
Last Updated: October 30, 2024
Introduction to the Endocannabinoids
The discovery of the endocannabinoids came about as a result of the identification of the major psychoactive component of marijuana, Δ9-tetrahydrocannabinol (Δ9-THC, or just THC). Marijuana is technically a preparation of the Cannabis plant intended for use as a medicine or as a psychoactive drug. Cannabis is a genus of flowering plants that includes three putative varieties, Cannabis sativa, Cannabis indica, and Cannabis ruderalis. Following the discovery of the bioactive compound in marijuana it was determined that THC worked by binding to specific plasma membrane proteins which were referred to as the “cannabinoid receptors”.
Although the existence of several receptors for THC and/or its synthetic analogs are suggested based on pharmacological data, to date only two cannabinoid receptors have been cloned and both are members of the G-protein coupled receptor (GPCR) family. These receptors are called the CB1 receptor and the CB2 receptor. CB1 is quite possibly the most abundant GPCR in the central nervous system. Although highly expressed in the CNS, CB1 receptors are also important for eliciting cannabinoid effects in peripheral tissues. Expression of CB2 receptor is abundant in several immune cells and many other tissues but is also seen in some regions of the brain. Whereas brain CB1 receptors are coupled, among other things, to inhibition of neurotransmitter release, CB2 receptors seem to participate in the regulation of cytokine release and function.
More than 60 different cannabinoids have been shown to be present in marijuana but only THC is psychoactive. There are, however, several compounds that have been shown to have well-documented biological effects and that have potential for therapeutic development. One such compound, cannabidiol exhibits anti-anxiety, anti-convulsive, anti-nausea, anti-inflammatory and anti-tumor properties. However, cannabidiol does not significantly interact with CB1 or CB2 receptors, and its biological activities are attributed to either inhibition of anandamide (an endocannabinoid) degradation, or to its antioxidant properties, or as a result of the interaction with as yet unidentified cannabinoid receptors. Another such compound with potential therapeutic benefits is tetrahydrocannabivarin, which has recently been shown to work as an antagonist of CB1 receptors.
The discovery of cannabinoid receptors suggested the existence of endogenous ligands capable of activating them. Indeed, this fact led to the discovery of the ligands which are termed the endocannabinoids. The two best studied endocannabinoids are anandamide (N-arachidonoylethanolamine; AEA) and 2-arachidonoylglycerol (2-AG). The synthesis and metabolism of anandamide and 2-AG are described below. These two endocannabinoids, as well as other proposed endocannabinoids, are derived from the non-oxidative metabolism of arachidonic acid, a biologically critical omega-6 polyunsaturated fatty acid (PUFA).
The cannabinoid receptors, the endocannabinoids and the proteins catalyzing endocannabinoid biosynthesis and inactivation constitute the endocannabinoid system (ECS). The discovery and characterization of the ECS has recently led to the development of an entirely new class of drugs for the treatment of obesity and related metabolic disorders.
The other characterized arachidonic acid-derived endocannabinoids are 2-arachidonoylglyceryl ether (2-AGE), N-arachidonoyl dopamine (NADA), and O-archidonoylethanolamine (O-AEA). O-AEA is more commonly called virodhamine. The attachment of ethanolamine to arachidonic acid in virodhamine is an ester linkage, whereas the attachment in anandamide is an amide linkage. Virodhamine functions as an antagonist of the CB1 receptor and as an agonist of the CB2 receptor.
Many other lipid-derived signaling molecules, that are structurally related to anandamide and 2-AG, are know to exist. One molecule of particular interest is oleoylethanolamide (OEA). Although structurally related to endocannabinoids, OEA does not activate cannabinoid receptors but instead binds the fatty acid-sensing GPCR identified as GPR119.
Additional endocannabinoid-like molecules are the fatty acid amides, palmitoylethanolaminde (PEA), stearoylethanolamide (SEA), linoleoylethanolamide (LEA), and docosahexaenoylethanolamide (DHEA), as well as the glycerol conjugated fatty acids, 2-oleoylglycerol (2-OG), 2-palimtoylglycerol (2-PG), and 2-linoleoylglycerol (2-LG).
Endocannabinoid Receptors
As indicated above, studies of Δ9-THC binding in tissue samples led to the identification of an orphan G protein-coupled receptor (GPCR) as the brain receptor for cannabinoids. This receptor was subsequently named the CB1 receptor. The CB1 gene, identified as CNR1, is located on chromosome 6q15 anis composed of 7 exons that generate 12 alternatively spliced mRNAs that collectively encode two protein isoforms. The major CB1 receptor is a protein of 472 amino acids. CB1 receptors are the most abundant receptors in the mammalian brain but are also present at much lower concentrations in a variety of peripheral tissues and cells.
Two splice variants of CB1 receptors have also been identified: CB1A, which has an altered amino-terminal sequence, and CB1B, which has an in-frame deletion of 33 amino acids at the amino terminus. Expression of both splice variants is seen at much lower levels than that of CB1.
A second cannabinoid GPCR, CB2, is expressed primarily in cells of the immune and hematopoietic systems but is also found to be expressed in the brain, in nonparenchymal cells of the cirrhotic liver, in the endocrine pancreas, and in bone. The CB2 gene, identified as CNR2 is located on chromosome 1p36.11 and is composed of 5 exons that encodes a protein of 360 amino acids.
In addition to binding to CB1 and CB2, anandamide can bind and activate the transient receptor potential vanilloid 1 (TRPV1) channel which is also known as the capsaicin receptor. TRPV1 activation is responsible for the sensation of heat when consuming spicy peppers. Anandamide is only a weak agonist of both the CB1 and CB2 receptors, whereas 2-AG is a full agonist of both receptors.
One interesting aspect of the cannabinoid receptors relates to the fact that there is stereospecific selectivity to the receptors. In addition, although some ligands bind to both the central and peripheral receptors, they display only peripheral and not centrally mediated cannabinoid-like bioactivity. These observations suggest that some ligands may act as antagonists rather than agonists within the CNS, but not peripherally. One of these ligands that displays central versus peripheral selectivity is ajulemic acid, a metabolite of THC that has been shown to have potent anti-inflammatory and analgesic properties without any overt behavioral or psychoactive effects. Ajulemic acid also binds to peroxisome proliferator-activated receptor γ (PPARγ) which accounts for its observed effects on adipocyte differentiation.
As indicated above both CB1 and CB2 receptors are GPCR. Although these two receptors share limited overall amino acid homology (48%), THC and most synthetic cannabinoids have similar binding affinities for the two receptors. The highest amino acid similarity between the two receptors (68%) is in their transmembrane domains which are thought to contain the binding sites for cannabinoids. The development of synthetic cannabinoid ligands that discriminate between CB1 and CB2 has been critically important for exploring the physiological functions of the endocannabinoids.
CB1 and CB2 receptors couple primarily to the Gi/o subtypes of G-protein. For more information on the types of heterotrimeric G-proteins coupled to GPCRs, and their activities go to the Signal Transduction Pathways: G-Proteins and GPCR page. CB1 receptors have been shown to efficiently couple and activate both Gi and Go type G-proteins. However, CB2 receptors only activate Go. In addition, the efficacy of a particular agonist is different whether CB1 receptors are coupled to Gi or Go type G-proteins. These data demonstrate that there is agonist-selective CB1 and CB2 receptor-associated G-protein signaling.
The signaling cascades initiated by cannabinoid binding to CB1 and CB2 receptors is remarkably complex. Normally, G-protein coupling to adenylate cyclase through Gi/o results in inhibition of cyclase activity via the release of Giα isoforms. However, cannabinoids can also activate isoforms of adenylate cyclase by stimulating the release of the βγ subunits from the receptor-associated G-proteins. Cannabinoids have also been shown activate several other signaling kinases. These include members of the mitogen-activated protein kinase family including JUN-terminal kinase (JNK), p38 MAPK, and extracellular signal regulated kinases 1/2 (ERK1/2).
In addition, cannabinoids are known to activate the phosphatidylinositol-3-kinase (PI3K) pathway. These additional activities of cannabinoids may be through G-protein activation or via pathways that are independent of G-proteins and involving various other adaptor proteins.
Another G-protein-independent pathway shown to be activated by cannabinoids involves activation of G-protein-coupled receptor kinase-3 (GRK3) and β-arrestin-2. These latter two signaling proteins are required for CB1 receptor desensitization, but not for receptor internalization, and the development of tolerance. Cannabinoids also regulate the activity of phosphatases.
Activation of CB1 receptors regulates the activity of protein phosphatase 2B (PP2B/calcineurin) and mitogen-activated protein kinase (MAPK) phosphatase-1 (MKP-1). MKP-1 is an important component of the anti-inflammatory actions of anandamide.
Cannabinoids have also been shown to inhibit voltage-dependent N-, P- and Q-type calcium channels and to activate inwardly rectifying potassium channels via the βγ subunits of Gi/o type G-proteins. The effects of cannabinoids at these ion channels underlie the neuromodulatory actions of the endocannabinoids in both the central and peripheral nervous systems. CB1 receptors can also couple to Gq/11 type G-proteins to enhance phospholipase C (PLC) activity, leading to increases in the concentration of intracellular calcium.
The development of potent and highly selective CB1 and CB2 receptor antagonists has had significant clinical implications. For example, the appetite-reducing effects of the CB1 antagonist rimonabant demonstrated that endocannabinoids are tonically active orexigenic (appetite inducing) compounds. With respect to CB1 and CB2 receptor antagonists developed to date it is important to note that these compounds function as inverse agonists. Thus, their effects do not necessarily reflect reversal of the tonic action of an endocannabinoid.
The definition of an inverse agonist is an agent that binds to the same receptor as an agonist but induces a pharmacological response that is opposite to that agonist. A prerequisite for an inverse agonist response is that the receptor must have a constitutive (intrinsic or basal) level of activity in the absence of any ligand. An agonist, by definition, increases the activity of a receptor above its basal level while the response to an inverse agonist is a decreases in receptor activity below the basal level. The development of CB1 and CB2 receptor-deficient mouse strains has allowed for the characterization of specific neuronal populations with well-defined cannabinoid-modulated behaviors.
Since their initial discovery and characterization pharmacological evidence has suggested the existence of more cannabinoid receptors than just CB1 and CB2 receptors. Of these additional additional cannabinoid responsive receptors the characteristics of two have been more extensively explored. One is a cannabinoid responsive receptor associated with the endothelium and is involved in vasodilation and endothelial cell migration. The second is found on presynaptic glutamatergic nerve terminals in the hippocampus and mediates the inhibition of glutamate release. That these are cannabinoid responses elicited through a receptor other than CB1 receptors was demonstrated in mice in which the CB1 receptor was knocked out yet responses were still observed.
In addition, the responses were sensitive to inhibition by a classic CB1 receptor antagonist. The endothelial and hippocampal sites of cannabinoid action are due to distinct receptors as defined by the fact that certain cannabinoid antagonists or agonists elicited effects upon one site but not the other. In addition, certain atypical cannabinoids with no affinity for CB1 or CB2 receptors behave as agonists or antagonists at the endothelial receptor but not at the hippocampal receptor. A brain cannabinoid receptor distinct from CB1 was also indicated by the ability of anandamide, but not other agonists, to stimulate GTPγS binding in brain plasma membranes from CB1 knockout mice.
Recent potentially clinically significant data has demonstrated that the orphan GPCR identified as GPR55 recognizes a variety of cannabinoid ligands. GPR55 couples to G12/G13 type G-proteins as well as the ρ (rho) kinase, which have been linked to vasoconstrictive responses. More information on the activity of GPR55 and its ligands can be found in the Bioactive Lipids and Lipid Sensing Receptors page.
Synthesis of Endocannabinoids
Anandamide and 2-AG are not prestored in secretory vesicles of resting cells. They are synthesized de novo and released only when and where necessary in response to physiological or pathological stimuli. These stimuli result in an increase in the intracellular concentration of calcium. Indeed, the formation of the two direct and distinct biosynthetic precursors of anandamide and 2-AG, as well as the conversion of these precursors into the two endocannabinoids, is catalyzed by calcium-sensitive enzymes.
The level of endocannabinoid synthesis in a given cell, in response to a given stimulus, will ultimately depend on the availability of arachidonic acid on the sn-1 or sn-2 position of phosphoglycerides for anandamide and 2-AG, respectively. Following stimulated synthesis, the endocannabinoids are released from the cell, activate their target cell, and are then rapidly removed from the extracellular space by a selective cellular reuptake mechanism followed, ultimately by intracellular enzymatic hydrolysis.
There are several additional endocannabinoid compounds that have been identified and characterized for biological activity. These include virodhamine, noladin, and N-arachidonoyldopamine (NADA). However, thus far no conclusive data has been obtained relating to the mechanisms of their biosynthesis.
N-arachidonoylethanolamine (AEA, anandamide) Synthesis
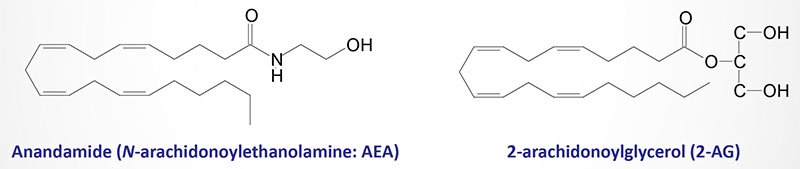
The in vivo biosynthesis of anandamide has been shown to occur via several distinct, yet related, pathways that depend in part on the cellular origin. Anandamide belongs to the family of N-acyl-ethanolamines (NAE). Prior to the discovery of anandamide, the synthesis of NAE was shown to be the result of a phospholipid-dependent pathway consisting of the enzymatic hydrolysis of the corresponding N-acyl-phosphatidylethanolamines (NAPE). The enzyme catalyzing this reaction was identified as a phospholipase D (PLD) selective for NAPE (NAPE-PLD). NAPE-PLD exhibits catalytic properties different from other PLD enzymes.
NAPE-PLD is encoded by the NAPEPLD gene located on chromosome 7q22.1 and is composed of 8 exons that generate two alternatively spliced mRNAs, both of which encode the same 393 amino acid protein.
The phospholipid precursors of NAE are in turn produced via the enzymatic transfer of an acyl group from the sn-1 position of phospholipids to the N-position of phosphatidylethanolamine (PE). This latter reaction can be catalyzed by a Ca2+-dependent N-acyltransacylase (NAT) or a Ca2+-independent N-acyltransferase (iNAT). Both the NAPE-PLD and the NAT/iNAT involved in the synthesis of anandamide do not appear to be selective for a particular fatty acid moiety. It is not yet known whether NAPE-PLD is obligatory for anandamide synthesis. However, this NAPE-PLD does not recognize phosphatidylcholine nor phosphatidylethanolamine as substrates.
There are several parallel pathways for the generation of anandamide from NAPE. A secretory PLA2 (sPLA2) has been identified that can catalyze the hydrolysis of NAPE to lyso-NAPE. These lyso-NAPE can then serve as substrates for a lysophospholipase D (lyso-PLD) or for members of the glycerophosphodiester phosphodiesterase family (e.g. GDE4 or GDE7) which generates N-acyl-ethanolamides, including anandamide.
Another alternative parallel pathway for anandamide synthesis has been identified in macrophages. This pathway involves hydrolysis of NAPE to phosphoanandamide by a PLC, followed by dephosphorylation via the action of phosphatases where the protein tyrosine phosphatase non-receptor type 22 (PTPN22) is the major enzyme. The inositol 5′-phosphatase (originally called SHIP1) encoded by the INPP5D gene can also carry out the dephosphorylation reaction. This latter pathway was discovered in the course of studies aimed at defining the synthesis of anandamide in macrophages in response to bacterial endotoxin. The existence of this pathway may also account for the recent finding that the tissue levels of anandamide are unchanged in NAPE-PLD knockout mice. Of significance is the fact that mutations in the PTPN22 gene are strongly correlated to the development of type 1 diabetes.
2-Arachidonoylglycerol (2-AG) Synthesis
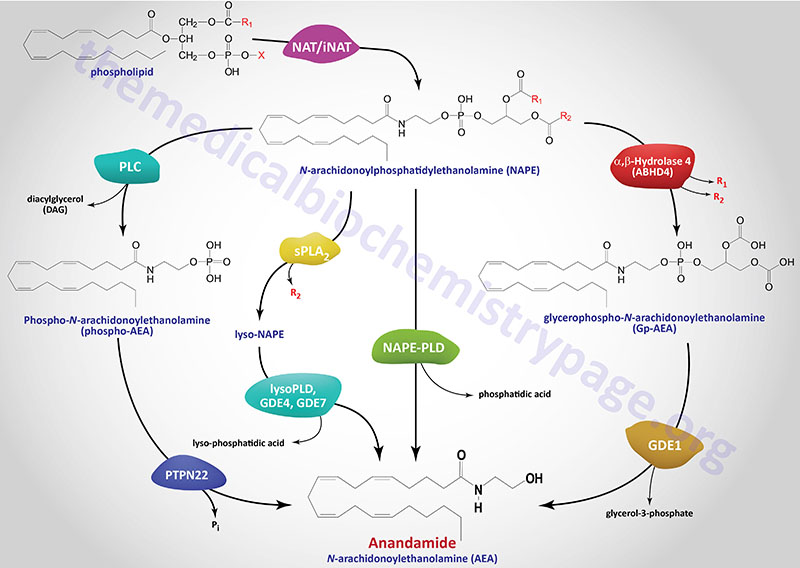
Several different types of stimuli have been shown to result in the synthesis of 2-AG in cells. These stimuli include lipopolysaccharides (LPS), platelet-activating factor (PAF), and thrombin as examples. In most cases, 2-AG is produced from the hydrolysis of diacylglycerols (DAGs) containing arachidonic acid in the sn-2 position. This hydrolysis is catalyzed by DAG lipases that are selective for hydrolysis of the acyl group attached at the sn-1 position. The DAG substrates for DAG lipases can be produced via hydrolysis of phosphatidylinositides (PI) where phosphatidylinositol-4,5-bisphosphate (PIP2) is the major precursor.
Formation of a DAG from PIP2 is predominantly catalyzed by a phospholipase C (PLC) family member with PLC-β1 being the predominant enzyme. This enzyme is active in macrophages, platelets and cortical neurons in the brain. Two sn-1-selective DAG lipase isozymes (DAGLα and DAGLβ) have been identified and characterized with both being proposed to be responsible for the formation of 2-AG in intact cells.
The DAGLα enzyme is derived from the DAGLA gene which is located on chromosome 11q12.2 and is composed of 25 exons that encode a 1042 amino acid protein.
The DAGLβ enzyme is derived from the DAGLB gene which is located on chromosome 7p22.1 and is composed of 15 exons that generate two alternatively spliced mRNAs.
The pattern of expression of these two DAG lipases correlates with the proposed function of 2-AG either as a mediator of neurite growth, during brain development, or as retrograde messenger mediating depolarization-induced suppression of inhibitory (DSI) or excitatory (DSE) neurotransmission in the adult brain. The two DAG lipases do not appear to be selective for arachidonate-containing DAGs but will hydrolyze the acyl group at the sn-1 position of numerous DAGs. The major DAG lipase involved in 2-AG synthesis is the DAGLα isoform.
Another substrate for DAG lipases is the DAG generated from the hydrolysis of phosphatidic acid (PA) via the activity of any of several phosphatidic acid phosphatases. The role of phosphatidic acid phosphatases in overall 2-AG synthesis was originally demonstrated in neuroblastoma and microglial cells. Humans express nine enzymes that belong to the phosphatidic acid phosphatase (PAP) family. Three of these enzymes are derived from members of the lipin (LPIN) gene subfamily with the other six family members belonging to the phospholipid phosphatase (PLPP) subfamily. The lipin proteins, specifically lipin-1, are involved in triglyceride synthesis.
Synthesis of Omega-3 Endocannabinoids
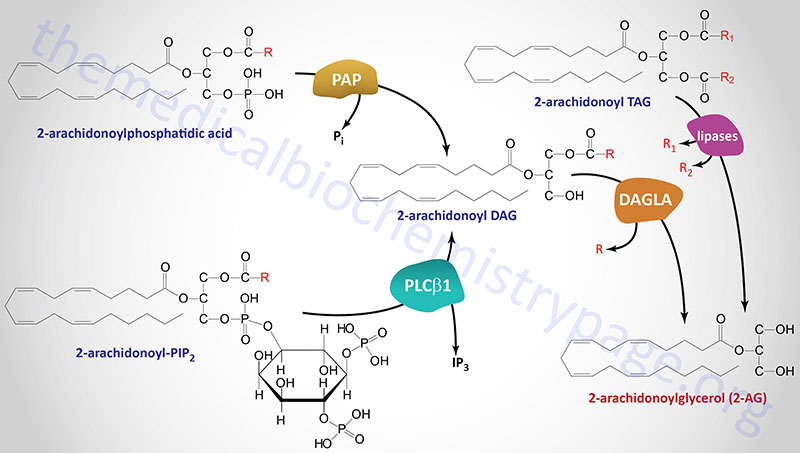
The omega-3 fatty acids, docosahexaenoic acid (DHA) and eicosapentaenoic acid (EPA), serve as substrates for non-oxidative synthesis of the endocannabinoid-like ethanolamides. DHA is the precursor for the synthesis of , N-docosahexaenoyl ethanolamine (DHEA) which is commonly termed synaptamide. EPA is the precursor for the synthesis of eicosapentaenoyl ethanolamide (EPEA).
Both synaptamide and EPEA exhibit anti-inflammatory properties. As its name implies, synaptamide promotes neural cell growth and synaptogenic properties. Synaptamide also possesses anticancer activity.
The metabolism of synaptamide and EPEA, via epoxygenases of the CYP family, yields additional bioactive lipids identified as omega-3 endocannabinoid epoxides. The major CYP family enzyme carrying out the oxidation of synaptamide and EPEA is encoded by the CYP2J2 gene.
The primary synaptamide derived epoxide is epoxydocosapentaenoic acid-ethanolamide (EDP-EA) and that derived from EPEA is epoxyeicosatetraenoic acid-ethanolamide (EEQ-EA). The primary regioisomer of EEQ-EA is 17,18-EEQ-EA and that of EDP-EA is 19,20-EDP-EA. Both 17,18-EEQ-EA and 19,20-EDP-EA exhibit potent anti-inflammatory activity as well as modulation of platelet activation.
Endocannabinoid Metabolism
In order to control the biological actions of the endocannabinoids, there are mechanisms for their rapid removal from their sites of action as well as pathways for their subsequent degradation. Being lipophilic the endocannabinoids can easily diffuse through the cell membrane. However, to ensure rapid, selective, and regulated diffusion, this process needs to be facilitated by a carrier or to be driven by a mechanism capable of rapidly reducing their intracellular concentration, or both.
Anandamide appears to be taken up by several different types of cells via a facilitated transport mechanism, known as the anandamide membrane transporter (AMT). Although the AMT has yet to be isolated or cloned, indirect evidence suggests that it can also mediate 2-AG, noladin, virodhamine and NADA uptake. Currently there is some controversy as to the method of anandamide uptake. Some evidence suggests that the enzyme responsible for anandamide hydrolysis, fatty acid amide hydrolase (FAAH), may be involved in the cellular uptake of anandamide. However, cell types that do not express FAAH are still able to rapidly take up anandamide and synthetic compounds that inhibit anandamide uptake show no effect on its enzymatic hydrolysis. What can be deduced from studies of anandamide uptake and hydrolysis is that FAAH activity can influence facilitated anandamide uptake, but that other mechanisms distinct from the intracellular metabolism of endocannabinoids must be involved to enhance the membrane transport rate of endocannabinoids.
FAAH was originally purified and cloned from rat liver and has been shown to catalyze the hydrolysis of long chain primary fatty acid amides and glycerol esters. FAAH is primarily found in microsomal membranes. The enzyme has an optimal activity under alkaline pH conditions. That FAAH is critical for the in vivo degradation of anandamide has been well documented. When the FAAH gene is knocked-out in mice they have 10 to 15 times elevated levels of anandamide across the brain resulting in supersensitivity to the actions of exogenous anandamide. These mice show tonic signaling by endogenous anandamide, resulting in CB1 receptor-mediated hypoalgesia, reduced anxiety, antidepressant activity, and lowering of blood pressure in different models of experimental hypertension.
Differentiating these effects of anandamide to peripheral versus central nervous system effects was made possible by the generation of peripheral tissue-specific knockout mice. These mice do not display the hypoalgesia seen in global FAAH knockout mice, but they do exhibit similar anti-inflammatory phenotype. This latter fact indicates the effects are mediated by elevated fatty acid amides in peripheral tissues. Another amidohydrolase, present in the lysosomes and active at an acidic pH, has been identified that catalyzes the same reaction as FAAH. This enzyme is structurally unrelated to FAAH yet it has been shown to contribute to the physiological degradation of anandamide in macrophages but not in the brain.
Although FAAH can catalyze 2-AG hydrolysis under in vitro conditions, in vivo it is not a substrate of FAAH, as indicated by the unchanged brain levels of 2-AG in wild-type and FAAH knockout mice. Other 2-AG hydrolases, known as monoglyceride lipases (MGLs), present in both membrane and cytosolic subcellular fractions, can catalyze the in vivo hydrolysis of 2-AG. Examination of the distribution of FAAH and MGL in the hippocampus, cerebellum, and amygdala, shows that FAAH is located postsynaptically, whereas MGL is localized in presynaptic axon terminals, including terminals of GABAergic interneurons. MGLs recognize other unsaturated monoglycerides as substrates which in some cases results in competition for 2-AG inactivation. Functional studies in the hippocampus demonstrate that depolarization-induced suppression of inhibition (DSI) is unaffected by pharmacological blockade of FAAH, but it is potentiated by blocking MGL. These results support other studies indicating that 2-AG, but not anandamide, is involved in synaptic plasticity in the hippocampus.
Endocannabinoids in the CNS and the Control of Feeding Behaviors
Regulation of feeding behavior has long been associated with the cannabinoid system. Initially these effects were thought to occur only via modulation of central nervous system signaling processes. Cannabis in its various forms increases appetite, particularly for palatable foods, and can also result in significant weight gain. The classic response to pharmacologic stimulation of CB1 receptors by systemic administration of plant (e.g. THC) or endogenous cannabinoids is the stimulation of food intake even in satiated animals. It is worth noting that the ability of THC to promote food intake is only observed with relatively low doses. This latter fact is most likely because the significant sedation and motor impairment seen with higher doses interferes with an animals’ ability to initiate feeding.
Similar to the effects of THC, when low doses of the endocannabinoid, anandamide, are administered either systemically or into the ventromedial hypothalamus (VMH) there is a CB1 receptor-dependent increase in feeding behavior. In addition, increases in food intake can be elicited by 2-AG administration systemically or into the lateral hypothalamus. Conversely, pharmacological antagonism of CB1 receptors attenuates the stimulatory effects of agonists, such as THC, on food intake. Of significance, this antagonism strongly reduces both the consumption of palatable food (such as sweet foods) by animals fed ad libitum and the intake of normal food, but not water, by animals deprived of food. However, antagonism of CB2 receptors does not block the feeding responses elicited by THC or anandamide.
The first selective CB1 receptor antagonist to be developed was the compound SR141716, commonly called rimonabant. Rimonabant blocks food consumption during both the appetitive (instinctive physical desire for food) and consumption phases of feeding behavior in pre-fed animals. However, it does not block the Pavlovian response to a palatable stimulus. These results suggests that endocannabinoids do not reinforce the ability of the stimulus (in this case food) to elicit a response to approach and consume food, but instead they are responsible for the maintenance of stimulus-induced goal-directed behaviors.
Other CB1 receptor antagonists exert identical effects with some exerting their effects for up to 6 days following a single dose. When the CB1 receptor is knocked out in mice they eat less than their wild-type littermates. Food intake in these CB1 receptor deficient mice is unaffected by rimonabant treatment whereas in wild-type mice rimonabant treatment reduces food intake to the levels seen in the knockout mice. These data, together with the established neuromodulatory role of endocannabinoids through CB1 receptors, strongly indicates that the brain endocannabinoid system controls food intake at two levels. First, it tonically reinforces the motivation to find and consume food. This effect is likely to involve an interacting with the mesolimbic pathways involved in reward mechanisms.
The dual action of cannabinoids in mesolimbic and hypothalamic regions is demonstrated by the observation that injection of endocannabinoids into these brain areas stimulates food intake. For more details on the role of the hypothalamus in the control of feeding behaviors go to the Gut-Brain Interrelationships and Cntrol of Feeding Behavior page. Second, it is activated on demand in the hypothalamus after short-term food deprivation and then transiently regulates the levels and/or action of other orexigenic and anorectic mediators to induce appetite.
The level of endocannabinoids varies in both the hypothalamus and the limbic forebrain during different phases of feeding behavior in rats. Their levels are highest during food deprivation and lowest during food consumption, as would be expected from orexigenic mediators. Within the hypothalamus, changes in endocannabinoid levels are inversely correlated with the changes in serum levels of leptin. Leptin is a key regulator of orexigenic and anorexigenic signaling within various hypothalamic nuclei. Indeed, leptin decreases endocannabinoid levels in the hypothalamus, similarly to its effects on other orexigenic mediators. In obese rodents with defective leptin signaling there is a significantly higher level of hypothalamic endocannabinoids. The gut hormone ghrelin is also likely to be involved in the regulation of hypothalamic endocannabinoid levels since they increase as the serum level of ghrelin increases following food deprivation. The effects of ghrelin on endocannabinoid levels are CB1 receptor antagonism with rimonabant. With respect to the limbic system endocannabinoids are thought to increase the drive to eat by enhancing dopamine release in the nucleus accumbens shell or by synergizing with opioids through as yet undefined mechanisms.
The endocannabinoid system likely exerts its influence over feeding behaviors via regulation of the expression and/or action of several hypothalamic anorexigenic and orexigenic peptides. CB1 receptors have been shown to co-localize with corticotropin-releasing hormone (CRH) in the paraventricular nucleus (PVN), with melanin-concentrating hormone (MCH) in the lateral hypothalamic area (LHA), and with prepro-orexin in the ventromedial hypothalamus (VMH). The MCH-containing neurons are tonically inhibited by GABAergic interneurons. This inhibitory tone can be suppressed by the release of endocannabinoids from MCH neurons and their retrograde activation of presynaptic CB1 receptors on the GABAergic interneurons. The resulting increase in the activity of MCH neurons may be a contributing factor in the appetitive effects of endocannabinoids. Deletion of CB1 receptors in mice is correlated to increased expression of CRH indicating that endocannabinoids are involved in tonic inhibition of expression of this anorexigenic peptide. Conversely, when CB1 receptors are activated, orexin-1 receptors (OX1R) become sensitized when these two receptors are expressed in the same cell. The result is an enhancement of the appetite-inducing action of orexins.
Unlike the above mentioned hypothalamic peptides, CB1 receptors are not found in cell types that express the orexigenic peptide, neuropeptide Y (NPY). However, evidence suggests that endocannabinoid activation downstream of NPY may mediate some of the orexigenic effects of this peptide. Pharmacologic antagonism or deletion of CB1 receptors results in an attenuated NPY response. For example mice lacking CB1 receptors are resistant to overeating induced by the orexigenic effects of NPY. Rimonabant is equally effective at suppressing appetite and feeding in both wild-type as in NPY knockout mice. This indicates that the induction of food intake by endocannabinoids is not mediated by NPY.
Rimonabant also inhibits the hyperphagia seen in leptin-deficient mice even if the mice were not starved first. This latter finding suggests that the absence of leptin results in increased endocannabinoid activity. Indeed, hypothalamic levels of endocannabinoids were elevated in leptin-deficient mice and reduced after leptin treatment. Obesity is associated with hyperleptinemia, yet the ability of leptin to inhibit food intake is blunted. Anandamide levels in the hypothalamus, limbic forebrain, and amygdala are significantly lower in obese mice compared with their lean controls. These results suggest that the endocannabinoids are an integral part of the leptin-regulated neural circuitry involved in appetite regulation.
An additional effect of endocannabinoids within the hypothalamus involves the melanocortin peptides. Rimonabant inhibits the feeding response induced by blocking melanocortin receptor 4 (MCR4) which is the receptor for α-melanocyte stimulating hormone (αMSH). However, α-MSH does not affect THC-induced feeding. This suggests that CB1 receptors are downstream from MCR4 receptors and have an obligatory role in α-MSH effects on food intake. The anorexigenic effects of α-MSH are blocked by the orexigenic peptide Agouti-related peptide (AgRP) which is an antagonist (inverse agonist) of MC4R.
There is likely to be cross talk between the endocannabinoid system and the endogenous opioid system in the regulation of feeding behaviors. Inhibition of food intake by μ-opioid receptor (MOR) antagonists and CB1 receptor antagonists is supra-additive, indicative of a synergism between these two systems in mediating the reinforcing effect of food. Indeed, CB1 receptor knock out mice fail to self-administer morphine suggesting that the site of this synergism is in the mesolimbic dopaminergic pathway that is involved in both drug and food reward. Rimonabant administration inhibits the orexigenic effect of morphine administered into the PVN which further indicates interactions between the two systems, unrelated to the hedonic aspects of feeding. MOR knockout mice are resistant to diet-induced obesity just as are CB1 receptor knockout mice, further demonstrating parallels between these two system in the regulation of feeding behaviors.
The endocannabinoid system also controls feeding behaviors via actions involving the vagal nerve the gut and the brain. When rats are deprived of food the levels of anandamide increase within the duodenum. At this location endocannabinoids are most likely reducing sensations of satiety via actions on the vagal nerve circuits. When rimonabant is administered peripherally it induces an anorexigenic response. In addition, destruction of the vagal nerve causes a reversal of the effects of peripheral anandamide administration. Food deprivation also enhances CB1 receptor expression in cholecystokinin (CCK)-1 receptor–expressing neurons that project to the duodenum. If CCK is administered, or if animals are re-fed, the expression of CB1 receptors increases in these neurons. From the results of these types of experiments it is likely that a reduction in endocannabinoid levels in the gut may mediate the induction of satiety by CCK. Also, the reduction in satiety in response to fasting may be medicated, in part, through increased levels of endocannabinoid in the small intestine and by a reduction in vagal CB1 receptor inhibition by CCK. The overall result is a disinhibition of the endocannabinoid system in within the axis of the vagal nerves.
Endocannabinoids in Peripheral Energy Homeostasis
Given that there was very strong evidence supporting the involvement of endocannabinoids in controlling food intake, it seemed only natural to attempt to treat obesity with drugs that functioned as antagonists of CB1 receptor. Rimonabant was the first such CB1 receptor-specific antagonist developed. Treatment of mice and rats with rimonabant reduces food intake even in animals that are genetic models of obesity such as leptin-deficient (ob/ob) and leptin receptor-deficient (db/db) mice.
As indicated above, the cannabinoids are associated with increased feeding behaviors. However, it is important to note that although marijuana smoking increases food intake, the increase in caloric intake will level off after a few days even though a person may continue to smoke. Although caloric intake declines with continual marijuana smoking there will be is a continual increase in weight gain. This suggests that cannabinoids exert independent effects on appetite and peripheral energy metabolism.
Peripheral targets of endocannabinoids include adipocytes, which express CB1 receptors. Stimulation of CB1 receptors on adipocytes affects lipid metabolism by regulating the level of adiponectin production, increasing lipoprotein lipase (LPL) activity, and by inhibiting AMP-activated protein kinase (AMPK). Inhibition of AMPK leads to increased fatty acid synthesis and a decrease in fatty acid β-oxidation due to reduced inhibitory phosphorylation of acetyl CoA carboxylase-1 (ACC1) which is the rate-limiting enzyme in fatty acid synthesis.
In CB1 receptor-deficient mice, the induced hypophagia accounts for the lean phenotype only in young and not in adult animals, which clearly indicated the involvement of peripheral metabolic target(s) in the latter. The additional documentation of functional CB1 receptors in primary cultured adipocytes and their role in regulating lipogenesis provided one of the likely peripheral targets for the anabolic effects of endocannabinoids. The lean phenotype of CB1–/– mice in this study was more prominent in male than in female animals, which could suggest that endocannabinoid regulation of adiposity may be subject to modulation by sex hormones.
Treatment of mice with cannabinoid agonists results in increased de novo hepatic lipogenesis and the expression of the lipogenic transcription factor SREBP1c as well as of its targets, ACC1 and fatty acid synthase (FAS). The role of CB1 receptors in these hepatic effects is demonstrated by the fact that CB1 receptor knockout mice or rimonabant treatment blocks the observed hepatic effects.
Within the brain fatty acid metabolism in the hypothalamus serves as a sensor of nutrient availability. Pharmacologic modulation of hypothalamic fatty acid metabolism influences food intake. Activation of CB1 receptors in the hypothalamus is associated with increased SREBP1c and FAS expression. Increased hypothalamic expression of these two genes is inhibited by rimonabant treatment. Modulation of AMPK activity by cannabinoids is observed not only in liver and adipose tissue but also in hypothalamus, where it has been linked to appetite control. Thus, the AMPK/ACC1/FAS pathway may represent a common molecular pathway involved in both the central appetitive and the peripheral metabolic effects of endocannabinoids.
Given that total caloric intake is similar in wild-type and CB1 receptor knockout mice fed a high-fat diet, the resistance of these mice to diet-induced obesity must be associated with increased energy expenditure. Treating mice with a CB1 receptor agonist decreases, whereas rimonabant treatment increases the activity of carnitine palmitoyltransferase-1 (CPT-1), the rate-limiting enzyme in fatty acid β-oxidation.
One of the factors involved in this effect is likely to be adiponectin which promotes fatty acid β-oxidation. Indeed, consumption of a high-fat diet causes a significant decline in serum adiponectin levels in wild-type but not in CB1 receptor knockout mice. Additionally, CB1 receptor activation in isolated adipocytes suppresses adiponectin expression. Adipose tissue expression of uncoupling protein-1 (UCP-1) is also down-regulated by CB1 activation. Conversely, rimonabant treatment increases adiponectin secretion by adipocytes and serum adiponectin levels in obese human subjects. Glucose uptake by skeletal muscle is also increased in response to rimonabant treatment, indicative of increased energy expenditure. This effect likely accounts for the increased glucose tolerance observed in obese individuals treated with rimonabant.
Endocannabinoids in Obesity
The possibility that an increase in the activity of the endocannabinoid system may contribute to at least some forms of obesity was suggested by three sets of findings. First, CB1 receptor antagonists were significantly more efficacious in reducing caloric intake and body weight in rodents with diet-induced or genetic obesity than in their respective lean controls.
Second, CB1 receptor knockout mice are resistant to diet-induced obesity. In both of these studies, overall caloric intake was not different between wild-type compared with CB1 receptor knockout mice receiving the high-fat diet, suggesting that peripheral mechanisms play a dominant role in the control of body weight by CB receptors. CB1 receptor knockout mice are also resistant to the metabolic changes that accompany diet-induced obesity in normal mice, including hypertriglyceridemia and elevated plasma leptin and insulin levels, indicative of leptin and insulin resistance, respectively. These metabolic changes, collectively referred to as the metabolic syndrome, could also be reversed by rimonabant treatment.
Third, endocannabinoids and CB1 receptors are up-regulated in the liver and adipose tissue in various forms of experimental as well as in human obesity. In wild-type mice on a high-fat diet the basal rate of de novo hepatic fatty acid synthesis is markedly increased, and the increase is partially reversed by rimonabant treatment. After 3 weeks on the high-fat diet, the mice were not yet overweight but showed significant hepatic steatosis. Their hepatic content of anandamide was increased 3-fold, and the level of CB1 receptor protein in liver plasma membranes was also markedly increased.
These findings indicate that intake of a high-fat diet activates the hepatic endocannabinoid system, which contributes to increased lipogenesis and the subsequent development of hepatic steatosis and, ultimately, the development of obesity. Feeding mice a high-fat diet also induces changes characteristic of the metabolic syndrome and also to rapidly induce the expression of SREBP1c and its downstream target lipogenic enzymes. CB1 receptor knockout mice are resistant to these diet-induced changes, which indicates that endocannabinoids have a major role in mediating them.
An up-regulation of CB1 receptors has been also reported in adipose tissue of genetically obese compared with lean mice, and elevated endocannabinoid levels have been detected in adipose tissue of obese compared with lean patients. In a study involving 40 women, circulating levels of anandamide and 2-AG were significantly increased in 20 obese versus 20 lean subjects, and remained elevated after a 5% diet-induced weight reduction. Although these plasma levels were much too low to exert hormone-like activity, they probably originate from overflow from tissues and thus may reflect functionally relevant changes in endocannabinoid content at or near sites of action. In the same study, FAAH expression was markedly reduced in the adipose tissue of obese subjects and correlated negatively with circulating endocannabinoid levels. Furthermore, the expression of both CB1 receptors and FAAH increased in mature adipocytes compared with preadipocytes. These findings suggest that the endocannabinoid system is activated in human obesity.
A genetic missense polymorphism in the FAAH gene predicting a proline to threonine substitution at position 129, which was reported to result in reduced cellular expression and activity of the enzyme, had been earlier found to be significantly associated with problem drug use. The same polymorphism has been linked to overweight and obesity in both Caucasian and African-American subjects. Interestingly, the elevated hepatic levels of anandamide in mice receiving a high-fat diet could be attributed to a decrease in FAAH activity, suggesting that FAAH may play a key role in regulating endocannabinoid tone in both experimental and human obesity. Although this finding could suggest the targeting of FAAH in the treatment of eating/metabolic disorders, such an approach will be complicated by the fact that oleoylethanolamide (OEA), an anorectic lipid that acts on PPARγ, is also a substrate for FAAH. The opposing effects of elevated levels of both anandamide and OEA after pharmacological blockade of FAAH may therefore result in no net change in appetite and energy metabolism.
That increased endocannabinoid activity may also contribute to obesity and its metabolic consequences in humans was indicated by the highly promising results of recent clinical trials with rimonabant. As in the animal models of diet-induced obesity, rimonabant was effective both in reducing body weight and in reversing many of the associated metabolic abnormalities in obese subjects.
In a multicenter study involving 1507 obese European subjects, rimonabant treatment for 1 year, combined with a moderately hypocaloric diet, not only reduced body weight but also reduced plasma triglycerides, increased HDL cholesterol, and decreased plasma insulin and insulin resistance.
In another study involving 1036 overweight/ obese subjects, rimonabant taken for 1 year significantly reduced body weight, weight circumference, and plasma triglycerides, increased HDL cholesterol, improved glucose tolerance, and significantly elevated plasma adiponectin levels, resulting in a 50% decrease in the prevalence of the metabolic syndrome in the study population.
Overall, the findings in clinical trials strongly support a pathogenic role of increased endocannabinoid activity in obesity and the associated metabolic abnormalities and highlight the unique therapeutic potential of CB1 receptor blockade. Additional benefits may be gained by combination therapies. The efficacy of statins to preferentially lower LDL cholesterol may be effectively complemented by the ability of rimonabant to increase HDL cholesterol. In the case of insulin, the ability of rimonabant to increase insulin sensitivity could reduce the dose requirement for insulin in obese diabetic subjects and could also counteract the tendency of insulin treatment to cause weight gain.