose metabolism and GABAergic currents since the mechanism depends on locally produced glycolytic ATP and GAPDH activity; cortical tissue isolated from epileptic patients contains GABRA1 subunits in a reduced phosphorylation state compared to tissue from non-epileptic individuals; mutations in the GABRA1 gene associated with susceptibility to juvenile myoclonic epilepsy |
| GABA-A alpha 2 (α2) | GABRA2 | polymorphisms in the GABRA2 gene associated with susceptibility to alcohol dependence; pharmacologic-specific activation of the GABA-A α2 subunit is highly effective against inflammatory and neuropathic pain without sedation typical of benzodiazepine activation of the α1 subunit |
| GABA-A alpha 3 (α3) | GABRA3 | similar to effects at the α2 subunit, pharmacologic-specific activation of the GABA-A α3 subunit is highly effective against inflammatory and neuropathic pain without sedation typical of benzodiazepine activation of the α1 subunit |
| GABA-A alpha 4 (α4) | GABRA4 | pentameric GABA-A receptors that contain the α4 subunit are insensitive to benzodiazepines; shunting inhibition involving GABA-A receptor complexes that contain the α4 subunit reduces NMDA receptor activation leading to impaired long-term potentiation, LTP |
| GABA-A alpha 5 (α5) | GABRA5 | variable numbers of a partial duplication in the GABRA5 gene are found in different individuals; the GABRA5 gene is located within the chromosome 15 imprinted region found deleted in Prader-Willi and Angelman syndromes; the duplication number is higher in individuals with cytogenetically detectable deletions in the 15q region |
| GABA-A alpha 6 (α6) | GABRA6 | cerebellar motor control is likely to be a distinct behavioral function associated with GABA-A receptors that contain the α6 subunit; disruption in expression of the GABRA6 gene leads to an associated loss of expression from the GABRD gene |
| GABA-A beta 1 (β1) | GABRB1 | defects in GABRB1 gene may contribute to schizophrenia as well as to excessive alcohol consumption |
| GABA-A beta 2 (β2) | GABRB2 | single nucleotide polymorphisms (SNP) and copy number variations (CNV) in GABRB2 gene associated with schizophrenia |
| GABA-A beta 3 (β3) | GABRB3 | the GABRA3 gene is located within the chromosome 15 imprinted region found deleted in Prader-Willi and Angelman syndromes; deletion of GABRB3 is found in both disorders and it is, therefore, suggested that loss of the β3 subunit plays a role in the pathogenesis of these syndromes |
| GABA-A gamma 1 (γ1) | GABRG1 | both the γ1 and γ2 subunits are important in the effects of the benzodiazepines on GABA-A receptor function; |
| GABA-A gamma 2 (γ2) | GABRG2 | both the γ1 and γ2 subunits are important in the effects of the benzodiazepines on GABA-A receptor function; polymorphisms in the GABRG2 gene are associated with susceptibility to epilepsy and febrile seizures; presence of the γ2 subunit results in a low sensitivity of GABA-A receptors to allosteric regulation by zinc ion |
| GABA-A gamma 3 (γ3) | GABRG3 | the γ3 subunit is critical to the allosteric regulation of GABA-A receptors by zinc ions whereas presence of the γ2 subunit results in a low sensitivity to zinc ion regulation |
| GABA-A delta (δ) | GABRD | polymorphisms in the GABRD gene are associated with susceptibility to epilepsy and febrile seizures; three variants of the GABRD protein are produced in the brain identified as GABRD-1A, -1B, and -1C; the δ subunit is involved in the tonic (continuous) currents elicited by GABA-A receptors which modifies the spatial and temporal integration of excitatory neurotransmission |
| GABA-A epsilon (ε) | GABRE | alternative splicing of the GABRE mRNA occurs at several positions depending upon the tissue of expression |
| GABA-A pi (π) | GABRP | expressed at highest levels in the uterus; presence of the π subunit in pentameric GABA-A receptors modifies the receptor sensitivity to steroidogenic compounds |
| GABA-A theta (θ) | GABRQ | |
| GABA-A rho 1 (ρ1) | GABRR1 | protein contains a chloride-sensitive anion channel |
| GABA-A rho 2 (ρ2) | GABRR2 | |
| GABA-A rho 3 (ρ3) | GABRR3 |
GABA-B Receptors
GABA also acts on GABA-B receptors that are members of the GPCR family of receptors. There are two GABA-B receptors subunits identified as GABA-B1 (GABAB1) and GABA-B2 (GABAB2). These two subunits heterodimerize to form the functional receptor that can be found on both pre- and post-synaptic membranes. Neither receptor subunit is functional as a GABA receptor independently.
The GABA-B receptors are coupled to G-proteins of the Gi type. The G-protein is linked to potassium channels (GIRK or Kir3) and activation of the G-protein results in increased conductance of the associated channel. GABA-B receptor activation on post-synaptic membranes generally leads to activation of the inwardly rectifying potassium channels which underlies the late phase of inhibitory postsynaptic potentials (IPSP). Activation of pre-synaptic GABA-B receptors decreases neurotransmitter release by inhibiting voltage-activated Ca2+ channels of the N or P/Q types.
Activation of GABA-B receptors also modulates the production of cAMP. This function leads to a wide range of actions on ion channels as well as other proteins that are targets of PKA. The cAMP modulation by GABA-B receptors effects modulation of both neuronal and synaptic functions.
Pharmacology of GABA Receptors
The anxiolytic/sedative effects of the barbiturates and benzodiazepines are exerted via their binding to subunits of the GABA-A receptors. Benzodiazepines bind to a site on the GABA-A receptor created by the association of the gamma (γ) subunit and one of the alpha (α) subunits.
There are two distinct subtypes of benzodiazepine receptors termed BZ1 (BZ1) and BZ2 (BZ2). The BZ1 receptor is formed by the interaction of γ and α1 subunits, whereas the BZ2 receptors is formed by the interaction of the γ and α2, α3 or α5 subunits.
The receptor for the barbiturates is the beta (β) subunit of the GABA-A receptor. When benzodiazepines bind to the GABA-A receptor they potentiate the actions of GABA and require the presence of GABA in order for activation of the ion channel. Barbiturates can induce GABA-A channel opening in the absence of GABA when administered at high dose and as a result they can be lethal due to the level of CNS suppression. The potential for lethal toxicity of a benzodiazepine requires an extremely large dose. This difference in toxicity between barbiturates and benzodiazepines is the major reason barbiturates are not often used clinically any longer.
The significance of the BZ1 receptor isoform is that it is solely involved in mediating the induction of sleep. This fact has led to the development of several classes of drug that specifically target this GABA-A receptor isoform, and more precisely, the site on the GABA-A complex that forms the BZ1 binding site. The non-benzodiazepine drug, zolpidem (Ambien®), exerts its hypnotic sleep inducing effects due to near selective binding to the BZ1 site. Another non-benzodiazepine drug used for its hypnotic sleep inducing effect is eszopiclone (Lunesta®). Although the precise mechanism of action of eszopiclone is not fully understood, it is believed to function similarly to zolpidem in binding to the BZ1 receptor site on GABA-A receptor isoforms.
Acetylcholine
Acetylcholine (ACh) is a simple molecule synthesized from choline and acetyl-CoA through the action of choline O-acetyltransferase. Neurons that synthesize and release ACh are termed cholinergic neurons.
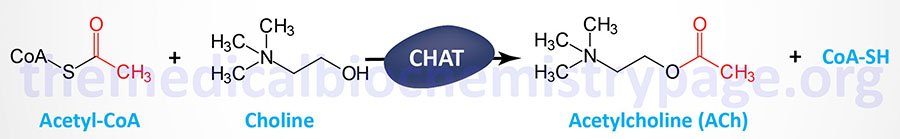
Choline O-acetyltransferase is encoded by the CHAT gene. The CHAT gene is located on chromosome 10q11.23 and is composed of 18 exons that generate seven alternatively spliced mRNAs, five of which encode the same 630 amino acid isoform of the enzyme.
Choline (a five-carbon quaternary ammonium cation) is primarily acquired in the diet and is transported into cells via the FLVCR1 (feline leukemia virus subgroup C receptor 1) and FLVCR2 encoded transporters. The transporters are members of the SLC family of transporters and as such are also identified as SLC49A1 and SLC49A2, respectively. Two isoforms of FLVCR1 have been characterized, FLVCR1a and FLVCR1b. Once inside cells, such as cholinergic neurons, free choline can be used for the synthesis of acetylcholine or it can be phosphorylated by choline kinase-α (encoded by the CHKA gene) which activates it for phospholipid synthesis. As a result of phospholipid synthesis, the choline for acetylcholine synthesis can also be derived from phosphatidylcholine.
Another source of choline for acetylcholine synthesis is glycerophosphocholine, GPC (also designated alpha-GPC). The action of phospholipases of the PLA2 family on PC yields a family of bioactive lipids termed the lysophosphatidylcholines, LPC (also designated lysoPC). One of the PLA2 family member enzymes, commonly identified as iPLA2 (encoded by the PNPLA6 gene), deacylates PC to yield GPC. Glycerophosphocholine is also derived via the actions of lysosomal PLA2 (encoded by the PLA2G15 gene) acting on LPC in the lysosomes. This lysosomal GPC is transported to the cytosol by the transporter identified as battenin (encoded by the CLN3 gene). Mutations in the CLN3 gene are the cause of the neuronal ceroid lipofuscinosis commonly identified as Batten–Spielmeyer–Sjogren disease.
Glycerophosphocholine (GPC) represents a major intracellular storage form of choline, particularly in cholinergic neurons. Release of choline from GPC involves the actions of the enzyme, glycerophosphodiesterase 1, which is encoded by the GPCPD1 gene. Expression of the GPCPD1 gene is high in brain and skeletal muscle.
De novo choline synthesis, although only a minor source of free choline, takes place in the liver and involves the conversion of phosphatidylethanolamine (PE) to phosphatidylcholine (PC) via the actions of the methyltransferase, phosphatidylethanolamine N-methyltransferase (PEMT). The choline can be released from PC through the actions of enzymes of the phospholipase D (PLD) family as well as in the conversion of PC to phosphatidylserine (PS) catalyzed by phosphatidylserine synthase 1 (encoded by the PTDSS1 gene).
When an action potential reaches the terminus of a presynaptic neuron a voltage-gated calcium channel is opened. The influx of calcium ions (Ca2+) stimulates the exocytosis of presynaptic vesicles containing ACh, which is thereby released into the synaptic cleft. This vesicle mobilization and ACh exocytosis occurs within a few hundred microseconds of the action potential reaching the pre-synaptic membrane. Vesicle mobilization results from the interaction of Ca2+ with proteins of the synaptotagmin family. The interaction of Ca2+ with synaptotagmins activates these membrane targeted proteins to interact with membrane phospholipids and other proteins of the exocytotic apparatus termed SNARE proteins. SNARE proteins are required for the process of membrane fusion that is a necessary step in the interaction of exocytotic vesicle with the plasma membrane.
Once released, ACh must be removed rapidly in order to allow repolarization to take place. The removal of acetylcholine is a hydrolysis reaction catalyzed by the enzyme, acetylcholinesterase (AChE). AChE is a highly active enzyme capable of hydrolyzing on the order of 25,000 molecules of ACh per second. The released choline is then taken back up by the presynaptic neuron where it can once again serve as a substrate for ACh synthesis via choline acetyltransferase.
Two different mammalian AChE isoforms are produced from the single ACHE gene (chromosome 7q22.1) in humans via alternative splicing and post-translational modification. These two AChE isoforms are termed T (tail) and H (hydrophobic). The T form (AChET, also known as the hydrophilic form) is the predominant enzyme in the brain and at the neuromuscular junction. The H form (AChEH) is the principal enzyme form found in erythroid cells. The AChEH isoform is anchored to red blood cell membranes via a GPI-linkage and this form constitutes the Yt blood group antigen.
Two main classes of ACh receptors have been identified on the basis of their responsiveness to the toadstool alkaloid muscarine and to nicotine, respectively. These receptors are, therefore, termed the muscarinic ACh receptors (mAChRs) and the nicotinic ACh receptors (nAChRs). The muscarinic ACh receptors are G-protein coupled receptors (GPCR) and are also referred to as metabotropic receptors. The nicotinic ACh receptors are ligand-gated ion channels which are also referred to as ionotropic receptors. Both receptor classes are abundant in the human brain.
Muscarininc Acetylcholine Receptors
The are five subtypes of muscarinic ACh receptors, identified as M1–M5, that were originally classified based upon their responses to various pharmacologic agents. The M1, M3, and M5 muscarinic ACh receptors are coupled to the Gq type G-proteins that activate PLCβ. The M2 and M4 muscarinic ACh receptors are coupled to Gi type G-proteins that inhibit adenylate cyclase. Muscarinic ACh receptor desensitization occurs in response to phosphorylation of the receptors by kinases that are members of the G-protein coupled receptor kinase (GRK) family. For example the M2 receptor is phosphorylated by GRK2 (originally called β-adrenergic receptor kinase-1, βARK1). More information on the GRK family can be found in the Signal Transduction Pathways: G-Proteins and GPCR page.
Table of the Muscarinic Acetylcholine Receptors
| Receptor Nomenclature | Gene Symbol | Expression Profile | Functions / Comments |
| M1 (M1) | CHRM1 | predominantly expressed in the forebrain, including the cerebral cortex, hippocampus and corpus striatum; ganglionic receptor; lungs; secretory glands | coupled to a Gq/11-type G-protein; M1 receptor agonists cause epileptic seizures; loss of M1 receptor function results in increased dopamine release from striatum, suggests that pharmacological blockade of this receptor may be useful in Parkinson disease |
| M2 (M2) | CHRM2 | predominant receptor in cardiomyocytes of the heart, specifically in sinoatrial and atrioventricular node cardiomyocytes and ventricular cardiomyocytes; also found in lungs and several smooth muscle cells | coupled to a Gi-type G-protein; activates K+-channel as well as decreasing cAMP; M2 receptor agonists induce analgesia with much less risk of addiction relative to opioid analgesics; M2 receptor responsible for cholinergic deceleration of cardiac rate in response to parasympathetic outflow from the brain |
| M3 (M3) | CHRM3 | broadly expressed throughout the brain at low levels; expressed in periphery in secretory glandular tissues and smooth muscle cells; expressed on parietal cells of stomach; expressed in vascular endothelial cells | coupled to a Gq/11-type G-protein; important for contraction of smooth muscle in the urinary bladder, ileum, stomach fundus, trachea and gallbladder; ACh binding to parietal cell M3 receptors induces mobilization of vesicles containing the proton (H+) pump to luminal membrane for gastric acid production in stomach |
| M4 (M4) | CHRM4 | abundantly expressed in striatum; lungs | coupled to a Gi-type G-protein; activates K+-channel as well as decreasing cAMP; locomotor activity increased by pharmacologic blockade of the M4 receptor |
| M5 (M5) | CHRM5 | abundantly expressed in dopamine-containing neurons of the substantia nigra par compacta; lungs | coupled to a Gq/11-type G-protein |
Nicotinic Acetylcholine Receptors
Nicotinic ACh receptors are divided into those found at neuromuscular junctions and those found at neuronal synapses. The nicotinic ACh receptors are composed of five types of subunits which are found in different combinations in different forms of nicotinic ACh receptors. There are 16 known nAChR subunit genes in the human genome that encode the alpha (α1–α7, α9, and α10), beta (β1–β4), delta (δ), epsilon (ε), and gamma (γ) subunits. The alpha subunit genes are designated CHRNA1–CHRNA7, CHRNA9, and CHRNA10. The beta genes are CHRNB1–CHRN B4, while the delta, gamma, and epsilon genes are CHRND, CHRNG, and CHRNE, respectively. Regardless of subunit composition or cellular location, all of the nAChRs are pentameric receptors. As indicated, all of the nAChRs are divided into two broad categories: neuromuscular-type and neuronal-type. Regardless of type, nAChRs that contain the α4 and β2 subunits are the highest affinity receptors.
There are two major types of neuromuscular nicotinic ACh receptors. One type is referred to as the embryonic receptor is composed of α1, β1, δ, and γ subunits in the form (α1)2β1δγ. The second type is referred to as the adult form and it is composed of α1, β1, δ, and ε subunits in the form (α1)2β1δε. There are five types of neuronal nicotinic ACh receptors, with one of the latter type also found in epithelial tissues. The neuronal nAChRs are only composed of various α and β subunits making up the pentameric receptor. For example, the ganglion nAChR is comprised of an (α3)2(β4)3 pentameric arrangement. Within the brain the most commonly expressed nicotinic ACh receptors are composed of the α2 through α7 subunits and the β2, β3, and β4 subunits. The most widely expressed neuronal nicotinic ACh receptors are those that contain the α7 subunit and those that contain the β2 subunit. Neuronal nicotinic ACh receptors that contain the β2 subunit have the highest affinity for acetylcholine.
The nicotinic acetylcholine receptors, as ligand-activated ion channels, are permeable to small monovalent and divalent cations. The main conducting ions under biological conditions are Na+, K+, and Ca2+. The activation of nicotinic acetylcholine receptors by the binding of ACh leads to an influx of Na+ into the cell and an efflux of K+ out of the cell, resulting in a depolarization of the postsynaptic neuron and the initiation of a new action potential or the depolarization of the innervated muscle fiber at the neuromuscular junction. Although the major ions contributing to the changes in membrane potential upon ACh binding to nicotinic acetylcholine receptors are Na+ and K+, the influx of Ca2+ ion is an important contributor. Desensitization of the nAChRs occurs, in part, as a result of phosphorylation by either PKA or PKC.
Cholinergic Agonists and Antagonists
Numerous compounds have been identified that act as either agonists or antagonists of cholinergic neurons. The principal action of cholinergic agonists is the excitation or inhibition of autonomic effector cells that are innervated by postganglionic parasympathetic neurons and as such are referred to as parasympathomimetic agents. The cholinergic agonists include choline esters (such as ACh itself) as well as protein- or alkaloid-based compounds. Several naturally occurring compounds have been shown to affect cholinergic neurons, either positively or negatively.
The responses of cholinergic neurons can also be enhanced by administration of cholinesterase (ChE) inhibitors. ChE inhibitors have been used as components of nerve gases but also have significant medical application in the treatment of disorders such as glaucoma and myasthenia gravis as well as in terminating the effects of neuromuscular blocking agents such as atropine.
Table of Several Natural Cholinergic Agonists and Antagonists
| Source of Compound | Mode of Action | |
| Agonists | ||
| Nicotine | alkaloid prevalent in the tobacco plant | activates nicotinic class of ACh receptors, locks the channel open |
| Muscarine | alkaloid produced by Amanita muscaria mushrooms | activates muscarinic class of ACh receptors |
| α-Latrotoxin | protein produced by the black widow spider | induces massive ACh release, possibly by acting as a Ca2+ ionophore |
| Antagonists | ||
| atropine (and related compound scopolamine) | alkaloid produced by the deadly nightshade, Atropa belladonna | blocks ACh actions only at muscarinic receptors |
| Botulinus toxin | eight proteins produced by Clostridium botulinum | inhibits the release of ACh |
| α-Bungarotoxin | protein produced by Bungarus genus of snakes | prevents ACh receptor channel opening |
| d-Tubocurarine | active ingredient of curare | prevents ACh receptor channel opening at motor end-plate |
Cholinergic Pharmacology
Pharmacological intervention in the functions of acetylcholine is effected by either of two routes. In the direct-acting class there are the acetylcholine mimicking drugs (cholinomimetics) and in the indirect-acting class are the acetylcholinesterase inhibitors. There are numerous cholinomimetic drugs which includes methacholine, carbachol, and bethanechol as prominent examples. Methacholine exerts its effects through the muscarinic acetylcholine receptors and is used primarily in the bronchial challenge test used to diagnose hyperactivity in the bronchial tree as it typical in asthma. Carbachol functions primarily by activating nicotinic acetylcholine receptors and can exert systemic effects in the gastrointestinal system and in the bladder. However, it is used primarily as a locally administered drug in the treatment of glaucoma. Bethanechol is a muscarinic acetylcholine receptor agonist used primarily in the treatment of urinary retention following anesthesia and in diabetic neuropathy.
Acetylcholinesterase (AChE) inhibitors are used to increase the effective level and action of acetylcholine acting at both muscarinic and nicotinic acetylcholine receptors. Acetylcholinesterase inhibitors that are used pharmacologically are principally of the reversible (competitive and noncompetitive) type. Irreversible AChE inhibitors exert toxic effects such as the effects of the toxic organophosphate pesticides and nerve agents. The reversible AChE inhibitors, such as donepezil, rivastigmine and galantamine, are commonly used in the treatment neurodegenerative disorders such as Alzheimer disease (AD) and Parkinson disease (PD). These reversible inhibitors are also used to treat the neuromuscular disorder, myasthenia gravis, that results from autoimmune destruction of nicotinic acetylcholine receptors (nAChR).
Reversible AChE inhibitors are also used as antidotes in the treatment of organophosphate pesticide intoxication. Although these AChE inhibitors are used to reduce the symptoms associated with AD they do not exert their effects in the long term (being effective for only 12-24 months) and they have no effects on the rate of cognitive decline in AD. The carbamates (derived from carbamic acid: NH2COOH) represent a large class of compounds, many of which are reversible AChE inhibitors (e.g. rivastigmine). Although reversible, the carbamates can exert acute toxic effects that are similar to those of the irreversible organophosphates. Indeed, several carbamate compounds are used as pesticides and parasiticides in the veterinary field. Clinically the carbamates are used in the treatment of myasthenia gravis, glaucoma, and neurodegenerative disorders such as AD and PD, similarly to donepezil and galantamine. Although the irreversible AChE inhibitors are quite toxic and have been used as deadly nerve agents (e.g. VX) and as insecticides, they do have pharmacologic utility. The drug echothiophate (phospholine) is administered locally in the treatment of glaucoma and metrifonate is used in the treatment of AD and PD.
As described above, the neurotransmitters and receptors of the parasympathetic nervous system are those of the cholinergic family. The principal neurotransmitter is acetylcholine (ACh) and the receptors are the muscarinic acetylcholine receptors M2 and M3. For example, the primary vascular response to ACh binding to M3 receptors on endothelial cells is the activation of nitric oxide synthase (NOS) and the production of nitric oxide (NO). However, it is important to note that the endothelial M3 receptor is not innervated by cholinergic nerve fibers, but responds to the binding of circulating ACh. Production of NO results in relaxation of the smooth muscle cells leading to vasodilation. Nicotinic ACh receptors are located postsynaptically in all autonomic ganglia and at the neuromuscular junction (NMJ). At the NMJ, nicotinic receptors function as the excitatory receptor for the postsynaptic cell.
As pointed out in the introduction to this page, neurotransmission within the sympathetic and parasympathetic ganglia involves the release of ACh from preganglionic efferent nerves. Once released, the ACh then binds to nicotinic receptors in the membrane of the cell bodies of the postganglionic efferent nerves. Ganglionic blockers (primarily nicotinic ACh receptor antagonists) are drugs that function by inhibiting autonomic activity via interference with the transmission of nerve impulses within autonomic ganglia. Therefore, ganglionic blockers reduce sympathetic outflow. With respect to cardiac tissue, ganglionic blockade results in decreased cardiac output due to both decreased chronotropic (heart rate) and inotropic (contraction strength) activity. Ganglionic blockers also lead to reduced sympathetic output to the vasculature resulting in decreased sympathetic vascular tone. This latter effect causes vasodilation and reduced systemic vascular resistance resulting in decreased arterial pressure. It is important to note that parasympathetic nerve transmission (outflow) is also reduced by ganglionic blocking drugs. For this reason, as well as the development of more highly selective drugs for the treatment of hypertension, ganglionic blockers (e.g. mecamylamine and hexamethonium) are not commonly used any longer in the treatment of hypertension.
Catecholamines: Dopamine, Epinephrine, Norepinephrine
The principal catecholamines are norepinephrine, epinephrine and dopamine (3,4-dihydroxyphenylethylamine). These compounds are formed from the amino acid tyrosine. Tyrosine is produced, primarily, in the liver from phenylalanine through the action of phenylalanine hydroxylase. The tyrosine is then transported to catecholamine-secreting neurons where a series of reactions convert it to dopamine, then to norepinephrine, and finally to epinephrine (see also Amino Acid Derivatives: Neurotransmitters, Nitric Oxide, and More).
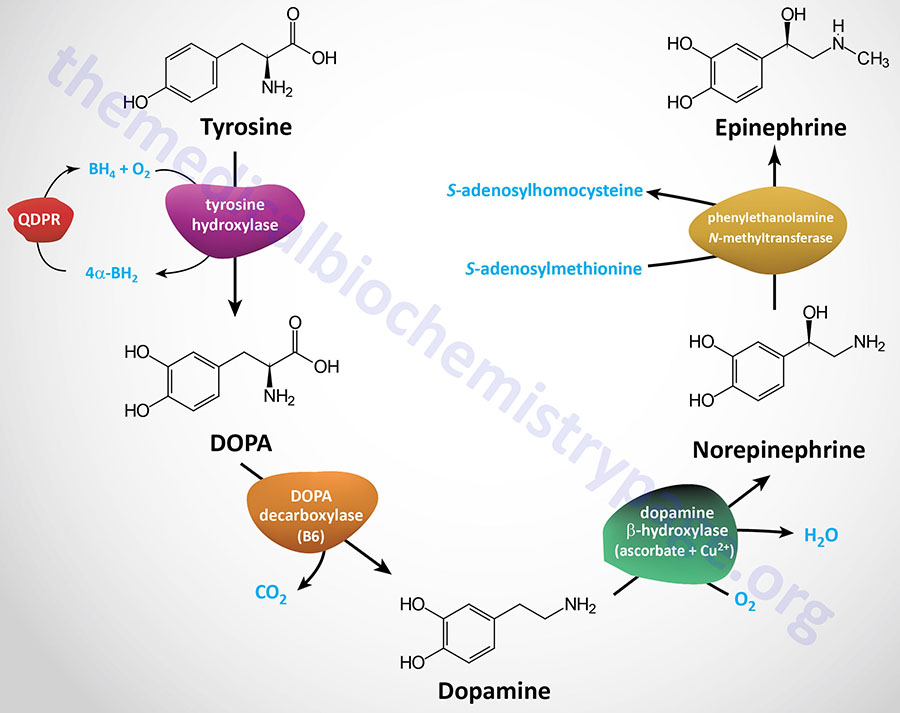
Within the substantia nigra locus of the brain, and some other regions of the brain, synthesis proceeds only to dopamine. Within the locus coeruleus region of the brain the end product of the pathway is norepinephrine. The presence of high concentrations of tyrosine in the locus coeruleus and the substantia nigra leads to increased melanin synthesis which confers on these brain regions a dark bluish coloration observable in brain sections. Indeed, these brain regions are so-called due to the dark bluish-black pigmentation. The Latin term, substantia nigra, means “black substance”. The Latin word coeruleus means “dark blue, blue, or blue-green”. Within adrenal medullary chromaffin cells, tyrosine is converted to norepinephrine and epinephrine.
Once synthesized, dopamine, norepinephrine, and epinephrine are packaged in granulated vesicles for secretion in response to the appropriate nerve impulse. Within these vesicles, norepinephrine and epinephrine are bound to ATP and a protein called chromogranin A. Norepinephrine is the principal neurotransmitter of sympathetic postganglionic nerves. Both norepinephrine and epinephrine are stored in synaptic knobs of neurons that secrete it, however, epinephrine is not a mediator at postganglionic sympathetic nerve impulses.
The major location, within the brain, for norepinephrine synthesis is the locus coeruleus of the brainstem. The major brain region for the synthesis of dopamine is the substantia nigra which is located below the posterior hypothalamus and next to the ventral tegmental area.
Outside the brain, the major site of norepinephrine and epinephrine synthesis is in adrenal medullary chromaffin cells. Outside the brain, dopamine is synthesized in several tissues including the gastrointestinal system where its actions reduce gastrointestinal motility, the pancreas where its actions inhibit insulin synthesis, and in the kidneys where its actions increase sodium excretion and urinary output.
Catecholamines exhibit peripheral nervous system excitatory and inhibitory effects as well as actions in the CNS such as respiratory stimulation and an increase in psychomotor activity. The excitatory effects are exerted upon smooth muscle cells of the vessels that supply blood to the skin and mucous membranes. Cardiac function is also subject to excitatory effects, which lead to an increase in heart rate and in the force of contraction. Inhibitory effects, by contrast, are exerted upon smooth muscle cells in the wall of the gut, the bronchial tree of the lungs, and the vessels that supply blood to skeletal muscle. In addition to their effects as neurotransmitters, norepinephrine and epinephrine can influence the rate of metabolism. This influence works both by modulating endocrine function such as insulin secretion and by increasing the rate of glycogenolysis and fatty acid mobilization.
The primary effects of the catecholamines are exerted as neurotransmitters upon their stimulated release from presynaptic nerve terminals in the appropriate target organ. However, release of the catecholamines from adrenal medullary cells to the systemic circulation allow them to function as hormones as well. Regardless of their site of release, the catecholamines exert their effects by binding to receptors of the G-protein coupled receptor (GPCR) family. The catecholamines are also known as adrenergic neurotransmitters and the neurons that secrete them are referred to as adrenergic neurons. Norepinephrine-secreting neurons are specifically termed noradrenergic neurons. Some of the norepinephrine released from presynaptic noradrenergic neurons is recycled in the presynaptic neuron by a reuptake mechanism similar to that responsible for regulating the CNS actions of serotonin.
Adrenergic Receptors
The actions of norepinephrine and epinephrine are exerted upon binding to and activating the adrenergic receptors of which there are nine distinct forms. As indicated, the adrenergic receptors are all members of the GPCR family. There are two distinct types of adrenergic receptor identified as the α (alpha) and β (beta) receptors. In addition, there are two functionally distinct classes of α adrenergic receptor identified as the α1 and α2 forms. Within each α-adrenergic receptor type there are several variants encoded by distinct genes as well as additional isoforms that result from alternative mRNA splicing.
The α1 receptors consist of the α1A, α1B, and α1D receptors. The α1 receptors are coupled to Gq-type G-proteins that activate PLCβ resulting in increases in IP3 and DAG release from membrane PIP2.
The α2 receptors consist the α2A, α2B, and α2C receptors. The α2 receptors are coupled to Gi-type G-proteins that inhibit the activation of adenylate cyclase and therefore, receptor activation results in reduced levels of cAMP and consequently reduced levels of active PKA.
The β adrenergic receptors are composed of three types: β1, β2, and β3 each of which couple to Gs-type G-proteins resulting in activation of adenylate cyclase and increases in cAMP with concomitant activation of PKA. However, the β2 and β3 receptors, dependent on tissue of expression, can switch from Gs to Gi/o signaling following modification of the receptors, such a phosphorylation of the receptor by PKA.
The highest levels of expression of the β3 receptor are found in adipose tissue, particularly in brown adipose tissue (BAT). In addition to Gs to Gi/o activation, the intracellular tail of the β3 receptor interacts with the SRC tyrosine kinase which promotes the activation of the ERK pathway.
Dopamine Receptors
Dopamine binds to dopaminergic receptors identified as D-type receptors and there are five receptors identified as D1, D2, D3, D4, and D5. All five dopamine receptors belong to the G-protein coupled receptor (GPCR) family. The D1 and D5 dopamine receptors are coupled to the activation of Gs-type G-proteins and, therefore, receptor activation results in activation of adenylate cyclase. The D2, D3, and D4 dopamine receptors are coupled to Gi-type G-proteins and, therefore, receptor activation results in the inhibition of adenylate cyclase. The D1 and D5 receptors constitute members of the D1-like receptor family. The D2, D3, and D4 receptors constitute members of the D2-like receptor family.
Table of Adrenegic and Dopaminergic Receptors
| Receptor Type | Gene Symbol(s) | Expression Profile | Functions / Comments |
| α1 | ADRA1A ADRA1B ADRA1D | predominates in vascular smooth muscles in the vessels of the skin; smooth muscle cells of the proximal ends of the major coronary arteries; liver; gastrointestinal system; kidneys; central nervous system (CNS); adipose tissue | three subtypes: α1A, α1B, α1D; coupled to Gq-type G-proteins; vasoconstrictor for coronary arteries and veins; decreases GI smooth muscle cell motility; induces contraction of smooth muscle in uterus, urethral sphincter, vas deferens, and ureter; increases hepatic glycogenolysis and gluconeogenesis; α1A receptor predominates in contraction of renal and caudal arteries; is also responsible for cardiac myocyte hypertrophy; α1B receptor likely to be responsible for contraction of small vessels; α1D receptor predominates in contraction of aorta, femoral, iliac, and superior mesenteric arteries; salt-induced hypertension involves functions of α1D receptor; α1D responsible for aortic smooth muscle hypertrophy; majority of α1 receptor agonists exert their effects via regulation of α1B receptor function; α1 receptor signaling linked to the MAPK pathway; within the CNS the α1 receptors are involved in motor control, locomotion, and cognitive functions |
| α2 | ADRA2A ADRA2B ADRA2C | widely distributed within the central nervous system (CNS); vessels; adipose tissue; kidneys; platelets | three subtypes: α2A, α2B, α2C; coupled to Gi-type G-proteins; coupled to the regulation of multiple signaling pathways in addition to adenylate cyclase such as the MAPK pathways, phospholipase D (PLD), activation of G-protein coupled inwardly-rectifying potassium channels (GIRK), and inhibition of Ca2+ channels; α2 receptors are essential feedback regulators of neurotransmitter release (norepinephrine and acetylcholine); receptors function within the CNS to decrease blood pressure and exert bradycardic effects, exert a hypothermic effect, and induce arterial and venous vasoconstriction; in the periphery α2 receptors inhibit insulin release and stimulate glucagon secretion, modulate gluconeogenesis and glycolysis, inhibit gastric acid secretion and gastric motility; involved in thrombus stabilization by inducing platelet aggregation; α2B subtype important during embryonic vascular development, also induces hypertension |
| β1 | ADRB1 | heart; liver; kidney; skeletal muscle; lung; colon; thyroid gland; adipocytes (preadipocytes only in BAT) | coupled to Gs-type G-protein; β1 receptors in the heart represent >80% of the total β receptor expression in this tissue; receptor activation leads to increased heart rate in the sinoatrial node (a chronotropic effect), increases atrial muscle contraction (an inotropic effect), increases ventricular muscle contractility; increases hepatic glycogenolysis and gluconeogenesis; increases fat mobilization from adipose tissue; increases renin release from kidneys; enhances sensation of hunger through release of ghrelin by the stomach; in heart failure the chronic sympathetic activation of β1 receptors contributes to pathologic remodeling in the heart |
| β2 | ADRB2 | adipose tissue but not brown adipocytes; liver; bronchioles; skeletal muscle; smooth muscle; platelets; lung; kidney; colon; thyroid gland; heart | coupled to Gs-type G-protein; switches to Gi following PKA-mediated phosphorylation of the receptor; bronchodilator and vasodepressor; induces relaxation of smooth muscle in bronchus, bronchioles, and uterus, relaxes detrusor muscle; induces the production of NO in platelets via activation of NOS3 (eNOS); inhibits release of insulin; stimulates adipose tissue lipolysis; stimulates hepatic glycogenolysis and gluconeogenesis |
| β3 | ADRB3 | abundant in adipocytes of BAT and omental fat; gallbladder and bladder; is not expressed in the heart, skeletal muscle, liver, kidneys, lung, or thyroid gland | coupled to Gs– and Gi-type G-protein, regulation of lipolysis, principal norepinephrine receptor in BAT, increase lipolysis in BAT and plays major role in adaptive thermogenesis |
| D1 | DRD1 | expressed at a high level of density in the nigrostriatal, mesolimbic, and mesocortical areas, such as the caudate-putamen (striatum), nucleus accumbens, substantia nigra, olfactory bulb, amygdala, and frontal cortex, lower levels in the hippocampus, cerebellum, thalamic and hypothalamic areas; kidney | coupled to Gs-type G-protein, together with D5 receptor forms the D1-like family; most abundant dopamine receptor in the CNS, regulates neuronal growth and development, involved in vital central nervous system functions that includes voluntary movement, regulation of feeding behavior, affect, reward, sleep, attention, reproductive behaviors, impulse control, working memory, and learning; renal receptors control renin secretion |
| D2 | DRD2 | within CNS high levels are found in the caudate-putamen, nucleus accumbens, olfactory tubercule, substantia nigra and ventral tegmental area; lower levels in septum, hypothalamus, and cortex; kidney, adrenal glands, sympathetic ganglia, gastrointestinal tract, blood vessels, heart | coupled to Gi-type G-protein, together with D3 and D4 receptors forms the D2-like family; like D1 receptors the D2 receptors are critically involved in working memory, reward and reinforcement mechanisms; regulation of locomotion, presynaptic receptors inhibit locomotion, postsynaptic receptors activate locomotion; regulation of renal function, blood pressure, vasodilation, and gastrointestinal motility |
| D3 | DRD3 | selectively associated with the limbic system of the CNS such as the shell of the nucleus accumbens and the olfactory tubercle; not expressed outside CNS | coupled to Gi-type G-protein, together with D2 and D4 receptors forms the D2-like family, limbic system receives dopamine inputs from the ventral tegmental area which is associated with cognitive, emotional, and endocrine functions; regulation of locomotor effects; modulation of cognitive functions |
| D4 | DRD4 | has the lowest level of brain expression of all the dopamine receptors, found in frontal cortex, amygdala, hippocampus, hypothalamus, globus pallidus, substantia nigra pars reticulata, and thalamus; kidney, adrenal glands, sympathetic ganglia, gastrointestinal tract, blood vessels, heart | coupled to Gi-type G-protein, together with D2 and D3 receptors forms the D2-like family; modulation of cognitive functions; regulation of renal function, blood pressure, vasodilation, and gastrointestinal motility |
| D5 | DRD5 | within the CNS expressed at low levels in multiple brain regions, including pyramidal neurons of the prefrontal cortex, the premotor cortex, the cingulated cortex, the entorhinal cortex, substantia nigra, hypothalamus, the hippocampus, and the dentate gyrus; kidney, adrenal glands, sympathetic ganglia, gastrointestinal tract, blood vessels, heart | coupled to Gs-type G-protein, together with D1 receptor forms the D1-like family, likely involved in affective, neuroendocrine, or pain-related aspects of dopaminergic functions |
Adrenergic Pharmacology
With respect to the sympathetic nervous system (see above), the principal neurotransmitters are norepinephrine and epinephrine and the receptors are α1, β1, and β2. Alpha-adrenergic receptors of the sympathetic nervous system play important roles in cardiac and vascular function. The presence of the α1 receptor in arteries causes them to constrict upon binding epinephrine or norepinephrine. This effect results in increased blood pressure and increased blood flow returning to the heart. Significantly, however, is the fact that the blood vessels in skeletal muscles lack α1 receptors so that they can remain open to utilize the increased blood pumped by the heart, particularly in response to stress.
Table of Adrenegic Receptor Drugs
| Drug Class | Major Target(s) | Effects / Comments |
| non-selective α blockers | both classes of α receptor; primary responses at a1 receptors | reduce vasoconstriction caused by both norepinephrine and epinephrine acting at α1 receptors; inhibition of α2 receptors exerted within sympathetic nervous system; phenoxybenzamine is most well known in this class, was once also used to treat benign prostatic hyperplasia (BPH); yohimbine is used as an herbal treatment for erectile dysfunction but excess usage can lead to tachycardia, dizziness, and anxiety |
| selective α blockers | α1 receptors | all drugs in this class end with the suffix ‘osin’ (e.g. prazosin); drugs in this class can induce orthostatic hypotension; |
| α1 agonists | α1 receptors | function systemically to increase blood pressure; methoxamine and phenylephrine increase peripheral vascular resistance resulting in increased blood pressure; these drugs are used in the treatment of hypotension and shock |
| α2 agonists | α2 receptors | function centrally (i.e. within the CNS) to lower blood pressure; methyldopa is longest used drug in this class, although not used often any longer it is still the drug of choice (DOC) for the treatment of hypertension in pregnancy; cause vasodilation and reduction in blood pressure; also used to treat BPH; although classified as a non-selective agonist, clonidine is widely used drug for the treatment of hypertension and hot flashes in menopausal females; clonidine also approved for the treatment of attention deficit hyperactivity disorder (ADHD) |
| non-selective β blockers | β1 and β2 receptors | some non-selective β blocker drugs have the ‘olol’ suffix (e.g. propranolol) like the β1-selective blockers; because of their non-selectivity these drugs affect the heart, kidneys, lungs, gastrointestinal tract, liver, uterus, vascular smooth muscle (VSM), and skeletal muscle |
| β1 blockers | β1 receptors | β1 blocker drugs (e.g. metoprolol) all have a name with the suffix ‘olol’; β1-specific blockers function to decrease heart rate (chronotropic effects) and contractility (inotropic effects) leading to decreased blood pressure; β blockers are also sometimes prescribed for the treatment of glaucoma, migraine headaches, and anxiety |
| β2 agonists | β2 receptors | β2 agonists are principally used to induce bronchodilation in asthmatics and others with pulmonary dysfunction; these drugs are divided into the short-acting and long-acting subtypes; salbutamol is the most common of the prescribed short-acting drugs; all of the long-acting drugs have a name with the suffix ‘terol’ (e.g. formoterol) |
When considering the effects of various adrenergic receptor agonist and antagonist effects within the vasculature it is important to understand that the contractile characteristics and the mechanisms that cause contraction of cardiac myocytes and vascular smooth muscle (VSM) are very distinct. The contractile properties of cardiac myocytes are fast and of extremely short duration. In contrast, VSM undergoes slow, sustained, tonic contractions. While both cardiac muscle and VSM contain actin and myosin, VSM do not express the regulatory troponin complex as do striated muscle cells such as cardiac myocytes. An additional difference between VSM and cardiac myocytes relates to the structural arrangement of actin and myosin. In heart muscle cells these proteins are organized into distinct bands, whereas, in VSM they are not. Although organized differently, the contractile proteins of VSM are indeed highly organized in order to allow for maintaining tonic contractions and reducing vascular diameter.
Contraction of VSM is initiated by by several distinct phenomena including mechanical, electrical, and chemical stimuli. Mechanical contraction refers to the passive stretching of VSM from the cell itself and is therefore termed a myogenic response. Electrical stimulation of VSM contraction involves depolarization of the membrane, most often as a result of the opening of voltage gated calcium channels (L-type calcium channels) leading to increased intracellular calcium concentrations. When discussing chemical stimuli, that initiate contraction in VSM, these signals are hormones and neurotransmitters such as epinephrine and norepinephrine, angiotensin II, vasopressin (anti-diuretic hormone, ADH), endothelin-1, and thromboxane A2 (TXA2). Each of these molecules binds to specific receptors on the VSM cell or to receptors on the endothelial cells adjacent to the VSM. The consequences of receptor activation are VSM contraction. Although each of these receptor-mediated VSM contraction processes are different, they converge at the point of increased intracellular calcium concentration.
Increases in free intracellular calcium result from either increased calcium influx into the VSM or via the release of sarcoplasmic reticulum (SR) stored calcium. Within the VSM cell, free calcium binds to the regulatory protein, calmodulin. Calcium-calmodulin activates myosin light chain kinase (MLCK) which then phosphorylates myosin light chains. Phosphorylation of myosin light chains induces the formation of cross-bridges between the myosin heads and the actin filaments leading to smooth muscle contraction. Conversely, relaxation of VSM cells occurs in response to reduced levels of myosin light chain phosphorylation.
Adrenergic receptor stimulation by epinephrine or norepinephrine involves G-protein-coupled signal transduction pathways that impinge upon levels of the PKA activating molecule, cAMP. Since α1 receptors are coupled to the activation of Gq proteins there is a resultant increase in release of intracellular calcium via the action of the second messenger IP3 binding to SR membrane receptors. The consequences of the released calcium are, therefore, VSM contraction.
Norepinephrine is the major activator of α1 receptors. Norepinephrine also activates α2 receptors which are Gi coupled receptors. The resultant inhibition of cAMP production due to the inhibition of adenylate cyclase leads to increased MLCK activity. The effects, therefore, of norepinephrine at α1 and α2 receptors are the same but elicited via different signaling pathways.
On the other hand, epinephrine activates β2 receptors which are coupled to Gs proteins which activate adenylate cyclase resulting in increased cAMP concentrations. In most cells an increase in cAMP leads to an increase in the activity of the kinase, PKA. Although it would seem counterintuitive for this pathway to be activated under conditions where VSM relaxation was needed, the increased cAMP levels induced by VSM β2 receptor activation result in inhibition of MLCK, thereby reducing myosin light chain phosphorylation. In addition, activated PKA phosphorylates a membrane potassium channel (KATP) in VSM resulting in hyperpolarization of the cell preventing the Ca2+ influx that is required for contraction. The net effect of both of these β2 receptor-medicated events is VSM relaxation.
Activation of the β1 receptor in the heart results in an increase in both the inotropic (heart rate) and the chronotropic (strength of contraction) activity of the heart muscle. Pharmacologic antagonism of the β1 receptor in the heart, such as with metoprolol (or any other of this drug class; identifiable by the ‘olol’ ending), results in decreasing heart rate and contractility. The overall effect is a decrease in blood pressure. This is the basis for the use of beta blocker drugs in the treatment of hypertension and to decrease the chance of a dysrhythmia after a heart attack.
The β2 receptors are prevalent in the smooth muscle cells of the bronchioles of the lungs and arteries of skeletal muscle. Activation of the β2 receptors in bronchioles causes them to dilate which allows more oxygenated air to enter the lungs. Simultaneously, activation of β2 receptors in the arteries of skeletal muscle causes them to dilate to allow increased blood flow into this tissue. Both of these receptor-mediated activities allow for an enhanced response to stress such as is typical of the fight-or-flight response.
It is important to note that norepinephrine also binds weakly to β2 receptors which results in vasodilation as for the case of epinephrine. This phenomenon is most noticeable pharmacologically when α1 blockers such as prazosin (drugs in this class all end in ‘osin’) are utilized. Under normal physiological conditions this vasodilator effect of norepinephrine is overwhelmed by α1 receptor-mediated vasoconstriction. Equally important is the fact that, although epinephrine binds with highest affinity to VSM β2 receptors to induce vasodilation, at high concentrations it will bind to α1 and α2 receptors which can override β2 receptor effects leading to vasoconstriction.
Catecholamine Catabolism
The catecholamines, DOPA, dopamine, norepinephrine, and epinephrine are catabolized to inactive compounds through the sequential actions several enzymes including catecholamine-O-methyltransferase (COMT) and monoamine oxidase (MAO). Compounds that inhibit the action of MAO have been shown to have beneficial effects in the treatment of clinical depression, even when tricyclic antidepressants are ineffective. The utility of MAO inhibitors was discovered serendipitously when patients treated for tuberculosis with isoniazid showed signs of an improvement in mood. This effect of isoniazid was subsequently shown to be due to the inhibition of MAO.
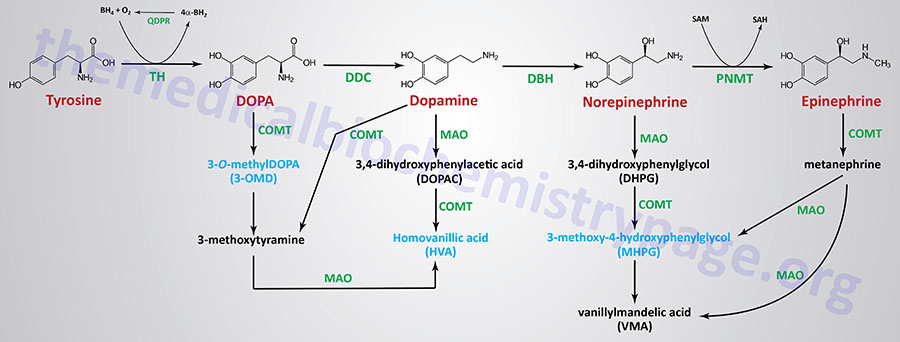
Dopamine: Reward Reinforcement and Feeding Behaviors
Overall control of feeding behavior is a complex process involving several well defined neural circuits. These circuits consist of interactions between the brainstem and the hypothalamus as well as interactions between the gut and the hypothalamus. For detailed information on the latter go to the Gut-Brain Interrelationships page. The control of feeding behavior also involves overlapping processes such as motivational drive, satiety and the anticipation of food. A major neurotransmitter involved in the coordination and reinforcement of these reward processes is dopamine. Indeed, every known type of reward, including food, results in increased levels of dopamine in the brain. Although the cell bodies of dopaminergic neurons are confined to only a few areas of the brain, these neurons send projections to numerous areas including those involved in the regulation of feeding behaviors such as the hypothalamus.
Dopamine mediates the motivational and rewarding aspects of food seeking behavior via specific dopaminergic projections from the ventral tegmental area (VTA) to the nucleus accumbens (NAc). The VTA is the origin of the dopaminergic cell bodies and the NAc is a brain region in the basal forebrain that sends projections to the basal ganglia situated at the base of the forebrain. The NAc is involved in reinforced learning, reward, pleasure, addiction, fear, aggression, and impulsivity. The reward pathways involving dopamine are also referred to as the mesolimbic or mesocorticolimbic system which also sends projections to the medial prefrontal cortex, hippocampus and amygdala. The mesolimbic dopaminergic circuits are involved in the motivation to earn food rewards but not for the triggering of actual food consumption.
Dopamine also mediates food consumption by sending projections from the substantia nigra to the dorsolateral striatum. Although mesolimbic dopamine circuits have clearly been associated with reward processes, the specifics of its involvement in the process are quite complex. It is important to be able to distinguish between the diverse aspects of motivational function that are differentially affected by dopamine activity.
Pharmacological manipulation of dopamine demonstrates that mesolimbic dopamine is indeed critical for many aspects of motivational function, but also that it is not critically involved in all aspects of motivational function. In addition, some of the effects of mesolimbic dopamine are linked to aversive motivation and learning. However, the studies on the fundamental characteristics of reinforcing stimuli have concluded that mesolimbic dopaminergic signals, acting as positive reinforcers, tend to be preferred and thus, elicit approach, goal-directed, and high demand behaviors characteristic of positive reinforcement.
In addition to direct dopaminergic neuronal actions, the activity of the mesolimbic system is modulated by peripheral hormones that are known to regulate feeding behaviors via hypothalamic circuits such as leptin and ghrelin. Leptin is an anorexigenic hormone (decreases desire for food) produced by adipose tissue whereas, ghrelin is an orexigenic hormone (increase desire for food) produced by the stomach. Leptin action in the VTA results in reduced firing of dopaminergic neurons and decreases food intake. Conversely, animal studies demonstrate that loss of leptin receptors in the VTA leads to increased food intake. Ghrelin receptors are present in the VTA and NAc and activation of these receptors leads to increased food intake. These observation reinforce the role of dopamine in feeding behavior and demonstrate the interconnections between peripheral and central neurotransmitter actions in overall regulation of feeding.
Serotonin
Serotonin (5-hydroxytryptamine, 5HT) is formed by the hydroxylation and decarboxylation of tryptophan (see also Amino Acid Derivatives: Neurotransmitters, Nitric Oxide, and More).
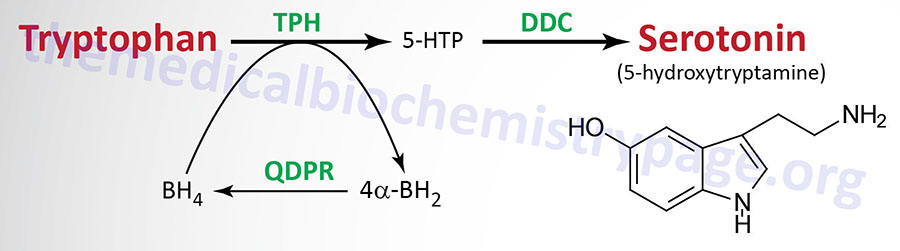
The greatest concentration of 5HT (90%) is found in the enterochromaffin cells of the gastrointestinal tract. Most of the remainder of the body’s 5HT is found in platelets and the CNS. Platelets themselves are incapable of synthesizing serotonin but acquire it from plasma via the action of the serotonin transporter (SLC6A4). The effects of 5HT are felt most prominently in the cardiovascular system, with additional effects in the respiratory system and the intestines. Vasoconstriction is a classic response to the administration of 5HT.
Neurons that secrete 5HT are termed serotonergic. Following the release of 5HT, a portion is taken back up by the presynaptic serotonergic neuron in a manner similar to that of the reuptake of norepinephrine.
Serotonin Receptors
The function of serotonin is exerted upon its interaction with specific receptors. At least 15 serotonin receptors have been cloned and are classified in seven families: 5HT1, 5HT2, 5HT3, 5HT4, 5HT5, 5HT6, and 5HT7. All of the serotonin receptor genes in these seven families are abbreviated as HTR followed by a numeral and letter designation. Within the 5HT1 group there are subtypes HTR1A, HTR1B, HTR1D, HTR1E (a putative 5HT receptor), and HTR1F. There are three 5HT2 subtypes, HTR2A, HTR2B, and HTR2C (was originally identified as the 5HT1C receptor). There are five 5HT3 subtypes, HTR3A, HTR3B, HTR3C, HTR3D, and HTR3E. The receptors HTR3C, HTR3D, and HTR3E are referred to as serotonin-like receptors. There are two 5HT5 subtypes, HTR5A and HTR5B in the human genome but the HTR5B gene is a pseudogene.
Most of these receptors are of the metabotropic family and thus, coupled to G-proteins that affect the activities of either adenylate cyclase or phospholipase Cβ (PLCβ). The 5HT3 class of receptors are ion channels (ionotropic receptors). Like other ionotropic receptors, the 5HT3 receptors are composed of five subunits and are either homopentameric or heteropentameric where in the heteropentameric form there is always at least one 5HT3A subunit..
Table of Serotonin Receptor Families
| Receptor Family | Associated G-Proteins | Result of Receptor Activation |
| 5HT1 | Gi/Go go to the Signal Transduction Pathways: G-Proteins and GPCR page for description of various G-proteins | inhibits cAMP production, inhibitory neurotransmission; 5HT1A induces K+ channel opening |
| 5HT2 | Gq/G11 | increased production of DAG and IP3, excitatory neurotransmission; 5HT2A induces closure of K+ channels |
| 5HT3 | ligand-gated Na+ and K+ channels | depolarizes axonal membrane, excitatory neurotransmission |
| 5HT4 | Gs | increases cAMP production, slow excitatory neurotransmission |
| 5HT5 | Gi/Go | inhibits cAMP production, inhibitory neurotransmission |
| 5HT6 | Gs | increases cAMP production, excitatory neurotransmission |
| 5HT7 | Gs | increases cAMP production, excitatory neurotransmission |
Some serotonin receptors are presynaptic and others postsynaptic. The 5HT2A receptors mediate platelet aggregation and smooth muscle contraction. The 5HT2C receptors are suspected in control of food intake as mice lacking this gene become obese from increased food intake and are also subject to fatal seizures. The 5HT3 receptors are present in the gastrointestinal tract and are related to emesis (vomiting). Also present in the gastrointestinal tract are 5HT4 receptors where they function in secretion and peristalsis. The 5HT6 and 5HT7 receptors are distributed throughout the limbic system of the brain and the 5HT6 receptors have high affinity for antidepressant drugs.
Table of the Serotonin Receptors
| Receptor Sub-Type | Gene Symbol | Sites of Expression | Functions/Comments |
| 5HT1A | HTR1A | vasculature, CNS | aggression, anxiety, blood pressure, appetite, memory, mood, cardiovascular tone, heart rate, respiration, pupillary dilation, nociception (pain sensation), sexual behavior, erectile function, emesis (vomiting), thermoregulation, sleep, addictive behaviors, thermoregulation |
| 5HT1B | HTR1B | vasculature, CNS | locomotion, aggression, anxiety, vasodilation of cerebral vessels (plays role in triptan class antimigraine drugs), memory, appetite (by modulating melanocortin pathways), mood, learning, sexual behavior, erectile function, addictive behaviors |
| 5HT1D | HTR1D | CNS | thalamic blood pressure regulation (triptan class antimigraine drugs act through this receptor and 5HT1B), locomotion, anxiety |
| 5HT1E | HTR1E | CNS | precise function unclear, inhibits adenylate cyclase |
| 5HT1F | HTR1F | CNS | involved in migraine headaches, anxiety and cognitive disturbances |
| 5HT2A | HTR2A | gastrointestinal tract, smooth muscles, vasculature, CNS, PNS, liver | liver regeneration, anxiety, blood pressure, appetite, learning, memory, mood, cognitive abilities, sexual behavior, sleep, addictive behaviors |
| 5HT2B | HTR2B | heart gastrointestinal tract, smooth muscles, vasculature, CNS, PNS, liver | receptors are mitogenic involved in cardiac tissue development and liver regeneration, gastrointestinal motility, blood pressure (vasoconstriction), appetite, anxiety, sleep |
| 5HT2C | HTR2C | gastrointestinal tract, smooth muscles, vasculature, CNS, PNS | anxiety, locomotion, gastrointestinal motility, blood pressure, appetite (by modulating melanocortin pathways), mood, sexual behavior, erectile function, thermoregulation, sleep, addictive behaviors; the small molecule 5HT2C-selective agonist, lorcaserin (Belviq), was approved by the US FDA in 2012 for the treatment of obesity but in 2020 that approval was withdrawn due to increased risk of cancer; lorcaserin-mediated activation of 5HT2C receptors in neurons of the NTS result in enhanced synthesis and release of GLP-1; pre-mRNA is edited at five sites [A, B, C’ (E), C, and D]; in victims of suicide, the level of C’ editing is much higher and the level of D editing is significantly decreased when compared with unaffected individuals, in mice treated with the antidepressant, fluoxetine, the pattern of C, C’, and D editing is the exact opposite to that observed in victims of suicide |
| 5HT3A | HTR3A | predominantly expressed in small intestine, colon, and CNS; low levels detected in spleen, thymus, and prostate | anxiety, gastrointestinal motility, emesis (vomiting), learning, memory, addictive behaviors; an alternatively spliced version is identified as 5HT3As |
| 5HT3B | HTR3B | kidney, CNS, skeletal muscle | |
| 5HT3C | HTR3C | adult brain, colon, intestine, lung, muscle, and stomach, fetal colon and kidney | do not form homomeric channels but form heteromeric channels with 5HT3A |
| 5HT3D | HTR3D | adult liver, adult and fetal kidney, adult and fetal colon | |
| 5HT3E | HTR3E | strong in small intestine, weak in spinal cord and retina; colorectal adenocarcinoma cell lines | do not form homomeric channels but form heteromeric channels with 5HT3A |
| 5HT4 | HTR4 | heart, gastrointestinal tract, CNS, PNS | respiration, appetite, gastrointestinal motility, learning, memory, mood, anxiety |
| 5HT5A | HTR5A | exclusive to CNS | locomotion, sleep |
| 5HT6 | HTR6 | CNS, highest in caudate nucleus | cognitive abilities, learning, memory, anxiety, mood; receptor polymorphisms associated with schizophrenia; high affinity for antidepressants and antipsychotics in particular clozapine |
| 5HT7 | HTR7 | predominates CNS; also found in coronary arteries, liver, kidney, spleen, and pancreas | blood pressure, respiration, thermoregulation, sleep, memory, mood, anxiety |
Adenosine
Adenosine is the nucleoside composed of ribose and the nucleobase adenine. Adenosine is found ubiquitously throughout the body and it is directly involved in numerous processes that are required for the maintenance of cellular viability in the context of energy charge (as ATP), redox control (in NAD+/NADH), DNA and RNA synthesis (as the precursors dATP and ATP, respectively), and in the regulation of the epigenetic modification of DNA and histones by methylation (in S-adenosylmethionine: SAM). Due to the fact that all cells utilize adenosine in the processes of intracellular metabolism, its levels are tightly regulated under normal physiological conditions.
Adenosine is also produced extracellularly where it is used in a controlled manner as an extracellular signal. All mammalian cells in the various different tissues have been shown to utilize adenosine as a paracrine hormone to coordinate cellular activity or as a neurotransmitter to coordinate neural activity within the central nervous system (CNS). One of the earliest observations of the extracellular activity of adenosine was its effects on coronary artery dilation which lead to increases in coronary blood flow and an elevation in myocardial oxygen tension. These initial observations were made over 45 years ago and since that time the roles of adenosine in the regulation of numerous critical physiological processes has greatly expanded.
Generation and Fates of Adenosine
The production of adenosine intracellularly occurs from its immediate precursor AMP (5′-AMP) via the action of the enzyme 5′-nucleotidase which is encoded by the NT5E (5′-nucleotidase ecto) gene. There are several proteins derived from the NT5E gene which includes membrane anchored and cytoplasmic versions. The membrane anchored form of 5′-nucleotidase is also known as CD37. The membrane anchored 5′-nucleotidase is responsible for the conversion of extracellular nucleotides to their corresponding nucleosides so that they can be transported into the cell. Intracellular adenosine is also derived from S-adenosylmethionine (SAM, AdoMet) following its conversion to S-adenosylhomocysteine (SAH, AdoHcy) in the course of transmethylation reactions. The release of adenosine from SAH is catalyzed by adenosylhomocysteinase (also called S-adenosylhomocysteine hydrolase: SAH hydrolase) which is encoded by the AHCY gene.
Intracellular adenosine is capable of undergoing various conversions for different metabolic and biosynthetic pathways depending upon the energy state and the physiological status of the cell. Metabolism of adenosine represents the classic purine nucleotide catabolism pathway. In this pathway adenosine is first deaminated to inosine via the action of adenosine deaminase (ADA). Inosine is then converted to the nucleobase, hypoxanthine, through the action of purine nucleotide phosphorylase (PNP) which removes the ribose-1-phosphate. Hypoxanthine is then catabolized to uric acid via the action of xanthine oxidase. Intracellular adenosine does not just have a catabolic fate as it can readily be converted back to AMP through the action of adenosine kinase and then subsequently to ADP and ATP via the actions of nucleotide monophosphate and nucleotide diphosphate kinases, respectively. Adenosine kinase (encoded by the ADK gene) phosphorylates adenosine using ATP as the phosphate donor.
Intracellular adenosine can also be transported out of the cell by members of the equilibrative nucleoside transporter (ENT) family of transporters. Humans express four members of the ENT family all of which are encoded by genes in the solute carrier (SLC) family, specifically the SCL29 family. The predominant ENT used for adenosine transport is ENT1, encoded by the SLC29A1 gene. Additional nucleoside transporters that are known to transport adenosine are members of the concentrative nucleoside transporter (CNT) family. The CNT are Na+-dependent nucleoside transporters. The CNT transporters are encoded by genes of the SLC28 family, of which there are three in humans.
Extracellular AMP is produced by degradation of ATP via the action of the plasma membrane localized enzyme, ectonucleoside triphosphate diphosphohydrolase 1 (encoded by the ENTPD1 gene) which represents the major mechanism for the production of adenosine outside the cell. The ENTPD1 gene encoded protein in also known as CD39. Another ATP degrading enzyme that is a type II transmembrane protein is ectonucleotide pyrophosphatase/phosphodiesterase 1 (encoded by the ENPP1 gene).
Extracellular AMP can also be generated from cyclic-AMP (cAMP) via the action of the membrane anchored form of 5′-nucleotidase. Extracellular ATP, ADP, and AMP are predominantly the result of release from cells experiencing stress. Under normal physiological conditions, adenosine is principally derived via the intracellular reactions described above. Extracellularly generated adenosine can be transported into the cell via the actions of the ENT and CNT transporters. The direction of adenosine uptake or release from cells is determined by the concentration difference across the membrane.
The primary role of the ENT are to transport adenosine into or out of cells on the basis of the adenosine concentration gradient. The role of the CNT are to facilitate adenosine transport into the cell against an adenosine concentration gradient. This is accomplished by the CNT transporters utilizing the energy of the Na+ gradient. Under normal physiological conditions the direction of adenosine flux is from extracellular to intracellular. However, during pathological conditions, for example during hypoxia, the flux direction is reversed. Following the uptake of extracellular adenosine, it is either catabolized to uric acid or converted to AMP as described above.
The majority of the pool of intracellular free nucleotides originates through the de novo synthesis pathways in the liver. The liver as the source of free nucleotides for other tissues is highly similar to the role of the liver in synthesizing many other compounds for export. In order to export nucleotides the liver dephosphorylates them to their nucleoside forms as well as phosphorolytically cleaving them into their constituent nucleobases and ribose-1-phosphate. This means that adenosine is synthesized predominantly in the liver as the product of dephosphorylation of AMP. Following transport to the blood the adenosine is picked up by other tissues through the actions of the ENT and intracellular the fate of adenosine depends primarily on the energy state of the cell. At low energy charge the conversion of adenosine to AMP results in the activation of the master metabolic regulatory kinase, AMPK. At high energy charge, ATP degradation mainly produces the purine catabolic byproducts inosine and hypoxanthine not adenosine. This is due to the fact that at high concentrations of ATP, AMP deaminase and the cytosolic form of 5′-nucleotidase are allosterically activated.
Adenosine as a Hormone and Neurotransmitter
Adenosine is known to exert numerous functions as an extracellular signaling molecule. However, its roles as a neurotransmitter in the brain and as a hormone acting in the coronary vessels are considered its most significant functions. The significance of adenosines function in the brain stems, in large part, from the fact that the highest density of adenosine receptors is within the brain when compared to all other tissues. Adenosine has been shown to exert its plethora of effects through interaction with, and activation of, four distinct adenosine receptors.
Adenosine receptors were initially defined as the P1 class of purinoreceptors where the P2 class of purinoreceptors are now identified as the P2X and P2Y receptors. The P1 receptors were subsequently divided into two subgroups of adenosine receptors defined as the A1 and A2 families. These receptors are now identified as the adenosine A1 (A1AR), A2A (A2AAR), A2B (A2BAR), and A3 (A3AR) receptors. All four receptors are members of the large G-protein coupled receptor (GPCR) superfamily.
Numerous neurotransmitter receptors undergo a process of desensitization in the presence of continued or repeated exposures to ligand. In the case of GPCR type receptors the process of desensitization is complex and often includes receptor phosphorylation by a family of G-protein-coupled receptor kinases (GRK). The result of GPCR phosphorylation is the preferential intracellular binding of the receptor to proteins of the arrestin family followed by clathrin-coated pit mediated internalization. The processes of receptor desensitization have been shown to occur with all four adenosine receptor subtypes. With respect to the internalization process the A1AR is internalized slowly, whereas with the A2AAR and A2BAR internalization is more rapid.
Table of Human Adenosine Receptors
| Receptor | Gene | Associated G-Protein | Comments / Activities |
| A1: A1AR | ADORA1 | Gi/o | ADORA1 gene is located on chromosome 1q32.1 and is composed of 4 exons that generate four alternatively spliced mRNA encoding three distinct protein isoforms; although the primary G-protein family coupled to the A1AR is the Gi/o type, activation of A1AR has also been coupled to Gq-type G-protein activation; highest level of expression is in the brain; within the CNS A1AR is found in neurons in the cortex, hippocampus and cerebellum, astrocytes, oligodendrocytes, and microglia; within neurons the receptor is highly localized to synaptic regions where they modulate the release of neurotransmitters, principally glutamate, acetylcholine, serotonin, and GABA; in addition to the A1AR-mediated modulation of neurotransmitter release, A1AR activation dampens neuronal excitability, controls sleep/wake states, decreases nociception (sensation of pain), and exerts sedative, anticonvulsant, anxiolytic, and locomotor depressant effects; outside the brain ADORA1 expression is high in the heart (atrial tissues), the kidney, adipose tissue, and the pancreas; in the heart activation of A1AR results in negative chronotropic, inotropic, and dromotropic effects; in the kidney A1AR activation results in reduced renal blood flow and renin release; activation of A1AR in adipose tissue results in inhibited lipolysis; in the pancreas A1AR activation results in reduced insulin secretion |
| A2A: A2AAR | ADORA2A | Gs | ADORA2A gene is located on chromosome 22q11.23 and is composed of 6 exons that generate five alternatively spliced mRNAs, four of which differ in the 5′-UTR; all five mRNAs encode the same 412 amino acid protein; highest levels of expression are in bone marrow and lymph nodes; ADORA2A is the predominant adenosine receptor subtype responsible for coronary blood flow regulation; receptor activation results in coronary artery dilation as a result of both endothelial cell-dependent and -independent mechanism; brain expression of the ADORA2A gene is seen in neurons, microglia, and oligodendrocytes of the striatum and olfactory tubercle; in the periphery expression of the ADORA2A gene is high in leukocytes, platelets, and the vasculature; in these tissues the activation of A2AR has been shown to mediate anti-inflammatory effect, inhibition of platelet aggregation, and induction of vasodilation, respectively |
| A2B: A2BAR | ADORA2B | Gs | ADORA2B gene is located on chromosome 17p12 and is composed of 6 exons that encode a 332 amino acid protein; highest levels of expression are in the colon, esophagus, and skin; the A2BAR protein has the lowest affinity for adenosine of the four receptors; A2BAR activation is proposed to have an indirect role in the responses to hypoxia and ischemia via the promotion of angiogenesis through increased endothelial cell functions; A2AR activation exerts positive nociceptive and inflammatory roles in the periphery; |
| A3: A3AR | ADORA3 | Gi/o | ADORA3 gene is located on chromosome 1p13.2 and is composed of 2 exons that generate three alternatively spliced mRNAs, each of which encode distinct protein isoforms; highest levels of expression are in adrenal glands and small intestine; although levels of expression of the ADORA3 gene is low in the brain the A3AR has been shown to play a protective role along with A1AR in the responses to cerebral ischemia |
The major effects of adenosine within the brain are exerted via its interactions with the A1AR and A2AAR receptors. Within the brain both A1AR and A2AAR are primarily located in synapses, in particular in excitatory glutamatergic synapses. Adenosine activation of A1AR in the brain functions to inhibit neurotransmission whereas activation of A2AAR functions to facilitate neurotransmission. In adult mammalian brain the removal of adenosine occurs via the actions of astroglia. Astroglia express the adenosine kinase ADK gene and several members of the SLC29A family that encode the ENT proteins, primarily ENT2 (SLC29A2) and ENT3 (SLC29A3). Through the actions of these proteins astroglia serve as “reservoirs” for adenosine via both release and uptake, where uptake is mediated, in part, by the activity of adenosine kinase. The significance of adenosine kinase activity to overall brain function is evident from the fact that deficiencies in the enzyme appear to be involved in several pathologies including epilepsy. When the ADK gene is knocked out in mice, the animals develop spontaneous seizures and exhibit profound defects in hippocampus-dependent learning and memory.
As indicated above, adenosine is required for the formation and homeostasis of S-adenosylmethionine (SAM; AdoMet) pools. SAM is the methyl donor in numerous transmethylation reactions including those of DNA methylation. Given this role of adenosine in DNA methylation it is not surprising that disruption in the adenosine pools in the brain have been shown to be associated with changes in the pattern and state of DNA methylation, in particular in the hippocampus. For example, the use of an adenosine kinase inhibitor drug in mice results in a decrease in DNA methylation in the hippocampus and the appearance of spontaneous recurrent seizures. However, it should be pointed out that the effect of intracellular adenosine acting through regulation of the SAM-mediated transmethylation pathway is not restricted to the nervous system and is evident in other cell types such as vascular endothelial cells.
Within the vasculature adenosine interacts with its receptors that are located on vascular smooth muscle cells and endothelial cells of the coronary arteries. The primary result of adenosine receptor activation in the coronary vessels is vascular relaxation and consequent vasodilation. All four adenosine receptors are found in coronary smooth muscle cells but only A2AAR and A2BAR have been shown to be present on coronary endothelial cells. Evidence in mice has shown that A3AR is localized on endothelial cells of the aorta, where their activation leads to contraction of the underlying smooth muscle.
Histamine
Histamine Synthesis and Catabolism
Histamine is referred to as a biogenic amine which is a potent neurotransmitter that binds to specific histamine receptors (see below). Histamine is synthesized by the enzymatic decarboxylation of the amino acid histidine by the enzyme L-histidine decarboxylase (HDC). Within the gastrointestinal tract bacteria also produce histamine via a similar decarboxylation reaction. The principal cells that synthesize and release histamine are mast cells and basophils of the immune system, enterochromaffin-like cells of the gastrointestinal system, and neurons. The synthesis and storage of histamine by mast cells and basophils represents the greatest store (>90%) of the neurotransmitter. Within the brain the neurons that synthesize histamine are within the tuberomammillary nucleus of the hypothalamus.
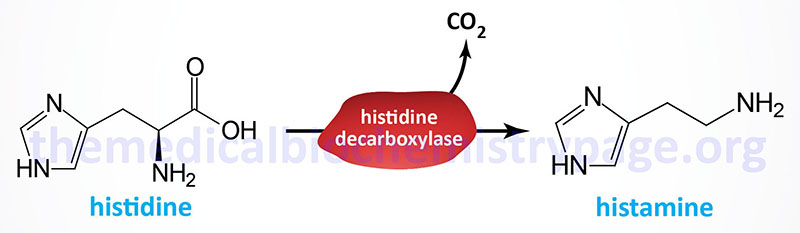
The histidine decarboxylase gene (symbol: HDC) is located on chromosome 15q21.2 and is composed of 14 exons that generate two alternatively spliced mRNAs encoding two distinct isoforms of the enzyme. The isoform 1 protein is composed of 662 amino acids and the isoform 2 protein is composed of 629 amino acids.
Following its release, histamine is metabolized by two alternative pathways, one of which is an oxidation and the other a methylation reaction. The oxidation of histamine is catalyzed by a diamine oxidase encoded by the AOC1 (amine oxidase, copper containing 1) gene. The product of diamine oxidase reaction is imidazole acetaldehyde which is itself further metabolized to imidazole acetate via the action of an aldehyde dehydrogenase family enzyme. In addition to the oxidation of histamine the diamine oxidase enzyme is involved in the metabolism of polyamine family neuromodulator, agmatine as well as the polyamine putrescine.
The methylation of histamine is catalyzed by the enzyme histamine N-methyltransferase encoded by the HNMT gene. The methyl group utilized by the HNMT encoded enzyme is derived from S-adenosylmethionine. The product of the methyl transfer reaction is N-methylhistamine which is then further metabolized to N-methylimidazole acetate via the action of monoamine oxidase, MAO. MAO is also critically involved in the metabolism of the catecholamines.
Histamine Receptors
Humans express four distinct histamine receptors identified as H1R, H2R, H3R, and H4R. All four histamine receptors are members of the G-protein coupled receptor superfamily. The H1R protein is encoded by the HRH1 gene, the H2R protein is encoded by the HRH2 gene, the H3R protein is encoded by the HRH3 gene, and the H4R protein is encoded by the HRH4. The HRH1 gene is located on chromosome 3p25 and is composed of 8 exons that generate four alternatively spliced mRNAs all of which encode the same 487 amino acid protein. The HRH2 gene is located on chromosome 5q35.2 and is composed of 8 exons that generate two alternatively spliced mRNAs. HRH2 isoform 1 is 397 amino acids and HRH2 isoform 2 is 359 amino acids. The HRH3 gene is located on chromosome 20q13.33 and is composed of 4 exons that encode a 445 amino acid protein. The HRH4 gene is located on chromosome 18q11.2 and is composed of 5 exons that generate three alternatively spliced mRNAs. Isoform 1 is 390 amino acids. Isoform 2 is 302 amino acids. Isoform 3 is 67 amino acids.
The H1R protein is coupled to a Gq-type G-protein. These receptors are expressed in a wide variety of tissues including the gastrointestinal tract, central nervous system (specifically cells of the tuberomammillary nucleus of the hypothalamus), both airway and vascular smooth muscle cells, endothelial cells, chondrocytes, monocytes, neutrophils, dendritic cells, T and B lymphocytes, adrenal medulla, and the cardiovascular and genitourinary systems. Expression of H1R in lymphocytes and monocytes is critical to the role of this receptor in mediating the modulatory effects of histamine within the immune system. With its distributed expression the H1R participates in ileum contraction, modulates circadian cycle, systemic vasodilatation, bronchoconstriction (allergy-induced asthma), and mediates pruritus (sensation of itchy skin).
The H2R protein is coupled to a Gs-type G-protein. Most immune cells that express the HRH1 gene also express the HRH2 gene. In addition to immune cells, the HRH2 gene is expressed in gastric parietal cells where the receptor plays a critical role in the regulation of gastric acid production in response to histamine. Indeed, histamine is the major molecule responsible for increased gastric acid production. Acetylcholine (ACh) and gastrin also contribute to gastric acid production but not to the degree of histamine. The HRH2 gene is also expressed in additional cells of the gastrointestinal tract, cells of the central nervous system, smooth muscle cells, endothelial cells, and cardiomyocytes. The principal responses to histamine binding to the H2R are stimulation of gastric acid secretion, increased sinus rhythm (cardiac rhythm initiated via depolarization from the sinus node), smooth muscle relaxation, inhibition of antibody synthesis, inhibition of T-cell proliferation and inhibition of cytokine production.
The H3R protein is coupled to a Gi-type G-protein. Expression of the HRH3 gene occurs nearly exclusively within the central nervous system, CNS. Regions of the brain that contain cell types expressing H3R include basal ganglia, cortex, hippocampus and strial area. Within the CNS the effects of histamine binding to H3R include decreases in acetylcholine (ACh), serotonin and norepinephrine production and release. Outside the CNS low level HRH3 expression has been found in the gastrointestinal and cardiovascular systems and in bronchial cells of the lungs.
The H4R protein is coupled to a Gi-type G-protein. Expression of the HRH4 gene is seen at high levels in the gastrointestinal tract, spleen, thymus, medullary cells, bone marrow and peripheral hematopoietic cells, including eosinophils, basophils, mast cells, T-cells, leukocytes and dendritic cells. Lower levels of expression of HRH4 are detected in the brain, heart, liver and lung.
Trace Amines
The trace amines represent a family of biogenic amines that are found in the brain at much lower concentrations than the more classical biogenic monoamines such as dopamine, norepinephrine, epinephrine, serotonin, and histamine. The trace amines are derived via the actions of biosynthetic and catabolic enzymes that are also utilized in the synthesis and catabolism of the classic biogenic amines.
The trace amines found to be active in humans include the tyramines [para-tyramine (p-tyramine), meta-tyramine (m-tyramine), 3-methoxytyramine, and N-methyltyramine], β-phenylethylamine (β-PEA), N-methylphenethylamine (NMPEA), the octopamines (m-octopamine and p-octopamine), synephrine, tryptamine, and 3-iodothyronamine. The thyronamines, which includes 3-iodothyronamine, are a family of decarboxylated and deiodinated metabolites of the thyroid hormones.
Trace amines were originally described as biogenic compounds that served as neurotransmitters in invertebrates. It is now known that the octopamines (so-called since octopamine was first isolated from the salivary glands of the octopus) and tyramine are the invertebrate sympathetic nervous system counterparts of norepinephrine and epinephrine in vertebrates. The significance of the trace amines in humans has been demonstrated since disturbances in the level of their synthesis and metabolism is correlated with numerous neuropsychiatric disorders, including Parkinson disease, depression, schizophrenia, and attention deficit hyperactivity disorder (ADHD).
Trace Amine Receptors
The presence of trace amines in humans, particularly in the brain, has been known for many years but it was not until the discovery a G-protein coupled receptor (GPCR) specific for trace amines (identified as TAAR1) that a clear understanding of the roles of these compounds in neurobiology began to be dissected. Following the characterization of TAAR1, additional members of the trace amine receptor family have been found to be expressed in humans. These receptors are identified as TAAR1, TAAR2, TAAR5, TAAR6, TAAR8, and TAAR9.
All of the human TAAR genes encode members of the class A rhodopsin-like GPCR family. Humans also possess three TAAR encoding pseudogenes identified as TAAR3P, TAAR4P, and TAAR7P. All of the human TAAR genes, including the pseudogenes are located in the same chromosome, region: 6q23.2. Within mammalian species there are over 50 identified trace amine receptors.
The role of TAAR1 is the most well characterized of the human TAAR gene family. Expression of the TAAR1 gene predominates in the major monoaminergic regions of the brain that includes the ventral tegmental area (VTA) and the dorsal raphe. One of the major effects of TAAR1 activation is negative control of dopaminergic neural activity. The trace amines, β-PEA, p-tyramine, p-octopamine, tryptamine, and 3-iodothyronamine have all been shown to bind to and activate TAAR1. Expression of TAAR2 is highest in the cerebellum. A nonsense mutation in the TAAR2 gene has been found to be correlated with schizophrenia.
Although the precise roles for trace amines in neuromodulation and neurotransmission are yet to be fully characterized in humans, accumulating evidence indicates that measurement for several of the trace amines in blood and urine can be useful for diagnostic purposes. Decreased production of both p-octopamine and tyramine have been found in patients suffering from depression. Conversely, increases in p-octopamine and tyramine have been shown to occur in patients suffering migraine headaches. Indeed, in individuals that suffer from migraines the consumption of foods rich in tyramine (e.g. chocolate, most cheeses, yogurt, shrimp, soybean condiments, teriyaki sauce, tempeh, miso soup, green bean pods) can trigger a migraine. In addition, increased plasma concentrations of p-octopamine have been correlated in several cases of chronic liver disease and hepatic encephalopathy.
In individuals who suffer from cluster headaches reduced levels of both p-octopamine and synephrine have been found in their plasma. The concentration of β-PEA in the human brain is highest in regions of dopaminergic innervation. Recent data indicate that measurement of reduced urine concentration of β-PEA is associated with children suffering from attention deficit hyperactivity disorder, ADHD.
Although 3-iodothyronamine is a derivative of the thyroid hormones it does not activate the thyroid hormone receptor but instead is a ligand for members of the TAAR family, specifically TAAR1, TAAR2, and TAAR5, with highest affinity for TAAR1. One effect of 3-iodothyronamine is to modulate adrenergic neurotransmission, in particular in the major norepinephrine releasing region of the brain, the locus coeruleus. Studies have also indicated that 3-iodothyronamine may play a role in the modulation of feeding behaviors through actions within the arcuate nucleus of the hypothalamus.
Agmatine
Agmatine (1-amino-4-guanidinobutane) is a member of the polyamine family that functions as a neuromodulator. The neuromodulatory function of agmatine and is exerted by its ability to modulate the activity of NMDA receptors, α2-adrenergic receptors, nicotinic acetylcholine receptors, the serotonin receptors 5-HT3 and 5-HT2A, and imidazoline receptors. The imidazoline receptors (I1, I2, and I3) are plasma membrane receptors that were identified by their binding clonidine and other imidazole drugs.
Agmatine Synthesis and Catabolism
Although agmatine was discovered in the early 1900’s, study of its synthesis and functions in humans began in earnest in the early 1990’s. Decarboxylation of arginine yields agmatine which is catalyzed by an enzyme identified as arginine decarboxylase, ADC. Whereas ADC has been characterized in plants, bacteria, and other mammals, the definitive ADC encoding gene in humans has yet to be characterized. An original assessment that ADC activity was associated with one of the antizyme inhibitor proteins, specifically the AZIN2 encoded protein, has subsequently been shown to be erroneous. Nonetheless, agmatine synthesis from arginine in humans is known to occur, particularly within the brain.
The major sites of agmatine synthesis are the brain, liver, and kidneys. Agmatine catabolism contributes to the synthesis of the polyamines as well as to the inhibitory neurotransmitter, GABA.
Catabolism of agmatine can proceed via two distinct pathways, primarily determined by the location of the catabolic pathway. Within the brain of mammals, such as rats and mice, the metabolism of agmatine occurs through the actions of an enzyme of the ureohydrolase family identified as agmatinase. Humans express a gene (AGMAT) with suspected agmatinase activity but as yet this has not been definitively demonstrated. Agmatinase releases urea from agmatine generating the polyamine, putrescine. This putrescine can go on to be converted to spermidine and then spermine via the normal polyamine biosynthesis pathway.
Within the periphery, agmatine is metabolized by an enzyme of the diamine oxidase (DAO) family that in humans is encoded by the AOC1 gene. The product of DAO activity on agmatine is the generation of γ-guanidinobutyraldehyde (4-guanidinobutyraldehyde). Gamma-guanidinobutyraldehyde is oxidized to γ-guanidinobutyrate (4-guanidinobutyrate) by an enzyme of the aldehyde dehydrogenase family, likely the ALDH1A1 encoded enzyme. Gamma-guanidinobutyrate is predominantly excreted but it can also serve a s substrate for an enzyme of the ureohydrolase family (possibly agmatinase) to yield the inhibitory neurotransmitter GABA (γ-aminobutyrate).
Biological Effects of Agmatine
As indicated above, agmatine modulates the activities of numerous receptors. With respect to the α2-adrenergic receptors, nicotinic acetylcholine receptors, serotonin receptors, and imidazoline receptors agmatine functions as an agonist. Conversely, agmatine is an antagonist of the NMDA receptors.
Within the brain the neuromodulatory effects of agmatine serve to exert anticonvulsant, antineurotoxic, and
antidepressant effects. Outside the brain agmatine has been shown to exert protective effects on the heart and the kidneys, as well as to exert a generalized cytoprotective effect.
In addition to its neuromodulatory functions agmatine can affect various metabolic and physiological processes. Additional biological activities associated with agmatine include the modulation of insulin release from the pancreas, inhibition of all three forms of nitric oxide synthase (NOS), and regulation of the expression of the rate-limiting enzyme of polyamine synthesis, ornithine decarboxylase, ODC.
Within the liver agmatine reduces the expression of several genes whose encoded enzymes function in hepatic lipogenesis, the consequences of which are decreased intrahepatic triglyceride accumulation. Agmatine has also been shown to increase the rate of urea production in hepatocytes.
Within the kidneys agmatine increases renal filtration rates by enhancing vasodilation, an effect that is associated with enhanced sodium excretion.