

Last Updated: May 24, 2026
Introduction to Gut-Brain Interactions
The term, gut-brain interrelationships, refers to the processes by which there are back-and-forth interactions between the gastrointestinal system and the central nervous system (the brain). These interactions involve the release of hormones and neurotransmitters that act via the blood and via specialized nerves such as the vagus. These interrelationships constitute a major mechanism for the control of feeding behaviors.
The brain, in particular the hypothalamus, plays highly critical roles in the regulation of energy metabolism, nutrient partitioning, and the control of feeding behaviors. The gastrointestinal tract is intimately connected to the actions of the brain in metabolic and appetite control, in a large part, through interactions with the hypothalamic-pituitary axis. These gut-brain interactions occur via the release of gut peptides that exert responses within the brain as well as through neuroendocrine and sensory inputs from the gut. Although a complete discussion of the interrelationships between the gut and the brain in the control of energy homeostasis and regulation of feeding behavior is beyond the intended scope of this page, focus will be placed on briefly reviewing the current literature.
The primary centers in the brain involved in the control of appetite are the hypothalamic-pituitary axis and the brain stem. The role of these brain regions in appetite control are discussed in the section below on Hypothalamic Control of Feeding Behavior.
The consumption of food initiates a cascade of neuronal and hormonal responses within and by the gastrointestinal system that impact responses in the central nervous system. The brain initiates responses to feeding even before the ingestion of food. The very sight and smell of food stimulates exocrine and endocrine secretions in the gut as well as increasing gut motility. Ingestion of food stimulates mechanoreceptors leading to distension and propulsion to accommodate the food. As the food is propelled through the gut various regions of the intestines secrete numerous hormones that circulate to the brain and impact hypothalamic responses as discussed in the sections below.
The mechanoreceptor responses are transmitted via afferent nerve signals along the vagus nerve to the dorsal vagal complex in the medulla and terminating in the nucleus of the solitary tract (NTS, for the Latin term nucleus tractus solitarii). Projections from the NTS enter the visceral sensory complex of the thalamus which mediates the perception of gastrointestinal fullness and satiety.
Several hormones released from the gut, in response to food intake, exert anorexigenic (appetite suppressing) responses in the brain, particularly in the hypothalamus. These hormones include glucagon-like peptide-1 (GLP-1), cholecystokinin (CCK), peptide tyrosine tyrosine (PYY), pancreatic polypeptide (PP), and oxyntomodulin (OXM or OXY). A single orexigenic (appetite stimulating) hormone, ghrelin, is known to be released by cells of the stomach.
Hypothalamic Control of Feeding Behavior
The hypothalamus is located below the thalamus and just above the brain stem and is composed of several domains (nuclei) that perform a variety of functions. The hypothalamus forms the ventral portion of the region of the brain called the diencephalon. Anatomically the hypothalamus is divided into three broad domains termed the posterior, tuberal, and anterior regions. Each of these three regions is further subdivided into medial and lateral areas.
The various nuclei of the hypothalamus constitute the functional domains of the various hypothalamic areas. The primary nuclei of the hypothalamus that are involved in feeding behaviors and satiety (the sensation of being full) include the arcuate nucleus of the hypothalamus (ARC, also abbreviated ARH), the dorsomedial hypothalamic nucleus (DMH or DMN), and the ventromedial hypothalamic nucleus (VMH or VMN). All three of these nuclei are located in the tuberal medial area of the hypothalamus. The ARC is involved in the control of feeding behavior as well as in the secretion of various pituitary releasing hormones. The DMH is involved in stimulating gastrointestinal activity. The VMH is involved in neural signals that elicit the sensations of satiety.
Early experiments involving lesions in the hypothalamus demonstrated that the lateral hypothalamic area (LHA) is responsible for transmitting orexigenic signals (desire for food intake) and loss of this region results in starvation. The medial hypothalamic nuclei (VMH and to a lesser extent the DMH) are responsible for the sensations of satiety and lesions in these regions of the hypothalamus result in hyperphagia (excessive hunger) and obesity.
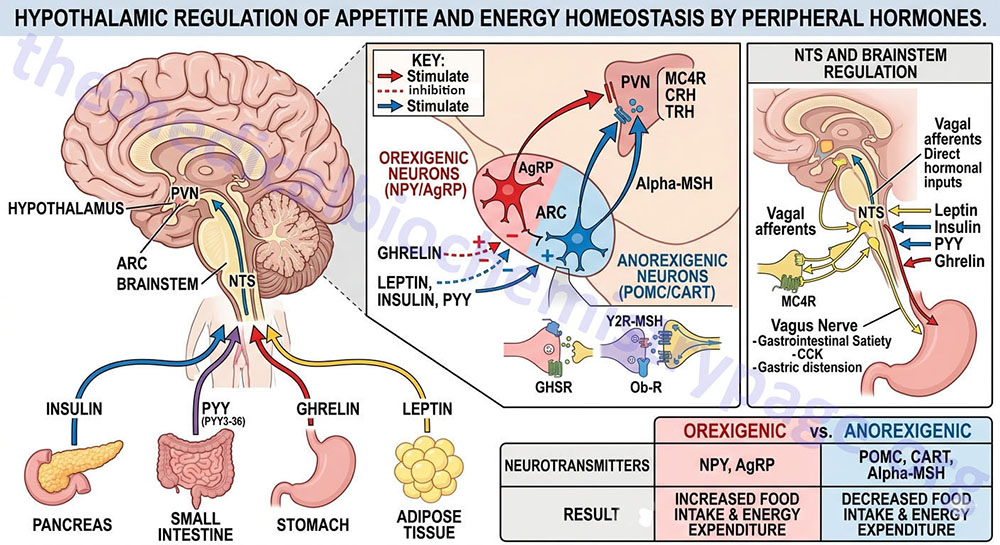
Appetite is a complex process that results from the integration of multiple signals at the hypothalamus. The hypothalamus receives neural signals, hormonal signals, such as leptin, cholecystokinin (CCK), and ghrelin, and nutrient signals such as glucose, free fatty acids, amino acids, and volatile (short-chain) fatty acids. These effects are processed by a specific sequence of neurotransmitters beginning within domains of the ARC that consist of orexigenic neurons expressing neuropeptide Y (NPY) and Agouti-related peptide (AgRP) as well as domains that consist of anorexigenic neurons expressing pro-opiomelanocortin, POMC (yielding the neurotransmitter α-MSH) and cocaine and amphetamine-regulated transcript (CART).
These ARC neurons are referred to as first order neurons and they release their neurotransmitters to act on second order orexigenic neurons (containing either melanin concentrating hormone, MCH or orexin) or anorexigenic neurons (expressing corticotropin releasing hormone, CRH) to alter feed intake. In addition, satiety signals from the liver and gastrointestinal tract signal through the vagus nerve to the nucleus of the solitary tract (NTS, for the Latin term nucleus tractus solitarii) to cause meal termination, and in combination with the hypothalamus, integrate the various signals to determine the feeding response. The activities of these neuronal pathways are also influenced by numerous factors such as nutrients, fasting, and disease to modify appetite and hence to exert impacts on growth and reproduction.
Hypothalamic Orexigenic Peptides
Neuropeptide Y, NPY
NPY is a hypothalamic neuroendocrine protein that is a member of a family of structurally related proteins identified as the pancreatic polypeptide (PP) family of hormones. In addition to NPY this family is composed of two gut hormones, pancreatic polypeptide (PP) and peptide tyrosine-tyrosine (PYY) both of which are discussed below. Each of these peptide hormones contains 36 amino acids consisting of numerous tyrosines (hence the Y peptides nomenclature) and an α-amidation at the C-terminus. The three-dimensional structure of these hormones includes a hairpin-like motif referred to as the pancreatic polypeptide fold (PP-fold). The PP-fold is required for interaction of the hormones with specific G-protein coupled receptors (GPCRs).
The PP family of proteins bind to a family of receptors that were originally characterized as NPY receptors. There are four NPY receptors in humans and they are designated as Y1, Y2, Y4, and Y5. An additional receptor identified as Y6 is found in mice and rabbits. Comparisons of the amino acid sequences of the four human Y receptors show that receptors Y1, Y4 are more closely related to each other than to the receptors Y2 and Y5. Receptors Y1, Y2, and Y5 preferentially bind NPY and PYY, whereas, Y4 exhibits highest affinity for PP. Although the Y5 receptor is expressed and binds ligand it is a truncated protein. The Y2 receptor is involved in anorexigenic responses (suppression of appetite) whereas the Y1 and Y5 receptors have been shown to induce orexigenic responses (stimulation of appetite). The Y2 receptors are thus, referred to as inhibitory receptors with respect to the activity of NPY and they are abundantly expressed on NPY neurons in the arcuate nucleus (ARC) of the hypothalamus.
NPY is expressed throughout the mammalian brain with highest levels found in the ARC of the hypothalamus. NPY is one of the most potent orexigenic factors produced by the human body. Within the ARC there are two neuronal populations that exert opposing actions on the desire for food intake. Neurons that co-express NPY and another neuropeptide called agouti-related peptide (AgRP) stimulate food intake, whereas, neurons that co-express POMC and cocaine and amphetamine-regulated transcript (CART) suppress the desire for food intake. The role of NPY in appetite control can be demonstrated by central administration of NPY which results in a markedly increased desire for food intake. The Y1 and Y5 receptors mediate the bulk of the effects of NPY on the hypothalamic-pituitary-thyroid axis. Within the ventromedial nucleus (VMN) of the hypothalamus, binding of NPY to the Y1 receptor results in inhibition of neuronal function (via hyperpolarization) which results in interference with the satiety role of the VMN. The majority of hypothalamic Y2 receptors are found on NPY-containing neurons. Conversely, Y2 receptor activation in the ARC results in inhibition of the actions of NPY which accounts for the anorexigenic actions associated with Y2 activation.
Of significance to dieting and weight loss is the fact that when people lose excess weight the level of NPY increases which likely is a contributing factor to the inability of most people to keep the weight off. This phenomenon has been demonstrated in mice fed a high-fat diet. These mice will become obese, have increased fat mass, and increased circulating levels of leptin. When the animals are placed on a calorie-restricted diet they lose the excess fat and leptin levels decline. However, the level of expression of the NPY gene is observed to be significantly increased. This and other data indicate that NPY is also one of the most important hypothalamic-derived neuropeptides mediating the effects of leptin on overall energy homeostasis. Whereas, losing excess weight is associated with increased expression of NPY, the levels of the anorexigenic peptides, POMC and CART, do not change.
Agouti-related peptide, AgRP
As the name implies, agouti-related peptide (AgRP), is a protein with sequence homology to the agouti protein which controls coat color in rodents. AgRP is a protein of 132 amino acids encoded on chromosome 16q22. AgRP is expressed primarily in the ARC and is found to co-localize with neurons that also produce NPY. Although expression of AgRP is restricted to the ARC, AgRP fibers project to several brain areas as well as to multiple areas within the hypothalamus, including the paraventricular nucleus (PVN or PVH) and perifornical lateral hypothalamus (PFLH). In addition, all of these AgRP nerve terminals contain NPY. The PVN is a region of the hypothalamus that integrates neuropeptide signals from numerous regions of the brain and hypothalamus (e.g. the ARC) as well as the brainstem. The perifornical lateral hypothalamus is a subdomain of the LHA that is involved in arousal and food-seeking behaviors.
AgRP together with NPY represent a distinct set of ARC-expressed orexigenic peptides. AgRP is classically referred to as a member of the central melanocortin system, which in addition to AgRP comprises α-melanocyte stimulating hormone, α-MSH (see below for description of α-MSH actions) and two melanocortin receptors identified as melanocortin receptor-3 (MC3R) and melanocortin receptor-4 (MC4R). Whereas, α-MSH is an agonist of both MC3R and MC4R, AgRP serves to antagonize the actions of α-MSH at these same receptors with highest antagonist activity on MC4R. In addition to antagonizing the effect of α-MSH at the MC3R and MC4R, AgRP suppresses the basal activity of the MC4R, thus defining AgRP as an inverse agonist.
The close functional relationship between AgRP and NPY is demonstrated by the fact that the expression of these two peptides is similarly modulated under identical physiological conditions such as negative energy balance or increased energy demand that occurs during food deprivation. During periods of fasting both AgRP and NPY levels rise and evidence indicates that this is due primarily to a drop in the level of the peripheral hormones leptin and insulin and a rise in ghrelin. Also, AgRP like NPY shows a strong circadian rhythm in its expression, rising at the onset of natural feeding cycles. As to be expected, the expression of both AgRP and NPY are conversely suppressed under conditions of positive energy balance. In studies in experimental animals manipulations in diet also result in alterations in the levels of AgRP expression. AgRP gene expression is higher in rats on a low-energy diet compared to a fat-rich diet or in conditions where glucose utilization is reduced. In fact both AgRP and NPY are suppressed by a single injection of glucose. On the other hand, injections of the compound Intralipid (which increases circulating lipids) does not result in changes in AgRP levels.
The strong orexigenic effects of AgRP can be demonstrated by injecting the peptide into the brain of experimental animals. Central injection of AgRP has a potent stimulatory effect on food intake which can also be seen using a MC4R antagonist. These results confirm the function of AgRP as an antagonist of α-MSH. The food intake stimulation exerted by injection of AgRP is similar to that seen by central injection of NPY with differences being that the duration of the effect with AgRP is much longer than that exerted by NPY. However, the long-term effect does not involve the MC4R, which indicates that AgRP likely induces long-term changes to the neural signaling pathways downstream of this receptor. Chronic administration of AgRP results in increased daily food intake while simultaneously decreasing oxygen consumption and the capacity of brown adipose tissue to expend energy. These chronic effects result in increased fat mass accumulation all of which are effects similar to those seen with chronic administration of NPY.
In complimentary studies using transgenic mice that overexpress the AgRP gene it has been shown that increased levels of AgRP result in increased feeding behavior and food intake. These mice exhibit hyperphagia and obesity, in addition to increased body length, hyperinsulinemia, late-onset hyperglycemia, and pancreatic-islet hyperplasia. These results are similar to those seen in mice that ectopically express the NPY gene and are also evident in mice that harbor a MC4R knock-out.
There exists an antagonism between the actions of AgRP (and NPY) and the melanocortins in controlling eating and body weight. This is evident from studies showing changes in endogenous AgRP that are opposite to those seen with the melanocortin peptides. In addition, brain mapping shows that AgRP neurons interact with POMC neurons in the ARC through the inhibitory neurotransmitter γ-amino butyric acid (GABA). Both AgRP and NPY axons that co-localize GABA project onto POMC expressing cells in the ARC and the AgRP-stimulated release of GABA results in inhibition of the activity of the POMC neurons, an effect also seen with NPY. Peripheral hormones that act on the ARC and thereby affect the actions of AgRP and NPY are leptin and ghrelin. Leptin binds to its receptor present on AGRP and NPY neurons and inhibits their firing resulting in reduced GABA release onto POMC neurons. This leptin-induced reduction in GABA action at POMC neurons is a disinhibition and is, in part, the mechanism by which leptin decreases feeding behaviors. Conversely, ghrelin binding its receptor activates AgRP and NPY neurons, resulting in an increase in GABA release with resultant inhibition of POMC neurons.
Given that AgRP and NPY, which are activated under similar conditions and have comparable effects, it is indicative that they very likely evolved to ensure the signaling of hunger during food scarcity and to enable the body to endure long periods of negative energy balance.
Melanin-Concentrating Hormone, MCH
Melanin-concentrating hormone (MCH) was originally identified as a 19 amino acid cyclic peptide that induced the lightening of the skin in fish. Subsequently the peptide was identified in rodents to be overexpressed in response to fasting and also elevated in genetically obese mice (ob/ob mice). In humans and other mammals MCH is expressed exclusively in the lateral hypothalamus and zona incerta (region of gray matter cells in the subthalamus below the thalamus). In humans there are two G-protein coupled receptors (GPCRs) that bind MCH identified as MCH1R and MCH2R. Rodents, however, do not possess the MCH2R gene. MCH1R is a typical GPCR that couples to the activation of both Gq and Gi type G-proteins. The Gq class of G-protein activates phospholipase C-β (PLCβ) resulting in increases in intracellular Ca2+. The Gi class represses adenylate cyclase activity resulting in decreased cAMP production. Expression of MCH1R is seen throughout the brain with highest levels in the cortex, hippocampus, amygdala, and nucleus accumbens. MCH1R expression is also seen in peripheral tissues such as adipose tissue, intestines, lymphocytes, and the pituitary. The pattern of central nervous system expression of MCH and MCH1R suggests that this peptide-receptor system is involved in a host of physiological functions within the brain.
Involvement of MCH in the regulation of feeding behaviors and energy homeostasis has been shown in mice where either the MCH gene or the MCH1R gene have been knocked out. Additional information on the function of MCH in feeding and energy consumption has been obtained with the use of selective pharmacological MCH1R antagonists. In mice lacking the MCH gene the prominent phenotype is hypophagia (reduced desire for food intake) and lean body mass. These results indicate that MCH is an important orexigenic (appetite stimulating) hormone. In contrast, central administration of MCH results in increased food intake in mice. When the MCH1R gene is knocked out mice are hyperphagic, hyperactive, and lean. When MCH1R antagonists are administered peripherally animals exhibit decreased MCH-induced food intake. In addition, if MCH1R antagonists are chronically administered there is observed a decrease in body weight in rats that had diet-induced obesity. MCH1R antagonists have been shown to modulate leptin secretion and insulin release which suggests that the weight loss associated with systemic antagonist administration is due to both central and peripheral effects. Many studies indicate that MCH1R antagonists modulate energy homeostasis and thus, the anti-obesity effects are due primarily to increased energy expenditure and not to suppression of feeding behaviors.
In addition to modulation of feeding behaviors and energy expenditure, the MCH system has been shown to be involved in affective disorders such as anxiety and depression. Mice in which the MCH1R gene has been knocked out exhibit less anxiety-like behaviors than wild-type mice. Administration of MCH1R antagonists have also been shown to have anxiolytic (reducing anxiety) properties. These studies indicate that the MCH system is important in the modulation of stress responses. MCH1R antagonists have also been shown to be efficacious in animal models of depressive behavior although the precise mechanism for the antidepressant effects are not yet clearly defined.
The Orexins
The orexins constitute two neuroendocrine peptides derived from the same gene. These peptides are designated orexin A and orexin B. The orexins are also called the hypocretins as the peptides were independently isolated. One group used a subtractive cDNA screening approach to enrich for cDNAs from the hypothalamus and as such identified the peptides as hypocretins. Another group was screening for ligands that activated orphan G-protein coupled receptors (GPCRs) that would induce transient calcium currents in cells expressing the orphan GPCRs. The latter group demonstrated that the identified ligands induced orexigenic (appetite stimulating) responses and thus named the peptides orexin A and orexin B. Orexin A corresponds to hypocretin 1 (HCRT-1) and orexin B corresponds to hypocretin 2 (HCRT-2).
Isolation of the human orexin gene demonstrated that the encoded protein was a preproprotein that contained the amino acid sequences of both orexin A and orexin B. The orexin gene (HCRT: hypocretin neuropeptide precursor) is located on chromosome 17q21.1 and is composed of 2 exons that encode a 131 amino acid precursor protein. The preproprotein contains a 32 amino acid leader sequence typical of secreted proteins. Within the context of the 131 amino acid precursor the 33 amino acid orexin A peptide encoded by amino acids 33–65 and the 28 amino acid orexin B peptide encoded by amino acids 69–96. Both orexin A and orexin B peptides are C-terminally amidated. The N-terminal glutamine residue of orexin A is cyclized into a pyroglutamyl residue and the peptide contains two intrachain disulfide bonds. The amino acid sequences of vertebrate orexin A peptides are 100% conserved and those of vertebrate orexin B peptides are 93% conserved. Overall the full-length preproproteins are 75% conserved across various vertebrate species.
Structurally, both orexin A and orexin B are highly conserved and this structural conservation explains the ability of the orexin receptors to bind both peptides. There are two orexin receptors identified as OX1R and OX2R (also identified as HCRTR1 and HCRTR2, respectively). OX1R exhibits an order of magnitude higher affinity for orexin A compared to orexin B whereas OX2R has been shown to bind both peptides with equal affinity. The orexin receptors are typical GPCRs with OX1R coupling to the Gq subclass of G-proteins and OX2R coupling to both the Gq and Gi class of G-protein. The Gq class of G-protein activates phospholipase C-β (PLCβ) resulting in increases in intracellular Ca2+. The Gi class represses adenylate cyclase activity resulting in decreased cAMP production. The OX1R protein is encoded by the HCRTR1 (hypocretin receptor 1) gene which is located on chromosome 1p35.2 and is composed of 9 exons that encode a 425 amino acid protein. The OX2R protein is encoded by the HCRTR2 (hypocretin receptor 2) gene which is located on chromosome 6p12.1 and is composed of 9 exons that encode a 444 amino acid protein.
The cell bodies of orexin expressing neurons are found in the lateral and posterior hypothalamic areas with axonal projections throughout the brain. Expression of the orexin receptors is also widely distributed throughout the central nervous system. This distribution of orexin and orexin receptor expression is suggestive of important roles in the emotional and motivational aspects of feeding behavior. Indeed, as their name implies, injection of orexin peptides into the brain was shown to increase food consumption in rats. However, in addition to increased feeding behavior central administration of orexins increases wakefulness and suppresses REM sleep. These latter observations demonstrate that orexins play a causative role in the regulation of sleep-wake cycles. Subsequent research demonstrated that loss of orexin function results in a condition in animals that mimics the sleep disorder in humans known as narcolepsy.
When the orexin gene is knocked out in mice they exhibit increased REM sleep during dark periods when they are normally awake. In addition these mice have frequent episodes of sudden collapse, during dark cycles, that resembles cataplexy in humans. Cataplexy often accompanies narcolepsy in humans. In human narcolepsy patients there is a significant reduction in the amount of detectable orexin A and orexin B as well as an 80%–100% reduction in the number of neurons that contain detectable prepro-orexin mRNA. However, no mutation has been found in either the prepro-orexin or orexin receptor genes except for one rare early onset severe case where there was a mutation in the signal peptide of prepro-orexin that impaired protein trafficking and processing. There is an association between certain HLA alleles and narcolepsy that suggests the reduced orexin levels may result from selective autoimmune destruction of orexin neurons. Dogs that harbor a null mutation in the OX2R gene exhibit a narcolepsy phenotype that is highly similar to human narcolepsy.
Galanin, GAL
Galanin (GAL) is a 29 amino acid peptide whose name is derived from the fact that it contains an N-terminal glycine residue and a C-terminal alanine. GAL is expressed in the gut and the brain with wide distribution throughout the hypothalamus including the PVN, the PFLH, and ARC. The expression of GAL in the hypothalamus is directly correlated to its role in energy homeostasis and the control of feeding behaviors. In addition to regulating feeding, GAL serves as a growth and prolactin-releasing factor to the lactotroph, especially in states of high estrogen exposure, is involved in learning and memory through effects in the hippocampus, and is involved in pain and seizures. Additionally, GAL exerts affective responses such as mood disorders and anxiety.
GAL exerts these myriad effects via binding to three distinct G protein-coupled receptors (GPCRs) identified as GALR1, GALR2, and GALR3. The highest levels of expression of GALR1 are seen in hypothalamic nuclei that includes the PVN and supraoptic nuclei; GALR2 in the ARC, DMH, and PVN; and GALR3 in the PVN, VMH, and DMH. When GAL is injected into the third ventricle of the brain or into the PVN it elicits a strong orexigenic (increased feeding) response with a preference for fat over protein and carbohydrates. The feeding behavior responses to GAL exposure are primarily due to binding GALR1 in the hypothalamus.
Although GAL is an orexigenic peptide it has marked differences in its responses to food deprivation and intake and the signals it induces compared to those of NPY and AgRP in terms of their responsiveness to endocrine and physiological signals. Injection of GAL results in a stimulatory effect on feeding behavior yet the response is smaller and of shorter duration than that induced by NPY.
The feeding response elicited by GAL has little impact on food preference, whether for carbohydrate or fat, whereas the responses to NPY result in a preference for carbohydrates. In addition, GAL-induced feeding is greatly attenuated when fat is removed from the diet. The primary function of GAL when an animal is consuming a high-fat diet is to restore carbohydrate balance, through behavioral and metabolic actions, under conditions where carbohydrate intake and metabolism are suppressed.
The adrenal steroid, corticosterone, only transiently inhibits GAL gene expression in PVN neurons and has no effect on feeding responses of GAL. In contrast this steroid hormone has a potent stimulatory effect on NPY and AgRP and on NPY-induced feeding.
Although insulin suppresses both GAL and NPY expression, leptin strongly inhibits NPY gene expression and release, yet produces little or no change in basal GAL expression in the ARC and only a small suppression of GAL expression in the PVN. The differential responsiveness of GAL neurons to leptin is likely due to the low concentration of leptin receptors on GAL neurons. The low leptin receptor level on GAL neurons compared to NPY neurons also explains their different responses to food restriction, which reduces leptin levels.
Food restriction does little to the level of GAL expression while markedly enhancing NPY gene expression. Difference in GAL and NPY expression are also found under conditions of altered nutrient metabolism. Administration of inhibitors of fatty acid oxidation suppress GAL expression but have no effect on NPY. Conversely, administration of inhibitors of glucose oxidation do not alter GAL expression but result in elevated expression of NPY. These nutrient differences are also observed in experimental animals fed different diets.
GAL expression in the PVN is stimulated by a high-fat diet whereas, NPY expression in the ARC is unaffected or reduced by fat consumption. These differences in response to fat-rich diets are also seen with fat accumulation and are likely the result of differences in responses to leptin. The ability of GAL to exert its stimulation of feeding responses may be due to its interactions with other peptide systems.
The opioids are believed to have some role in mediating GAL-induced feeding, since the opioid receptor agonist naloxone attenuates the GAL feeding response. GAL may also induce feeding via an inhibition of the anorexigenic melanocortin system (POMC; see below). This is suggested by evidence that POMC neurons in the ARC are innervated by GAL expressing neurons as well as expressing the GALR1 gene. Evidence indicates that GAL has a direct inhibitory action on ARC neurons expressing GALR1, as well as modulating the secretory activity of POMC neurons.
Hypothalamic Anorexigenic Peptides
POMC-Derived Melanocortins
The POMC-derived melanocortin peptides include α-MSH, β-MSH, γ-MSH, ACTH1-24, and ACTH1-13–NH2 (desacetyl-α-MSH). The POMC-derived melanocortins belong to a family of peptides referred to as the melanocortin system. This system includes the POMC-derived melanocortins which exhibit agonist activities, the antagonist peptide AgRP, the melanocortin receptors (MCR), and the melanocortin receptor accessory proteins (MRAPs). The MCR family of receptors consists of five identified members termed MC1R through MC5R.
The melanocortin system has been shown to be critical in the regulation of food intake and energy expenditure via a number of different assay systems involving both humans and animals. It is important to note that although β-MSH and γ-MSH are found to be produced in human brain they are not found in rodent brain or pituitary. However, when administered into the brain of animals the melanocortins α-MSH, β-MSH, and ACTH1-24 inhibit the intake of food. The actions of MSH peptides in feeding behavior is exerted primarily via peptide binding to the MC4R and to a lesser extent to the MC3R.
Genetic mutations in humans as well as in animals that disrupt the expression and processing (this includes the proteases that process the POMC precursor) of POMC peptides and MCRs are associated with changes in energy balance and can lead to obesity and type 2 diabetes. In humans there have been mutations identified in prohormone convertase 1/3 (PC1/3) and carboxypeptidase E (CPE), as well as in the α-MSH degrading enzyme prolylcarboxypeptidase (PRCP) that are associated with energy imbalance and a propensity for obesity. In mice a mutation in the CPE gene also results in the development of late-onset obesity. In genome-wide screens for gene polymorphisms associated with an increased risk of developing type 2 diabetes the MC4R gene was identified. Mutations in the MC4R gene are the most frequent causes of severe obesity in humans.
In mice ablation of the POMC gene (POMC-null) results in viable animals, even though they have minimal adrenal tissue and undetectable glucocorticoids due to the lack of ACTH. These animals develop obesity but do not become diabetic. Although not diabetic these mice do have a disruption in the regulation of glucagon secretion in response to experimental hypoglycemia. This indicates that POMC-derived peptides are involved in the regulation of glucagon. In humans a rare POMC-null mutation has been described. Unlike the situation in the POMC-null mice, humans with a lack of POMC expression are unable to survive without glucocorticoid supplementation from birth. Individuals that survive have red hair, dramatically increased desire for food intake, and high propensity for obesity. These conditions are the same as those seen in POMC-null mice. Another POMC mutation that has been identified in humans resulting in obesity is a point mutation in the cleavage site between β-MSH and β-endorphin. The consequences of this mutation suggests that β-MSH may be a significant endogenous anorectic agonist that activates the MC4R.
Cocaine- and Amphetamine-Regulated Transcript, CART
The cocaine- and amphetamine-regulated transcript (CART) peptides are neuroendocrine peptides involved in feeding behavior, drug reward systems, stress, cardiovascular functions, and bone remodeling. Expression of the CART gene is essentially confined to hypothalamic neuroendocrine neurons and limbic system circuits involved in reward processes. CART peptides are found areas of the brain involved in the control of feeding behaviors including regions of the hypothalamus such as the VMN, lateral hypothalamus (LH), PVN, NTS, ARC, and the nucleus accumbens.
The human CART protein is encoded by the CARTPT (CART prepropeptide) gene. The CARTPT gene is located on chromosome 5q13.2 and is composed of three exons. The gene is transcribed into two alternatively spliced mRNAs that encode proCART peptides of different lengths identified as proCART1-89 and proCART1-102. However, only the proCART1-89 peptide is found in humans. The proCART protein contains a signal sequence typical of secreted proteins and is required for insertion of the protein into vesicles and subsequent processing.
The active portions of the CART peptides are located downstream of the alternatively spliced region and thus both proCART1-89 and proCART1-102 encode the same biologically active hormones. The proCART proteins contain several sites for post-translational processing by prohormone convertases. In humans, where only the short form proCART1-89 peptide is present, the processing yields CART peptides identified as CART 42-89 and CART 49-89. In rodents where both CART mRNA transcripts yield proCART proteins the nomenclature of the processed peptide reflects the amino acid numbering of the longer 102 amino acid pro-protein and are identified as CART 55-102 and CART 62-102. There is a high degree of interspecies conservation of the active CART peptide sequences with the human rat homology being 91%.
A definitive CART receptor has as yet not been isolated. However, several lines of evidence indicate that CART peptides bind with high affinity and specificity to a cell surface protein(s) that triggers signaling events typical of a GPCR class receptor. In cell culture assays addition of CART peptides results in phosphorylation of extracellular signal-regulated kinase (ERK) as well as the transcription factor, cyclic AMP response element binding protein (CREB). These results indicate that the CART receptor is a GPCR that activates the Gi/o class of G-proteins.
A role for CART peptides in the regulation of feeding behaviors was demonstrated in animal models where intracerebrovascular (icv) injection results in decreased food intake. These types of results indicate that CART peptides are anorexigenic (decrease appetite). Within the ARC of the hypothalamus CART-peptide containing neurons are surrounded by NPY expressing nerve terminals.
The distribution of CART and NPY in the ARC suggests that these two neuropeptides may exhibit cross-talk in the regulation of feeding since NPY is an orexigenic (appetite stimulating) hormone and CART is an anorexigenic hormone. When animals are food-deprived the level of CART mRNA in the ARC decreases. Conversely, when leptin is administered to animals the level of CART mRNA in the ARC increases. In addition, in animals with disrupted leptin signaling the level of CART mRNA is nearly undetectable in the ARC.
The functions of CART peptides in inhibiting the desire for food intake may involve circuits that include the serotonin-4 receptor and MDMA receptors. MDMA is 3,4-methylenedioxy-N-methyl-amphetamine more commonly known as “ecstasy”. When CART mRNA levels are experimentally reduced the anorectic effects of MDMA as well as serotonin-4 receptor activation are abolished. In addition, mice in which the CARTPT gene has been knocked out exhibit increased desire for feeding and gain weight.
Several human studies have also indicated that CART peptides function in appetite control. In an Italian family where several members are obese it was found that a missense mutation was present in their CART gene. This mutation changed a Leu at position 34 to a Phe (L34F) and resulted in deficiency of CART peptide in the blood. If this mutant human gene is expressed in a mouse pituitary tumor cell line the expressed protein is poorly processed and secreted. In another study of morbidly obese individuals in France, a single nucleotide polymorphism (SNP) was identified in the CART gene at position –3608 where a T was replaced with a C. In a study examining the promoter region of the CART gene from several hundred individuals it was found that a polymorphism resides approximately 156kb upstream that may be associated with obesity.
Galanin-like peptide, GALP
Galanin-like peptide (GALP) is a 60 amino acid peptide that is structurally related to galanin, hence the derivation of its name. Amino acids 9–21 of GALP are identical to the first 13 amino acids of GAL. The structural and sequence similarities between GALP and GAL explain the fact that GALP functions by binding with high affinity to GAL receptors. However, there are differences in affinities of the two peptides for the different GAL receptors. GAL binds all three receptor subtypes (GALR1, GALR2, and GALR3) with similar affinities whereas, GALP binds with highest affinity to GALR3 followed by GALR2 with GALR1 binding with least affinity. Expression of GALP is almost exclusively found in the hypothalamic ARC, and GALP neurons project to the PVN but not the lateral hypothalamus.
Central injection studies revealed the anorexigenic effects of GALP as well as its responsiveness to the effects of leptin. However, it should be noted that there are differences in the responses to GALP injection when comparing rats and mice. Injection of GALP into mice results in the inhibition of feeding responses and also leads to an increase in energy expenditure and fat oxidation in brown adipose tissue resulting in a hyperthermic effect. In contrast, injection of GALP into rat brains results in an orexigenic response.
The leptin receptor is expressed on most GALP neurons and expression of GALP in the ARC is induced by leptin. In contrast, GALP expression in the ARC is significantly reduced in leptin-deficient (ob/ob) and leptin receptor-deficient (db/db) mice. Food deprivation results in reduced circulating levels of leptin and this in turn reduces the rapid entry of circulating GALP into the brain. Fasting results in a decrease in both the level of GALP mRNA as well as the number of GALP expressing neurons. Leptin administration will restore GALP expression in fasted animals as well as in leptin-deficient (ob/ob) mice.
Corticotropin-releasing factor (CRF) and related peptides
Corticotropin-releasing factor (CRF, also known as corticotropin-releasing hormone, CRH) belongs to an interacting family of proteins that includes CRF, at least two different CRF receptor subtypes (CRF1 and CRF2), a CRF-binding protein (CRF-BP) and the urocortins which are endogenous CRF receptor ligands. There are three known urocortins identified as urocortin 1, 2, and 3 (Ucn1, Ucn2, Ucn3). The urocortins are also identified by Roman numeral designations urocortin I, II, and III. CRF is a 41 amino acid peptide that is found widely expressed in the brain. CRF-expressing neurons are abundant in the hypothalamic PVN where they control the pituitary-adrenal axis regulating the release of ACTH and glucocorticoids.
Ucn1 is a 40 amino acid peptide that is expressed primarily in the lateral hypothalamus and supraoptic nucleus, with urocortin-containing neurons projecting to the VMH. Ucn2 is a 43 amino acid peptide and is expressed in the mouse hypothalamus in the PVN and ARC. In humans, increased Ucn2 expression is also seen in cardiac myocytes during heart failure. Ucn3 is a 38 amino acid peptide whose expression is found in the rostral perifornical area lateral to the PVN with Ucn3 neurons projecting throughout the hypothalamus and the limbic system. Ucn3 expression is also high in pancreatic β-cells where it stimulates insulin as well as glucagon secretion.
CRF and the urocortins function through two G protein-coupled receptors, CRF1 and CRF2. CRF and Ucn1 bind with high affinity to the CRF1. In contrast, Ucn2 and Ucn3 bind with much higher affinity than CRF to CRF2 and are therefore, are likely to be the endogenous ligands of this receptor. In addition to binding to two receptors, CRF and urocortins also bind to CRF-BP, which is expressed in association with CRF-expressing neurons in many brain areas including the hypothalamus. CRF-BP acts as an inhibitor of both CRF and the urocortins, thereby, modulating the biological actions of these peptides.
Hypothalamic expression of CRF is negatively regulated by the circulating level of corticosterone such that CRF mRNA and protein levels are highest when corticosterone levels are declining. In rodents, who feed during the dark cycle, corticosterone levels rise at the onset of feeding and CRF levels decrease. Glucocorticoids also negatively regulate CRF expression. This effect of glucocorticoids on CRF expression supports a permissive role for the adrenal steroids in promoting body fat accrual. Diabetes leads to increased CRF expression in the PVN and this effect is can be further enhanced by the administration of insulin. Expression of CRF is also stimulated in states of positive energy balance and is reduced in states of negative energy balance, such as food deprivation. Circulating nutrients also affect the level of CRF expression. When glucose levels rise, CRF levels decline with the opposite occurring when glucose levels fall. In contrast to the changes in CRF levels in response to serum glucose changes, excess fat consumption does not appear to alter CRF expression.
Central administration of CRF results in suppression of spontaneous feeding responses demonstrating its anorexigenic properties. The CRF-mediated suppression of feeding occurs along with a stimulation sympathetic nervous system activity and resting oxygen consumption which results in increased fat mobilization and oxidation and raises blood glucose while inhibiting insulin secretion. Central administration of Ucn1 also suppresses feeding, and its effect is strongest in the PVN and more potent and longer-lasting than that of CRF. Ucn2 administration also suppresses food intake as well as resulting in delayed gastric emptying, and decreased heat-induced edema. Chronic administration of CRF, but not Ucn1, increases brown adipose tissue mass and raises circulating levels of corticosterone and lipids while reducing the levels of glucose. Experimental evidence indicates that CRF2 mediates the anorectic effects of these ligands, while CRF1 mediates their metabolic effects. Mice lacking the CRF2 receptor exhibit a blunted response to the feeding-inhibitory effects of urocortin and selective CRF2 receptor antagonists block the suppressive effects of urocortins and CRF on food intake and body weight.
The role of CRF as an anorexigenic hormone may involve the NPY, melanocortin and CART systems, acting in a downstream fashion. The CRF neurons in the hypothalamus co-localize with both the NPY Y5 receptor and the MC4R. In addition, CRF expression in the PVN is stimulated by central administration of a melanocortin agonist but is inhibited by an MC4R antagonist. There is an antagonistic relationship between CRF and NPY demonstrated by the fact that administration of CRF and Ucn1 result in a reduction in both NPY expression and NPY-mediated feeding. Also, administration of CRF antagonists result in increased NPY-induced feeding responses. In contrast to the CRF-NPY antagonism, central administration of CART activates CRF neurons in the PVN, indicating that CRF may mediate the anorectic effect of CART. Leptin is also involved in the effects of CRF and the urocortins as demonstrated by the fact that the anorexigenic actions of leptin are attenuated in the presence of CRF antagonists. Leptin also facilitates the uptake of Ucn1 into the brain, thereby potentiating its anorexigenic actions.
Hypothalamic Lipid Metabolism and Energy Homeostasis
Hypothalamic Fatty Acid Metabolism
Within the central nervous system the metabolism of fatty acids is primarily for the purposes of membrane function and the central regulation of energy metabolism. Fats do not serve as a major source of energy within the brain, this need is obtained from the oxidation of glucose and ketone bodies. Although ketone bodies are essentially derived from fatty acids (via the acetyl-CoA derived from β-oxidation), these compounds arrive in the brain from the blood being produced within the liver. Most of the fatty acids within the brain are synthesized de novo within the various cells of the CNS. While many saturated and unsaturated fatty acids do not generally cross the blood-brain barrier (BBB), polyunsaturated fatty acids, such as linolenic acid, are transported across the BBB. Most of the fatty acids delivered to the brain are released from lipoprotein-associated triglycerides via the action of endothelial cell lipoprotein lipase or the lipoproteins are endocytosed by the endothelial cells and the fatty acids released by lysosomal hydrolases. Albumin-bound fatty acids gain access into the brain by both passive diffusion and protein carrier-mediated transport via FAT/CD36 and FATP1. For information on fatty acid transport into cells visit the Lipolysis and Fatty Acid Oxidation page.
Once fatty acids are taken up by cells in the brain they are activated through the action the various fatty acyl-CoA synthetases. Although fatty acyl-CoAs are substrates for β-oxidation, the rates of mitochondrial fat oxidation are extremely low in the brain. The fates of fatty acyl-CoAs, in the brain, are conversion into various structural entities including phospholipids, triglycerides, diacylglycerides, and fatty acyl-carnitine species as well as lipid-derived signaling molecules. Studies using palmitic acid have demonstrated that the majority of palmitate is incorporated into the neutral lipid fraction, principally consisting of triglycerides and non-esterified free fatty acids. In addition, in contrast to non-neural tissues, the desaturation and elongation of palmitate within the brain represents a very minor fate of the fat. Instead, palmitate is converted into fatty acids of less than 16 carbon atoms. The largest pool of lipid derived from palmitate is phospholipid of which the primary species is phosphatidylcholine.
Although β-oxidation of fatty acids represents a minor, if at all, source of the ATP pool in neurons, it is an important metabolic pathway determining the ultimate fate of fatty acids that enter the brain. As a result of fatty acid oxidation, a number of aqueous byproducts are detected in the brain such as fatty acyl-CoAs, fatty acyl-carnitines, ketone bodies and various amino acids. When using in vitro assays with palmitate it is found that the majority of the carbons from this fatty acid end up in the amino acids glutamate, glutamine, aspartate, asparagine, and GABA. In addition, numerous organic acids including citrate, malate, β-hydroxybutyrate, and acetyl CoA result from palmitate oxidation. By far, citrate represents the largest byproduct of fatty acid oxidation in the brain. These fates of fatty acids that enter the brain have been worked out with the use of a selective carnitine palmitoyltransferase 1 (CPT-1) inhibitor.
As discussed in the Lipolysis and Fatty Acid Oxidation page, regulation of the rate of fatty acid entry into the mitochondria for oxidation is exerted by malonyl-CoA-mediated inhibition of CPT-1. Indeed, inhibition of mitochondrial CPT-1 is emerging as a viable target for the central regulation of feeding behaviors and energy homeostasis. Inhibition of brain CPT-1 decreases the entry of fatty acid carbons into the aqueous metabolic pools but does not alter the distribution into various long-chain acyl-CoA compounds and triglycerides.
Fatty acids, specifically long-chain fatty acids via the formation of long-chain fatty acyl-CoAs, have very recently been shown to exert anorexigenic effects via the hypothalamus. For example, when oleic acid is injected into cerebral ventricles there is a resultant decrease in feeding behavior in laboratory animals. The decreases in feeding behavior are associated with declines in the expression and release of NPY and AgRP in the hypothalamus. In addition to reducing feeding behaviors, central administration of oleic acid is associated with increased peripheral glucose homeostasis. Oleic acid must be converted to its CoA derivative for these effects to be exerted since it has been shown that blockade of fatty acyl-CoA synthetase activity abolishes the oleic acid-regulated glucose homeostasis. Mitochondrial CPT-1 activity has also been shown to play a role in the central effects of fatty acids. For example the 8-carbon fatty acid, octanoic acid, does not exert any anorexigenic effects and mitochondrial uptake of octanoyl-CoA does not require CPT-1. Additionally, inhibition of hypothalamic CPT-1 leads to an increase in cytosolic long-chain acyl-CoA content and results in anorexigenic effects.
Humans express three different CPT-1 genes identified as CPT-1A, CPT-1B, and CPT-1C. The CPT-1B gene is not expressed in the hypothalamus and the level of hypothalamic CPT-1A expression is minimal. In addition, the binding affinity of CPT-1A for malonyl-CoA is relatively low. The CPT-1C gene is brain specific and binding of malonyl-CoA to the CPT-1C protein is of similar affinity to that of the CPT-1B isoform. In experiments in mice it has been shown that deletion of CPT-1C results in reduced feeding behavior and lowered body weight similar to studies in which hypothalamic malonyl-CoA levels are elevated. These results point to the fact that hypothalamic malonyl-CoA levels likely play a major role in the control of feeding behaviors.
However, the precise mechanisms by which malonyl-CoA and cytosolic long-chain fatty acyl-CoA interactions result in reduced appetite are not fully characterized. In the CPT-1C knock-out mice experiments, although the animals exhibited reduced feeding behavior they became obese more quickly on a high-fat diet than control animals even though they ate less. Also, the brain specific CPT-1C isoform does not catalyze carnitine transferase activity. Therefore, precisely how the inhibition of hypothalamic CPT-1C leads to long-chain acyl-CoA accumulation remains unclear.
Hypothalamic Malonyl-CoA Regulation
As indicated above the brain does not readily oxidize fatty acids for energy production. However, the major enzymes of the fatty acid biosynthetic pathway, such as acetyl-CoA carboxylase (ACC) and fatty acid synthase (FAS), are expressed in neuronal regions of the hypothalamus that are involved in energy homeostasis. Various hormones and metabolic fuels affect hypothalamic malonyl-CoA levels and malonyl-CoA is considered a key satiety inducing signal in the brain. During periods of fasting, hypothalamic levels of malonyl-CoA rapidly decrease and act as a signal of hunger. Conversely, during feeding, hypothalamic levels of malonyl-CoA rapidly rise and act as a signal to stop eating. Therefore, an understanding of the regulation of hypothalamic malonyl-CoA levels is of significance especially with respect to efforts to control feeding behaviors in humans.
As described in the Synthesis of Fatty Acids page, the production of malonyl-CoA is catalyzed by ACC. There are two isoforms of ACC, ACC1 and ACC2 (sometimes referred to as the ACCα and ACCβ isoforms, respectively). Expression of ACC1 predominates in the liver, whereas, expression of ACC2 predominates in tissues with a high oxidative capacity. These differences in expression have led to the suggestion that the malonyl-CoA produced by ACC2 is preferentially involved in the regulation of fatty acid oxidation, whereas, malonyl-CoA produced by ACC1 is preferentially involved in the regulation of fatty acid synthesis.
Both isoforms of ACC are allosterically activated by citrate and inhibited by palmitoyl-CoA and other short- and long-chain fatty acyl-CoAs. Citrate triggers the polymerization of ACC1 which leads to significant increases in its activity. Although ACC2 does not undergo significant polymerization (presumably due to its mitochondrial association) it is allosterically activated by citrate. Glutamate and other dicarboxylic acids can also allosterically activate both ACC isoforms. ACC activity is also regulated by phosphorylation. Both ACC1 and ACC2 contain at least eight sites that undergo phosphorylation. The sites of phosphorylation in ACC2 have not been as extensively studied as those in ACC1. Phosphorylation of ACC1 at three serine residues (S79, S1200, and S1215) by AMPK leads to inhibition of the enzyme. Glucagon-stimulated increases in cAMP and subsequently to increased PKA activity also lead to phosphorylation of ACC where ACC2 is a better substrate for PKA than is ACC1.
The role of ACC in regulating malonyl-CoA synthesis in the hypothalamus leading to anorexigenic effects has been demonstrated in studies on leptin. The anorexigenic effects of leptin involve the activation of ACC leading to increased malonyl-CoA levels in the ARC. When the activity of ACC2 is inhibited in the ARC, leptin is unable to reduce food intake and subsequent body weight, because it is no longer able to alter malonyl-CoA content within the ARC. This latter effect occurs even though leptin still induces an inhibition of AMPK indicating that malonyl-CoA-mediated effects on appetite and feeding behaviors are downstream of any effects of AMPK. These experimental results in animals demonstrate that ACC activity is an important regulator of feeding behaviors via the ability of ACC2 to regulate hypothalamic malonyl-CoA levels. Potential differences in ability of ACC1 and ACC2 to elicit decreases in appetite are difficult to assess since deletion of ACC1 in mice results in embryonic lethality.
Hypothalamic Malonyl-CoA Decarboxylase (MCD)
The intracellular levels of malonyl-CoA represent a balance between its synthesis from acetyl-CoA by ACC and its utilization in fatty acid synthesis by FAS as well as by its degradation to acetyl-CoA via the action of malonyl-CoA decarboxylase (MCD). Indeed, MCD is involved in regulating malonyl-CoA levels in multiple tissues. Inhibition of MCD results in reduced rates of fatty acid oxidation in highly oxidative tissues such as the heart. As well, MCD inhibition leads to reduced triaglyceride content in lipid synthesizing tissues such as the liver. When hypothalamic MCD levels are experimentally increased in laboratory animals there is a dramatic increase in food intake, weight gain, and ultimately results in obesity.
Transcriptional regulation of the MCD gene is exerted by PPARα, a major transcription factor involved in the regulation of fatty acid oxidation. Hypothalamic PPARα has been shown to play a role in the regulation of appetite, presumably via enhanced expression of MCD, with the use of pirinixic acid which is a PPARα agonist. As discussed below, inhibition of fatty acid synthase (FAS), the rate-limiting enzyme in de novo lipogenesis, decreases appetite and promotes weight loss. Administration of pirinixic acid results in normalization of malonyl-CoA levels in mice that have hypothalamic-specific knock-out of FAS. The PPARα-mediated increase in MCD results in reduced levels of malonyl-CoA in the hypothalamus. This is associated with increased food intake in the FAS knock-out mice demonstrating that malonyl-CoA levels are indeed responsible for the hypophagic effects observed in the FAS knock-out mice.
Hypothalamic Fatty Acid Synthase (FAS)
As indicated above, the intracellular levels of malonyl-CoA will decline as it is incorporated into de novo synthesized fatty acids via the action of FAS. The role of FAS in the regulation of feeding behavior and appetite has been demonstrated with the use of the FAS inhibitors C75 and cerulenin as well as in FAS knock-out mice (as indicated above). Inhibition of FAS causes potent anorexigenic effects that are associated with a reduction in hypothalamic NPY expression and increases in intracellular malonyl-CoA content. Mice with FAS deleted specifically in the ARC and PVN regions of the hypothalamus exhibit reduced appetite and weigh significantly less than control animals.
The anorexigenic effects of FAS inhibition with the use of C75 may not be due entirely to alterations in malonyl-CoA content. The anorexigenic effects of C75 have also been associated with increases in glucose metabolism which results in increased levels of ATP. Increases in ATP lead to inhibition of AMPK activity which in turn, results in reduced AMPK-mediated phosphorylation and inhibition of ACC. Therefore, there will be increased levels of malonyl-CoA demonstrating that malonyl-CoA is likely to be the primary signaling molecule responsible for the hypophagic effects of C75. FAS inhibitors, such as C75, also activate the mechanistic target of rapamycin (mTOR), and since mTOR and AMPK activities are inversely related they are likely to be reciprocally involved in the regulation of appetite.
Gastrointestinal Enteroendocrine Cells and Gut Hormone Release
Within the epithelium of the gastrointestinal (GI) system resides a specialized subset of cells termed enteroendocrine cells (EEC). Enteroendocrine cells are dispersed throughout the GI epithelium yet they represent just 1% of the total cell population. Collectively these highly specialized cells for the largest endocrine system in humans. Enteroendocrine cells differ from other endocrine cells in that they function as single cells and have a very limited life span. Like other cells of the GI system epithelium enteroendocrine cells are being continuously renewed from stem cells that reside in the crypts of the mucosa.
A considerable amount of research has defined the organization of the enteroendocrine system so that it is now recognized that there are basically three distinct subsets of these cells: pan-GI tract, stomach selective and intestine selective enteroendocrine cell types. However, not all peptides are expressed in all cells at the same time throughout the GI system. Expression at the gene level and at the protein level can also be distinct. For example, within the proximal small intestine ghrelin expression is high but this is only detectable at the mRNA level. Although ghrelin peptide was originally thought to be exclusive to the stomach, it has been shown that a specialized enteroendocrine cell type of the small intestine co-expresses both motilin and ghrelin peptides.
Unlike the other gut-derived substance secreted from enteroendocrine cells, serotonin (5-hydroxytryptamin: 5-HT) is not a peptide but is derived from the amino acid tryptophan. The primary functions attributed to gut-derived serotonin were regulation of gastrointestinal motility and gastrointestinal secretory processes. However, recent evidence has shown that gut derived serotonin is capable of exerting multiple metabolic effects. These effects include adaptation to fasting by regulating lipolysis in adipose tissue and glucose uptake and gluconeogenesis in the liver. Gut derived serotonin also appears to be involved in processes of gastrointestinal inflammation, bone formation, and hepatic regeneration.
Table of Gastrointestinal Enteroendocrine Cells
| Cell Type | Secreted Factor(s) | Major Location of Cells and Functions of Secreted Factors |
| Stomach Enteroendocrine Cells | ||
| A (X-like) cells | ghrelin, nesfatin-1 | predominantly located in the stomach ghrelin: induces increased feeding behavior nesfatin-1: derived via proteolytic processing of the 396 amino acid precursor protein encoded by the NUCB2 (nucleobindin 2) gene; reduces food intake |
| Enterochromaffin-like (ECL) cells | histamine | specific to stomach; histamine binds to H2 type receptors to activate parietal cells of the stomach to secrete H+ for formation of HCl in lumen of stomach |
| G cells | gastrin | predominantly located in the stomach antrum, some cells detected in duodenum; gastrin binds to cholecystokinin 2 (CCK2) receptors to activate parietal cells of the stomach to secrete H+ for formation of HCl in lumen of stomach; stimulates pepsinogen release from chief cells of stomach; also stimulates pancreatic secretions |
| P cells | leptin | regulation of overall body weight by limiting food intake and increasing energy expenditure, regulation of the neuroendocrine axis, inflammatory responses, blood pressure, and bone mass; leptin is also a major white adipose tissue-derived adipokine |
| Intestinal Enteroendocrine Cells | ||
| D cells | somatostain | considered pan-GI cell types; found in the stomach antrum and corpus as well as small intestine; somatostatin inhibits gastric acid production via antagonism of histamine release; inhibits pancreatic enzyme and bile acid secretions; inhibits release and action of numerous gut peptides such as CCK, OXM, PP, gastrin, motilin, GIP; |
| Enterochromaffin (EC) cells | serotonin (5-HT) | considered pan-GI cell types; found in stomach, small intestine, and large intestine; acting primarily through 5-HT4 class receptors serotonin stimulates gastrointestinal motility and secretory processes |
| I cells | cholecystokinin (CCK) serotonin (5-HT) | predominantly found in the proximal small intestine; CCK stimulates gallbladder contractions and bile flow; increases secretion of digestive enzymes from pancreas; vagal nerves in the gut express CCK1 (CCKA) receptors |
| K cells | GIP serotonin (5-HT) | predominantly found in the proximal small intestine; glucose-dependent insulinotropic peptide (GIP) inhibits secretion of gastric acid; enhances insulin secretion; also referred to as gastric inhibitory peptide |
| L cells | GLP-1 PYY oxyntomodulin (OXM) GLP-2 serotonin (5-HT) | primarily located in ileum (distal small intestine) and colon; small numbers have been found in duodenum and jejunum GLP-1: potentiates glucose-dependent insulin secretion; inhibits glucagon secretion; inhibits gastric emptying PYY: reduced gut motility; delays in gastric emptying, and an inhibition of gallbladder contraction; exerts effects on satiety via actions in the hypothalamus OXM: contains all of the amino acids of glucagon (see Figure below); inhibits meal-stimulated gastric acid secretion similar to GLP-1 and GLP-2 action; induces satiety, decreases weight gain, and increases energy consumption; has weak affinity for GLP-1 receptor as well as the glucagon receptor, may mimic glucagon actions in liver and pancreas GLP-2: enhances digestion and food absorption; inhibits gastric secretions; promotes intestinal mucosal growth |
| M (Mo) cells | motilin | located in small intestine; distinct from the M cells of GI that are found in the lymph follicles termed Peyer’s patches; motilin initiates inter-digestive intestinal motility; stimulates release of pancreatic polypeptide (PP); stimulates gallbladder contractions |
| N cells | neurotensin | located in jejunum; neurotensin a 13-amino acid peptide derived from a precursor that also produces neuromedin; involved in satiety responses and slows gastric emptying; also involved in nociception (sensation of pain) |
| S cells | secretin | located in the duodenum and jejunum; secretin induces pancreatic bicarbonate secretion; inhibits gastric secretions; stimulates PP secretion; induces satiety |
Gastrointestinal Hormones and Peptides
There are more than 30 peptides currently identified as being expressed within the digestive tract, making the gut the largest endocrine organ in the body. The regulatory peptides synthesized by the gut include hormones, peptide neurotransmitters, and growth factors. Indeed, several hormones and neurotransmitters first identified in the central nervous system and other endocrine organs have subsequently been found in enteroendocrine cells and/or neurons of the gut. Visit the Peptide Hormones and Their Receptors page to see a more complete list of gastrointestinal peptides and hormones and their modes of action. The following discussion will focus on the gut peptides with the best demonstrated roles in the control of appetite and feeding behavior via their interactions with signals produced in the hypothalamic-pituitary axis.
Table of Gastrointestinal Hormones and Peptides
| Hormone | Location | Major Action |
| Cholecystokinin (CCK) | enteroendocrine I cells predominantly in the duodenum, jejunum; also expressed in enteric nervous system and the brain | released from I cells in response to fat and/or protein ingestion; stimulates gallbladder contraction and bile flow, increases secretion of digestive enzymes from pancreas, vagal nerves in the gut express CCK1 receptors |
| Enterostain | derived from N-terminal end of pancreatic colipase; pentapeptide existing in three forms in mammals: APGPR (human), VPDPR, and VPGPR | regulates fat intake, peripheral or central administration inhibits consumption of a high-fat diet but not a low-fat diet |
| Famsin | 178 amino acids; cleaved from the membrane bound protein encoded by the C17orf78 gene | release is induced by fasting and promotes fasting-induced metabolism upon binding to the olfactory family receptor encoded by the OR10P1 (olfactory receptor family 10 subfamily P member 1) gene |
| FGF19 | gallbladder, duodenum, ileum | member of the large FGF family of growth factors; expression of FGF19 gene activated by transcription factor FXR, FXR is activated when ileal enterocytes absorb bile acids, when released to the portal circulation FGF19 stimulates hepatic glycogen and protein synthesis while inhibiting glucose production; reduces the expression and activity of CYP7A1 which is the rate-limiting enzyme in bile acid synthesis; acts in the gallbladder to induce relaxation and refilling with bile acids |
| Ghrelin | primary site is X/A-like enteroendocrine cells of the stomach oxyntic (acid secreting) glands, minor synthesis in intestine, pancreas and hypothalamus | regulation of appetite (increases desire for food intake), energy homeostasis, glucose metabolism, gastric secretion and emptying, insulin secretion |
| Glucagon-like peptide-1 (GLP-1) | enteroendocrine L cells predominantly in the ileum and colon | potentiates glucose-dependent insulin secretion, inhibits glucagon secretion, inhibits gastric emptying; is rapidly degraded by dipeptidylpeptidase 4 (DPP4) |
| Glucose-dependent insulinotropic polypeptide (GIP), originally called gastric inhibitory polypeptide | enteroendocrine K cells of the duodenum and proximal jejunum | secreted in response to food intake; inhibits secretion of gastric acid; is considered an incretin since it enhances insulin secretion; is rapidly degraded by dipeptidylpeptidase 4 (DPP4) |
| Nesfatin-1 | primarily expressed in enteroendocrine X/A-like cells in stomach and in white adipose tissue | proteolytic product of the 420 amino acid precursor protein encoded by the NUCB2 (nucleobindin 2) gene; following removal of the 24 amino acid signal peptide the 396 amino acid peptide is cleaved by proprotein convertases 1/3 (PC 1/3) and PC2; nesfatin-1 consists of amino acids 1–82; proteolytic processing also generates nesfatin-2 (amino acids 85–163) and nesfatin-3 (amino acids 166–396); stimulates reduced feeding via actions in the hypothalamus |
| Obestatin | primary site is stomach, minor synthesis in intestine | derived from pro-ghrelin protein, acts in opposition to ghrelin action on appetite |
| Oxyntomodulin | enteroendocrine L cells predominantly in the ileum and colon | contains all of the amino acids of glucagon (see Figure below); inhibits meal-stimulated gastric acid secretion similar to GLP-1 and GLP-2 action; induces satiety, decreases weight gain, and increases energy consumption; has weak affinity for GLP-1 receptor as well as glucagon receptor, may mimic glucagon actions in liver and pancreas |
| Pancreatic polypeptide: PP | pancreas | inhibits pancreatic bicarbonate and protein secretion |
| Peptide tyrosine tyrosine: PYY | enteroendocrine L cells predominantly in the ileum and colon | reduced gut motility, delays gastric emptying, inhibition of gallbladder contraction, induces satiety via actions in the arcuate nucleus (ARC) of the hypothalamus |
| Secretin | S-cells of duodenum | 27 amino acid peptide whose secretion is stimulated by the drop in pH in the lumen of the duodenum that follows gastric emptying; functions by binding to the secretin receptor (SCTR) which is a GPCR of the class B secretin-like family of receptors; stimulation of exocrine secretion of water and bicarbonate by the bile ducts, pancreas, and/or kidneys; inhibits gastric acid secretion; inhibits gastric emptying; induces satiety through effects in the ventromedial hypothalamus (VMH) |
| Vasoactive intestinal peptide (VIP) | gut, pancreas, brain | 28 amino acid peptide that belongs to the glucagon and secretin superfamily; VIP is closely related to a protein identified as pituitary adenylate cyclase-activating polypeptide type 1 (PACAP); initiates smooth muscle relaxation; stimulates pancreatic bicarbonate secretion; increases glycogen breakdown; within the brain VIP exerts effects on circadian rhythms; exerts effects by binding to three different GPCR identified as VPAC1 [VIP/PACAP (pituitary adenylate cyclase activating peptide 1) receptor type1], VPAC2, and PAC1 (pituitary adenylate cyclase-activating polypeptide type 1 receptor); VPAC1 and VPAC2 are encoded by the VIPR1 and VIPR2 genes, respectively; PAC1 is encoded by the ADCYAP1R1 gene |
Glucagon-Like Peptide-1: GLP-1
The glucagon-like peptides (principally glucagon-like peptide-1, GLP-1) and glucose-dependent insulinotropic peptide (GIP; originally identified as gastric inhibitory peptide) are gut hormones that constitute the class of molecules referred to as the incretins. Incretins are molecules associated with food intake-stimulation of insulin secretion from the pancreas.
GLP-1 is derived from the product of the glucagon (GCG) gene. The GCG gene is located on chromosome 2q24.2 and is composed of 6 exons. This gene encodes a preproprotein that is differentially cleaved (see Figure below) dependent upon the tissue in which it is synthesized. For example, in pancreatic α-cells the actions of proprotein convertase subtilisin/kexin type 2 (PCSK2) leads to the release of glucagon. PCSK2 is historically referred to as prohormone convertase 2 (PC2). In the gut the actions of proprotein convertase subtilisin/kexin type 1 (PCSK1) lead to release of several peptides including GLP-1. PCSK1 is historically referred to as prohormone convertase 1/3 (PC1/3).
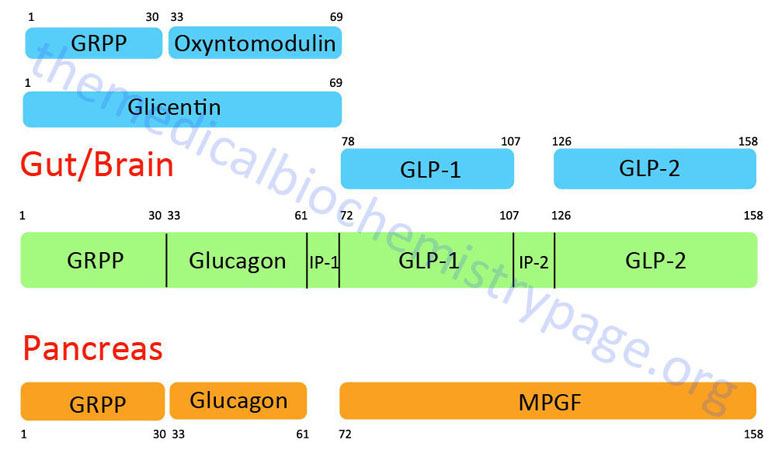
Upon nutrient ingestion GLP-1 is secreted into the blood from intestinal enteroendocrine L-cells that are found predominantly in the distal ileum and proximal colon with some production from these cell types in the duodenum and jejunum. Processing of preproglucagon in enteroendocrine L-cells results in the production of four forms of GLP-1 [GLP-1(1-36)NH2, GLP-1(7-36)NH2, GLP-1(1-37), and GLP-1(7-37)], GLP-2, glicentin, and oxyntomodulin. Bioactive GLP-1 consists of two forms; GLP-1(7-37) and GLP-1(7-36)amide, where the latter form constitutes the majority (80%) of the circulating hormone.
In addition to the gut, GLP-1 is produced within the brain, specifically in neurons located in the nucleus of the solitary tract (NTS). Neurons projecting from the NTS interact with numerous areas of the brain that express the GLP-1 receptor.
GLP-1 Receptor
All of the effects of GLP-1 are mediated following activation of the GLP-1 receptor (GLP-1R). The GLP-1 receptor is encoded by the GLP1R gene. The GLPR1 gene is located on chromosome 6p21.2 and is composed of 14 exons that generate three alternatively spliced mRNAs, only one of which encodes a functional 463 amino acid protein. The other two mRNAs are non-coding and likely degraded via the nonsense-mediated decay (NMD) pathway.
The GLP-1R is a GPCR of the class B secretin-like family of receptors. Specifically GLP1R is one of six members of the glucagon receptor subfamily that, in addition to GLP1R, includes the glucagon receptor (GCGR), the GLP-2 receptor (GLP2R), the secretin receptor (SCTR), the glucose-dependent insulinotropic peptide receptor (GIPR), and the growth hormone releasing hormone receptor (GHRHR).
GLP-1 binding to GLP-1R is coupled to the activation of a Gs-type G-protein resulting in increased cAMP production and activation of PKA. However, there are also PKA-independent responses initiated through the GLP-1R that involve the activation of a Gq-type G-protein. The Gq-mediated effects include the activation of the ERK1/2 pathway of signal transduction. Effects of GLP-1R activation have also been shown to include the PI3K pathway and the activation of AKT/PKB.
The GLP-1R is expressed on α- (glucagon secreting), β- (insulin secreting), and δ- (somatostatin secreting) cells of pancreatic islets, as well as in the heart, lung, kidney, stomach, small intestine, skin, and nerves in the peripheral and central nervous systems (PNS and CNS). Within the CNS, GLP-1R is found in neurons, astrocytes, and oligodendrocytes. Responses to GLP-1 are also exerted in the liver, adipose tissue, and muscle but these are indirect responses not due to direct receptor binding since the GLP1R gene is not expressed in these tissues. Within the mouse liver there are certain populations of immune cells [gamma-delta(γδ) T cells] that express the GLP1R gene and these may be the responding cells that exert the effects of GLP-1 in the liver, however, whether these cells are exerting GLP-1 effects in human liver remain to be elucidated.
The primary physiological responses to GLP-1 are glucose-dependent insulin secretion, inhibition of glucagon secretion, and inhibition of gastric acid secretion and gastric emptying. The latter effect will lead to increased satiety with reduced food intake along with a reduced desire to ingest food.
Within the pancreas, GLP-1R activation also results in β-cell proliferation and expansion concomitant with a reduction of β-cell apoptosis (programmed cell death). In addition, GLP-1 activity in the pancreas results in increased expression of the genes encoding glucose transporter-2 (GLUT-2) and glucokinase.
Within the CNS, intestinal GLP-1 exerts potent appetite suppression via effects exerted directly through GLP-1R activation within the hypothalamus. In addition, GLP-1 is produced by specific neurons in the nucleus of the solitary tract (NTS) that project GLP-1 effects to numerous regions of the brain including the hypothalamus, specifically the lateral hypothalamus. Indirectly, GLP-1 activity results in reduced hepatic gluconeogenesis while at the same time leading to increased glucose uptake and storage in skeletal muscle and adipose tissue.
The action of GLP-1 at the level of insulin and glucagon secretion results in significant reduction in circulating levels of glucose following nutrient intake. This activity has significance in the context of diabetes. The glucose lowering activity of GLP-1 is highly transient as the half-life of this hormone in the circulation is less than two minutes. Removal of bioactive GLP-1 is a consequence of N-terminal proteolysis catalyzed by dipeptidyl peptidase IV (DPP IV or DPP4). DPP4 is also known as the lymphocyte surface antigen CD26 and has numerous activities unrelated to incretin inactivation (see the Dipeptidyl Peptidase 4: DPP4 page for more information on DPP4).
Within the cardiovascular system the actions of GLP-1 are exerted cardiac endothelial cells and cardiomyocytes, vascular endothelial cells and smooth muscle cells (VSC), as well as VSC within the glomerulus of the kidney. In the heart the effects of GLP-1 include increased uptake of glucose, increased cardiac contractility, and increased cardiac protection from stressors. Within the vasculature GLP-1 actions result in vasodilation and increased production of nitric oxide, NO. In the kidney, GLP-1 action increases sodium and water excretion which contributes to a reduction in blood pressure.
Glucose-Dependent Insulinotropic Peptide: GIP
Glucose-dependent insulinotropic peptide (GIP) is derived from a 153-amino acid preproprotein encoded by the GIP gene and circulates as a biologically active 42-amino acid peptide identified as GIP(1-42). GIP is synthesized by intestinal enteroendocrine K-cells whose locations are primarily in the duodenum and proximal jejunum. Although the proximal small intestine is the major source of GIP synthesis, it is also produced by the stomach, the submandibular salivary glands, and in the central nervous system, CNS. Within the CNS the production of GIP is observed in the olfactory bulb, the hippocampus, the Purkinje cells in the cerebellum, the cerebral cortex, the amygdala, the substantia nigra, the hypothalamus, and the hindbrain.
The original activity associated with GIP was the inhibition of gastric acid secretion and the protein was originally called gastric inhibitory peptide. The ability of tissue extracts containing GIP to inhibit gastric acid secretion and gastric emptying were subsequently shown to not be associated with GIP itself.
The GIP gene is located on chromosome 17q21.32 and is composed of 6 exons that encodes the 153 amino acid preproprotein. Following synthesis the GIP precursor protein is cleaved in two internal locations by proprotein convertase subtilisin/kexin type 1 (PCSK1), which is historically identified as prohormone convertase 1/3 (PC1/3). Evidence suggests that following the actions of PCSK1, carboxypeptidase E removes the amino terminal Gln (Q) residue generating the bioactive GIP(1-42) protein. Evidence, albeit somewhat controversial, has shown that in pancreatic α-cells, proprotein convertase subtilisin/kexin type 2 (PCSK2; historically identified as PC2) cleaves the GIP(1-42) to generate GIP(1-30) which is subsequently processed by peptidylglycine α-amidating monooxygenase (encoded by the PAM gene) to generate GIP(1-30)NH2.
Subsequent to its initial characterization, research demonstrated that purified GIP stimulated glucose-dependent insulin secretion defining the protein as an incretin, characteristic of GLP-1. In humans, the ability of GIP to promote insulin secretion is very nearly the same as that exhibited by GLP-1. In the setting of obesity and type 2 diabetes the ability of GIP to promote insulin secretion is reduced.
In addition to its incretin activity, GIP has significant effects on fat metabolism exerted at the level of adipocytes. These actions include stimulation of lipoprotein lipase activity leading to increased uptake and incorporation of fatty acids by adipocytes.
Whereas GIP exerts positive effects on pancreatic β-cell proliferation and survival similar to that shown for GLP-1, the hormone does not affect glucagon secretion nor gastric emptying. Like GLP-1, GIP is inactivated through the action of dipeptidylpeptidase, DPP4.
The plasma half-life of GIP is less than 10 minutes in normal healthy adults. The removal of GIP is the function of both the kidneys and the consequences of the protease, DPP4 similar to the role of the kidneys and DPP4 in the removal of GLP-1 from the blood.
The actions of GIP are exerted following its binding to a specific receptor. The GIP receptor is a G-protein coupled receptor (GPCR). The GIPR has been shown to activate both Gs– and Gq-type type G-proteins. Expression of the GIP receptor is found in the pancreas, adipose tissue, blood vessels, bone, heart, hematopoietic cells, immune cells, and in the central nervous system (CNS). Within the CNS, GIPR expression is found in neurons, oligodendrocytes, and pericytes. Like the GLP-1 receptor, the GIP receptor is not expressed in skeletal muscle, hepatocytes, nor adipocytes.
The GIP receptor is encoded by the GIPR gene. The GIPR gene is located on chromosome 19q13.32 and is composed of 13 exons that generate two alternatively spliced mRNAs encoding precursor proteins of 466 amino acids (isoform 1) and 430 amino acids (isoform 2).
Activation of the GIPR in pancreatic β-cells results in activation of a Gs-type G-protein which subsequently results in the activation of PKA as well as the exchange protein activated by cAMP 2 (EPAC2). EPAC2 is encoded by the RAPGEF4 (RAP guanine nucleotide exchange factor 4) gene and as a result of alternative promoter usage and alternative splicing there are four functional EPAC2 isoforms. The activation of PKA leads to the phosphorylation of the SUR1 (encoded by the ABCC8 gene) subunit of the KATP channel that controls insulin secretion.
The GIPR is also expressed on pancreatic α-cells and activation of the receptor by GIP binding leads to increased glucagon release. The clinical significance of this GIP effect in the pancreas is that in type 2 diabetics the effects of GIP on β-cells is impaired but its effects in α-cells is not. The combined effect in type 2 diabetes is reduced insulin secretion and enhanced glucagon secretion which leads to exacerbation of the hyperglycemia typical in diabetics.
Despite the GIPR gene not being expressed in the liver, GIP does exert effects in this organ that includes increases in lipid content. In mice, the knock-out of the GIPR gene is correlated with reductions in hepatic lipid content. These results indicate that the primary indirect effect of GIP on the liver is at the level of lipid homeostasis.
Responses to GIP have been shown to be defective in type 2 diabetic patients. Interestingly, in gene knock-out mice it has been shown that loss of the GIPR is correlated to resistance to obesity even if the animals are fed a high fat diet.
Oxyntomodulin
Oxyntomodulin (OXM) is so called given that is was originally discovered from work examining the inhibition of the activity of oxyntic glands (gastric acid secreting) of the stomach. OXM is a 37 amino acid peptide derived from the pre-pro-glucagon gene and contains the entire 29 amino acids of the pancreas-derived glucose regulating hormone glucagon. The OXM protein contains an additional 8 amino acids at its C-terminus relative to glucagon. Synthesis and release of OXM occurs in the enteroendocrine L cells of the distal gut. These are the same cell populations that secrete GLP-1 and PYY. The secretion of OXM occurs within 5-10 minutes following ingestion of food and peaks within 30 minutes. The amount of OXM that is released is directly proportional to caloric intake. In addition to stimulated release in response to food intake, OXM exhibits diurnal variation in its concentration in the blood with highest levels detected in the evening and lowest levels in the morning.
Thus far there has been no specific OXM receptor identified. Evidence suggests that the anorexigenic effects of OXM are in fact exerted via the GLP-1 receptor (GLP-1R). In mice in which the GLP-1R gene has been knocked out the anorectic responses to injected OXM are abolished. Like GLP-1, OXM has demonstrated incretin activity (incretins stimulate insulin release in response to food intake) and this activity is abolished in the GLP-1R knock-out mouse. In addition, OXM exerts a protective effect on pancreatic β-cells similar to that exerted by GLP-1. Although the affinity of OXM for the GLP-1R is at least 50 fold less than that of GLP-1 itself, the ability of OXM to exert inhibition of food intake is equal to that of GLP-1. With respect to effects on food intake exerted via the hypothalamus when either OXM or GLP-1 are administered peripherally they exert differential neuronal activation within the hypothalamus. This suggests that these two hormones act via different hypothalamic pathways involved in appetite control. When OXM is administered into the brain the response is suppression of the effects of circulating ghrelin. These results suggest that part of the appetite suppressing effects of OXM are mediated by reduced ghrelin as well as increased hypothalamic release of anorexigenic peptides.
Of potential significance to the treatment of obesity, when OXM is administered intravenously to human subjects there is an observed reduction (19.3%) in food intake at mealtime. Additionally significant is the fact that this reduction in desire for food intake persisted over the course of 12 hours. When OXM is administered subcutaneously to overweight and obese subjects over a period of 4 weeks there is a significant reduction in body weight. The average weight loss in the volunteers was 2.3kg (1 lb) compared to only 0.5kg in untreated control subjects. The weight loss observed in these studies was likely due to a combination of reduced desire for food intake as well as an increased metabolic expenditure. When OXM was administered over a 4 day period to human subjects there was an observed 10% increase in total energy expenditure. Although these results prove promising for the potential for OXM in the treatment of obesity it is important to note that OXM is a target for inactivation by DPP-4 just as is GLP-1. Therefore, any OXM agonist must be resistant to inactivation by DPP-4.
Cholecystokinin, CCK
Cholecystokinin (CCK) is derived via post-translational modification of the pro-cholecystokinin product of the CCK gene. The CCK gene is located on chromosome 3p22.1 and is composed of 6 exons that generate two alternatively spliced mRNAs, both of which encode the same 115 amino acid preproprotein. Expression of the CCK gene predominates in the enteroendocrine I cells found in the duodenum and jejunum as well as within neocortical neurons in the brain.
CCK was the first gut hormone to be identified as having an effect on appetite. There are several bioactive forms of CCK that are designated based upon the number of amino acids in the peptide. The four major forms are CCK-8, CCK-22, CCK-33, and CCK-58. The predominant form that is found in human plasma is the CCK-33 form. The CCK-8 isoform is the primary form found in neurons. The CCK isoforms are also found as sulfated and non-sulfated variants. The tyrosine at position 77 (Y77) is sulfated and this post-translational modification is critical for the binding of CCK to the CCK1 receptor.
The level of CCK in the blood rises within 15 minutes of food ingestion and reaches a peak by 25 minutes. The elevation in plasma CCK levels remains for approximately 3 hours following a meal. The most potent substances initiating a release of CCK from enteroendocrine I cells are fats and proteins. Conversely duodenal bile acids are potent suppressors of the secretion of CCK.
CCK exerts its biological actions by binding to specific G-protein coupled receptors (GPCR). There are two CCK receptor types identified as CCK-1 and CCK-2. The CCK receptors are also identified as CCKA and CCKB whose designations referred to their location of prominent expression with CCKA referring to the alimentary tract (the gut) and CCKB referring to the brain. However, both receptors are found widely expressed in the CNS as well as the periphery. The CCKB receptor exhibits equal affinity for CCK and another gut peptide, gastrin. Gastrin is involved in the production and release of H+, thereby generating gastric acid.
The CCK-1 receptor is encoded by the CCKAR gene. The CCKAR gene is located on chromosome 4p15.2 and is composed of 5 exons that encode a 428 amino acid protein.
The CCK-2 receptor is encoded by the CCKBR gene. The CCKBR gene is located on chromosome 11p15.4 and is composed of 4 exons that generate three alternatively spliced mRNAs, each of which encodes a distinct protein isoform.
Upon binding its receptors in the gut, CCK induces contractions of the gallbladder and release of pancreatic enzymes and also inhibits gastric emptying. Within the brain (specifically the median eminence and ventromedial nucleus of the hypothalamus, VMH) CCK actions elicit behavioral responses and satiation. The CCK-1 receptor is believed to be the receptor primarily responsible for alterations in feeding behavior. Evidence for this comes from the Otsuka Long Evans Tokushima Fatty rat which has a null mutation in the CCK-1 receptor. These rats are hyperphagic and develop obesity even on a fat restricted diet. The situation is not entirely conclusive since knock-out of the CCK-1 receptor in mice does not result in a similar phenotype. However, the ability of CCK to regulate food intake has been clearly demonstrated in numerous studies. For example central administration of CCK results in reduced food intake. Of significance to appetite control, this effect is enhanced with co-administration of leptin. The synergistic effects of CCK and leptin may be due to the fact that their receptors are co-localized to the same sensory vagal afferent neurons.
In humans the correlations between CCK and obesity are growing. Studies have shown that the fasting levels of CCK are lower in morbidly obese individuals than in lean individuals. In addition, the post-feeding responses to CCK in morbidly obese individuals is attenuated compared to lean individuals. The primary appetite suppressing effects of CCK are exerted via its inhibitory actions on NPY neurons (orexigenic neurons) in the DMH (dorsomedial hypothalamic nucleus) and the NTS (nucleus of the solitary tract).
Using genome wide screens for polymorphisms in genes associated with obesity and/or increased feeding behaviors have shown a correlation to the CCK_H3 haplotype. Polymorphisms in the CCK-1 receptor gene may also predispose an individual to obesity. Although CCK is known to be involved in satiation it may have limited use as a therapeutic for the treatment of obesity at least when used alone. This is due to the fact that when studied in laboratory animals, administration of CCK resulted in reduced meal size but the animals increased their frequency of food intake such that the overall outcome was no net change in body weight. However, given the synergistic actions of CCK and leptin, combination therapies of these two hormones may prove useful.
Ghrelin
Growth hormone secretagogues (GHS) were originally characterized as molecules that exerted effects upon the pituitary and hypothalamus leading to amplification of the pulsatile release of growth hormone. The initial GHS was identified as a derivative of Met-enkephalin. Subsequent to this initially characterized GHS, numerous peptide and non-peptide compounds were developed and shown to exert related GHS activity.
These peptides and compounds were shown to bind to and activate a G-protein coupled receptor (GPCR) in the pituitary and hypothalamus. This receptor is identified as the growth hormone secretagogue receptor type 1a, GHS-R1a. GHS-R1a is encoded by the GHSR gene.
The GHSR gene is located on chromosome 3q26.31 and is composed of 2 exons that generate two alternatively spliced mRNAs. The 1a mRNA results from a splicing event that removes the intron and the resultant mRNA encodes the functional 366 amino acid protein. The 1b mRNA does not undergo splicing and as a result does not encode a functional receptor protein. However, the GHS-R1b protein plays a regulatory role in overall growth hormone secretagogue activity, such as that of ghrelin.
The stomach derived hormone identified as ghrelin was first discovered based upon its ability to interact with, and activate, GHS-R1a stimulating the release of growth hormone. Indeed, ghrelin was found to be the endogenous ligand for the GHS-R1a. The name ghrelin is derived from growth-hormone release. Ghrelin is produced and secreted by the A (X-like) enteroendocrine cells of the stomach oxyntic (acid secreting) glands. Because the A (X-like) cells express ghrelin they are also sometimes referred to as ghrelin cells or Gr cells. A (X-like) cells express the receptor for gastrin (gut hormone that stimulates gastric acid secretion by the stomach) and, therefore, it is believed that gastrin may directly stimulate ghrelin release. Smaller amounts of ghrelin are released from the small intestine and even less from the colon.
Ghrelin is encoded by the GHRL gene. The GHRL gene is located on chromosome 3p25.3 and is composed of 6 exons that generate 10 alternatively spliced mRNAs that collectively encode five distinct preproprotein isoforms. Five of the alternative mRNAs encode preproproteins that are processed into both the ghrelin-28 and obestatin proteins, while two of the mRNAs encode preproproteins that are processed into the ghrelin-27 and obestatin proteins, and the remaining three mRNAs encode preproproteins that lack the signal peptide and the ghrelin-28 sequences.
Obestatin exerts its effect in exact opposition to that of ghrelin. The name obestatin is derived from a contraction of obese and statin (to suppress). Whereas, treatment of animals with ghrelin results in increased appetite and food intake, obestatin treatment suppresses food intake.
As a result of the alternative splicing and also due to post-translational cleavage, the primary 117 amino acid preproghrelin protein can be processed into ghrelin-28 (28 amino acids corresponding to amino acids 24–51 of the preproprotein), obestatin (23 amino acids corresponding to amino acids 76–98 of the preproprotein) and desacyl-ghrelin (also called ghrelin-27: 27 amino acids). The formation of desacyl-ghrelin is the consequence of alternative splicing due to an intron that reside between the glutamines at positions 13 and 14 (Q13 and Q14) of the preproghrelin sequence.
Bioactive ghrelin is first acylated on the serine at position 3 with n-octanoic acid. This acylation is followed by the processing of ghrelin from proghrelin which involves cleavage by proprotein convertase subtilisin/kexin type 1, PCSK1. PCSK1 is also known as prohormone convertase 1/3 (PC1/3).
The attachment of octanoic acid to Ser3 of ghrelin is accomplished by the acyltransferase originally identified as ghrelin O-acyltransferase, GOAT. GOAT is encoded by the MBOAT4 gene which is a member of the membrane-bound O-acyltransferase (MBOAT) family of acyltransferase encoding genes. Expression of the MBOAT4 gene predominates in the stomach and the pancreas. The highest levels of MBOAT4 expression are in the ghrelin producing A (X-like) cells of the stomach. Evidence indicates that the non-acylated form of ghrelin-28 may act as an antagonist of the acylated hormone.
The desacyl-ghrelin (ghrelin-27) protein is also acylated on Ser3 and that acylation is required for its activity as for full-length ghrelin (ghrelin-28). The level of acylated ghrelin decreases during long-term fasting, despite the fact that total ghrelin levels are increased under these conditions. The reduction in ghrelin acylation during fasting correlates to a reduction in expression of the MBOAT4 gene.
GHS-R1a Signaling
GHS-R1a is a GPCR belonging to the rhodopsin-like family of GPCR. The associated G-proteins activated by ligand binding to GHS-R1a are of the Gq/11 family. Activation of GHS-R1a by ghrelin results in increased activity of phospholipase C beta (PLCβ) with the consequent production of diacylglycerol (DAG) and inositol-1,4,5-trisphosphate (IP3; Ins-1,4,5-P3) from membrane-associated phosphatidylinositol-4,5-bisphosphate (PIP2; PtdIns-4,5-P2). The production of IP3 stimulates Ca2+ release from the endoplasmic reticulum, ER. Ghrelin activation of GHS-R1a also results in activation of plasma membrane N-type Ca2+ channels (Cav2.2) resulting in further increases in intracellular free Ca2+. Termination of GHS-R1a signaling occurs following recruitment of β-arrestin2 to the receptor.
The truncated form of the GHS-R, identified as GHS-R1b, is composed of 289 amino acids and contains only five transmembrane domains. GHS-R1b does not bind ghrelin but functions as a regulatory protein by forming heterodimers with GHS-R1a. Expression of the GHS-R1a mRNA is found in several nuclei of the hypothalamus, hippocampus, substantia nigra, ventral tegmental area (VTA), dorsal and median raphe nuclei, anterior pituitary gland, pancreatic islets, adrenal gland, thyroid, lung, liver, kidney, immune cells, intestine, adipose tissue, and myocardium.
Signal transduction processes initiated as a result of GHS-R1a activation play roles in a variety of behaviors, including learning and memory, reward, impulsivity, anxiety, and vulnerability to stress. The wide array of activities associated with GHS-R1a activation is explained, in part, via the interaction of the receptor with other GPCR. These other GPCR include the dopamine D1 receptor (D1R) subtype, the melanocortin 3 receptor (MC3R), the serotonin 2C receptor (5HT2C), and the somatostatin receptor subtype 5 (SST5). The consequences of heterodimerization with GHS-R1a can lead to altered signaling such as is the case for the D1R. The D1R is normally coupled to the activation of a Gs-type G-protein, but in the GHS-R1a heterodimer, dopamine activation of D1R results in activation of a Gq-type G-protein. This results in exocytosis of glutamate receptors and synaptic reorganization. In the context of a GHS-R1a:D1R heterodimer the inactivation of the GHS-R1a component results in inhibition of D1R-mediated hippocampal behavior and memory.
Ghrelin Activities
Within the stomach the release of ghrelin leads to increased gastric acid secretion, increased gastric motility, and increased gastric emptying.
The major effect of ghrelin released to the blood is exerted within the central nervous system at the level of the arcuate nucleus where it stimulates the synthesis and release of neuropeptide Y (NPY) and agouti-related protein (AgRP). The highest levels of GHS-R1a expression within the hypothalamus are in NPY/AgRP expressing neurons.
Within NPY/AgRP neurons ghrelin activation of GHS-R1a results in increased activation of the mTOR signaling pathway leading to activation of expression of the NPY and AgRP genes. The actions of NPY and AgRP enhance appetite and thus, food intake.
Within the ventromedial hypothalamic nucleus (VMH) ghrelin activation of GHS-R1a results in activation of AMPK via the sirtuin (SIRT1) pathway leading to reduced intracellular levels of long-chain fatty acids. AMPK phosphorylates and inhibits acetyl-CoA carboxylase (ACC) leading to reduced levels of malonyl-CoA. Malonyl-CoA inhibits the import of fatty acids into the mitochondria, thus reduced levels of malonyl-CoA lead to increased mitochondrial oxidation of long-chain fatty acids as described above. The reduction in fatty acid levels appears to be the molecular signal leading to increased expression of NPY and AgRP. In addition, the ghrelin-mediated changes in fatty acid oxidation in VMH neurons result in increased release of glutamate. Glutamate then binds to NMDA and AMPA receptors on NPY/AgRP neurons resulting in an additional level of activated NPY and AgRP gene expression. However, it is important to note that the signaling events triggered by ghrelin binding to GHS-R1a are complex as discussed earlier.
Ghrelin-mediated effects on feeding behavior are also exerted via the vagal afferent nerve. The GHS-R1a gene is expressed in ganglia of the vagal afferent nerve and is then transported to the stomach where ghrelin can bind to it. Binding of ghrelin results in inhibition of electrical signaling which is then sensed by the nucleus of the solitary tract (NTS). This signal is then relayed to neurons that directly project to the hypothalamus where they make synapses with NPY/AgRP neurons activating the production and release of these orexigenic neuropeptides. The vagal pathway of ghrelin action in the hypothalamus is integrated into the systemic (peripheral) pathway of ghrelin action. This is demonstrated in vagotomized humans who do not increase their desire for food intake when receiving peripheral injections of ghrelin.
Within the hypothalamus the activation of GHS-1Ra in NPY and AgRP neurons is required for the orexigenic responses exerted by ghrelin release from A (X-like) cells in the stomach. Ghrelin signaling in the hypothalamus also activates Ca2+/calmodulin (CaM)-dependent protein kinase kinase beta (CaMKKβ). The activation of CaMKKβ results in the phosphorylation and activation of AMPK. Activation of AMPK in the hypothalamus results in altered fatty acid metabolism and increased mitochondrial fatty acid oxidation which ultimately results in increased release of NPY and AgRP leading to an increased desire for food intake.
Ghrelin effects on feeding behavior are also exerted by its ability to induce the release of endocannabinoids. As indicated, the ghrelin-mediated activation of GHS-1Ra leads to activation of PLCβ with the release of DAG and IP3 from membrane PIP2. The increase in DAG, not only stimulates signal transduction pathways, but also serves as a substrate for the synthesis of 2-arachidonoylglycerol (2-AG). Increases in feeding behavior result from the activation of the cannabinoid receptor 1 (CB1) by 2-AG.
The secretion of ghrelin is the inverse of that of insulin. The primary mechanisms that are coupled to production of ghrelin are fasting, hypoglycemia, and leptin. Conversely, inhibition of ghrelin production is exerted by food intake, hyperglycemia, and obesity. The action of ghrelin, at the level of increasing the release of NPY, is the exact opposite to that of leptin which inhibits NPY release.
Additional effects of ghrelin include inhibition of the expression of pro-inflammatory cytokines, modulation of the exocrine and endocrine functions of the pancreas, controlling gastric acid secretion and gastric motility, influencing sleep patterns, memory and anxiety-like behavioral responses.
Although the major direct physiological effects of ghrelin are exerted within the central nervous system, which in turn affects processes in numerous organ systems, there are direct effects of ghrelin in many of the same organ systems. Within adipose tissue ghrelin action results in increased triglyceride storage and decreased lipid oxidation. The effects of ghrelin on lipogenesis are also observed within the liver.
Role of LEAP2 in Ghrelin Functions
Recent (2018) data demonstrated that an endogenous protein functions as an antagonist of the actions of ghrelin exerted through GHS-1Ra. This protein was originally identified as a secreted protein found in the blood at elevated levels following gastric bypass surgery, specifically vertical sleeve gastrectomy (VSG). This secreted protein is identified as liver expressed antimicrobial peptide 2 (LEAP2). LEAP2 was originally characterized as liver-specific cysteine-rich cationic antimicrobial peptide. Subsequent studies have shown that LEAP2 is expressed in the liver and the small intestines.
The expression and release of LEAP2 is reciprocal to that of ghrelin. Fasting results in decreased LEAP2 levels whereas feeding results in increased levels. LEAP2 has been shown to completely block the feeding stimulus effects of ghrelin. LEAP2 exerts its effects by functioning as a GHS-1Ra antagonist.
Obestatin
Obestatin exerts its effect in exact opposition to that of ghrelin. Release of obestatin suppresses food intake and gastric emptying activity. Like ghrelin, which is post-translationally modified, obestatin is also modified but its modification is an amidation.
One report indicated that obestatin bound to an orphan G-protein coupled receptor (GPCR) identified as GPR39. Evidence indicates that GPR39 is also the zinc-sensing receptor (ZnR) that responds to Zn2+ in the processes of tissue repair. GPR39 is expressed in liver, gastrointestinal tract, adipose tissue, and pancreas. Activation of the receptor results in increased cAMP and consequent activation of PKA mediated signaling pathways.
Within the pancreas GPR39 has been shown to regulate the expression of insulin receptor substrate-2 (IRS-2) and pancreatic and duodenal homeobox-1 (PDX-1) and that activation of the receptor is required for increased insulin secretion. It is now clear that GPR39 is not the receptor for obestatin and the original observation was likely the result of zinc in the preparations of obestatin.
Although obestatin has clear demonstrated activity as an anorectic hormone, inducing decreased food intake, reduced body weight gain, as well as being involved in the regulation of sleep patterns and cell proliferation, no definitive receptor for this hormone has been identified.
Famsin
Famsin is a recently (2023) identified highly glycosylated gut-derived hormone that plays a role in the metabolic adaptations to fasting. The name famsin was chosen due to the protective effect of the protein during fasting (famine) conditions. Famsin is derived by cleavage of a 178 amino acid protein from the 275 amino acid single pass transmembrane receptor protein encoded by the C17orf78 gene. The cleavage of the C17orf78 protein occurs N-terminal to the predicted transmembrane domain and at the peptide bond between R178 and Q179. The likely protease responsible for the release of famsin is furin, an enzyme of the proprotein convertase subtilisin/kexin (PCSK) type family and as such is also identified as PCSK3.
The C17orf78 gene name refers to the fact that it was originally identified as an uncharacterized open reading frame found on chromosome 17, specifically at 17q12. The gene is composed of 7 exons that generate two alternatively spliced mRNAs where the isoform 1 mRNA encodes the 275 amino acid protein from which famsin is derived. Expression of the C17orf78 gene is highest in the intestines, specifically in the duodenum, with much lower levels in the liver. Expression of famsin is not detected in any other adult tissues.
As the name and function implies, the level of circulating famsin increases during the early period of fasting and then decreases after prolonged fasting (>18 hours) as well as after re-feeding. The primary responses to famsin are exerted in the liver where the hormone enhances gluconeogenesis and ketogenesis. Famsin exerts these effects in the liver by binding to a receptor of the olfactory receptor family, specifically OR10P1 (olfactory receptor family 10 subfamily P member 1). This receptor is identified as OLFR796 in mice.
The activation of the OR10P1 receptor by binding famsin results in enhanced expression the gluconeogenic genes encoding glucose-6-phosphatase (G6PC) and the cytosolic version of phosphoenolpyruvate carboxykinase (PCK1), the lipolytic genes encoding carboxy ester lipase (CEL), pancreatic lipase (PNLIP), pancreatic lipase-related protein 2 (PNLIPRP2), and colipase (CLPS), the ketogenic gene encoding mitochondrial HMG-CoA synthase (HMGCS2), and the lipid oxidation gene encoding carnitine palmitoyltransferase 1 (CPT1A). Famsin also increases the activity of adipose triglyceride lipase (ATGL) in hepatocytes.
Expression of the OR10P1 gene also occurs in α-cells of the pancreas. The binding of famsin to this receptor on these pancreatic cells result in increased Ca2+ mobilization. The increase in cytosolic Ca2+ leads to increased exocytosis of vesicles containing the hormone glucagon. The glucagon then binds to its receptors on hepatocytes activating the cascade of events resulting in increased gluconeogenesis. Coupled with the direct effects of famsin in the liver, the net effect is a significant increase in delivery of glucose to the blood during the early stages of fasting.
Pancreatic Polypeptide, PP
Pancreatic polypeptide was the first member of the PP-fold family to be isolated and characterized. It was originally found as an impurity in preparations of insulin from chicken pancreas. Subsequently it was shown to be produced and secreted by type F cells within the periphery of pancreatic islets. The stimulus for the release of PP is the ingestion of food and the level of release is proportional to the caloric intake. Increased circulating levels of PP can be detected in the blood for up to 6 hours following ingestion of food. Humoral signals that are involved in food intake-mediated secretion of PP include ghrelin, cholecystokinin (CCK), motilin, and secretin. Additionally, adrenergic stimulation secondary to either hypoglycemia or exercise results in increased release of PP. The actions of PP include delaying gastric emptying, inhibition of gallbladder contraction, and attenuation of pancreatic exocrine secretions. These gut actions of PP are associated with the mechanism referred to as the “ileal brake” which is manifest with the slowing of the passage of nutrients through the gut.
PP induces an anorexigenic response within the brainstem (area postremus, AP) and vagus. These responses are mediated via activation of the Y4 receptor which binds PP with highest affinity. In addition to its expression in the AP, the Y4 receptor is also expressed in regions of the hypothalamus including the ARC. Therefore, additional anorexigenic responses to PP can be induced within the hypothalamus. Thus, PP plays an important role in the regulation of satiety (the sensation of being full). In obese individuals there is a reduced level of PP secretion in response to food intake, whereas, in anorexia nervosa there is increased PP release following consumption of food. PP may also play a role in the pathogenesis of Prader-Willi syndrome (PWS). This disorder is characterized by short stature, reduced intellect, and hyperphagia (excessive hunger and abnormally large food intake). In patients with PWS there is a reduced secretion of PP in response to food intake as well as a reduced basal level of circulating PP.
Protein Tyrosine Tyrosine, PYY
PYY is produced by, and secreted from, intestinal enteroendocrine L-cells of the ileum and colon. Additional gut hormones that are secreted along with PYY include GLP-1 and oxyntomodulin (OXM). Both of these latter gut hormones are discussed above. Secreted forms of PYY include PYY1-36 and PYY3-36. PYY1-36 is full-length protein and is more commonly referred to as just PYY. PYY3-36 is generated via the actions of dipetidylpeptidase 4 (DPP4 or DPP-IV) on PYY. DPP4 is the same enzyme that inactivates GLP-1. Within the gastrointestinal tract the highest detectable levels PYY are found in the rectum with low levels found in the duodenum and jejunum. Within the central nervous system (CNS) PYY is detectable in the hypothalamus, medulla, pons, and spinal cord. As indicated above in the discussion of NPY, PYY binds to members of the Y receptor family. PYY is a potent agonist of both Y1 and Y2 receptors, whereas PYY3-36 is a Y2-specific agonist. The affinity of PYY3-36 for the Y2 receptor is approximately 1,000-fold higher than for the Y1 receptor.
The release of PYY results in reduced gut motility, a delay in gastric emptying, and an inhibition of gallbladder contraction. All of these actions are, like that of PP, associated with the ileal brake. Given that PYY is secreted from cells of the distal gut there must be signals associated with the response of the proximal gut to food intake that lead to PYY release. Indeed humoral factors such as CCK and gastrin are thought to mediate the rapid release of PYY in response to eating. The amount of PYY released in response to the ingestion of food is proportional to the caloric intake.
Animal and human studies of anorectic conditions indicate that PYY has a critical role in satiety. Within the CNS, PYY exerts its effects on satiety via actions in the hypothalamus, specifically the ARC of the hypothalamus. The ARC is in close proximity to the deficient blood-brain barrier of the median eminence of the hypothalamus, thus allowing this region to respond rapidly to the release of a gut hormone into the circulation.
Evidence confirming the role of PYY3-36 in inducing anorexia has been obtained in mice by direct injection of the peptide into the ARC. PYY3-36 has been shown to exert the inhibition on food intake in a Y2-dependent manner. Although it was proposed that PYY3-36 might exert its anorexigenic effects by activating Y2 receptors on POMC neurons in the ARC, the PYY3-36-induced inhibition of food intake has been shown to still occur in POMC knockout mice.
Given the role of PYY in appetite suppression it is thought that disturbances in PYY release in response to food intake may play a role in the development of obesity. Indeed, in obese humans there is a blunted PYY response following food intake compared to lean humans. Current therapeutic interventions designed to combat obesity involve studies of the efficacy of PYY at suppressing appetite.
Enterostatin
Dietary triglycerides are hydrolyzed by pancreatic lipase in conjunction with the co-activator protein, colipase. The importance of the role colipase in pancreatic lipase mediated triglyceride digestion was demonstrated in colipase knockout mice, who displayed severely reduced fat digestion and fat absorption when fed a high-fat diet.
Studies on the function of colipase demonstrated that the protein is secreted as a procolipase which is activated by trypsin cleavage. Not only is active colipase released from procolipase upon trypsin digestion but an additional peptide of five amino acids (pentpeptide) is also released. This pentapeptide was termed enterostatin and was shown to have no role in fat digestion and absorption.
However, and quite strikingly, it was subsequently shown that enterostatin exhibited appetite regulating properties. The appetite regulating effects of enterostatin are specific to high-fat or fat diets but not towards diets rich in protein or carbohydrate. These effects were demonstrated after intraperitoneal, intraduodenal, and intracerebrovascular injection of enterostatin.
From the intestine, the anorexigenic effects of enterostatin require intact vagal afferent connections to the brain. Based on these experimental observations it has been proposed that enterostatin functions in a negative feedback mechanism to control fat intake. In addition to the negative regulation of fat intake, enterostatin increases energy expenditure through both central and peripheral pathways. The functions of enterostatin involve effects exerted through the opioid systems of the intestines and the brain.
There are three forms of enterostatin found in vertebrates with the Ala-Pro-Gly-Pro-Arg (APGPR) pentapeptide present in humans. The Val-Pro-Asp-Pro-Arg (VPDPR) form is found in rodents and canines, and Val-Pro-Gly-Pro-Arg (VPGPR) is also found in rodents. Common to all three forms of enterostatin is the bridged proline structure, P-X-P. The significance of the P-X-P motif in the various forms of enterostatin is demonstrated by the binding targets of this peptide.
Studies aimed at identifying potential receptors/binding sites for enterostatin demonstrated that membranes from neural tissues possessed both high-affinity and a low-affinity binding sites. The presence of two binding sites with different affinities explains the variable dose-response characteristics observed with enterostatin. At low doses the peptide suppresses fat intake while at high doses it exhibits either no effect or in some experiments high doses stimulate fat intake.
The significance of the opioid system to enterostatin function was discovered during competitive binding studies. The specific high-affinity binding of enterostatin to membranes can be displaced by β-casomorphin1-5 and met-enkephalin. Both of these peptides are known to have high affinity binding to the μ-opioid receptors, MORs. The enkephalins represent two distinct pentapeptides, met-enkephalin (YGGFM) and leu-enkephalin (YGGFL), which are derived from the proenkephalin gene.
The casomorphins are small peptides derived from the proteolytic digestion of the milk protein casein. Bovine β-casomorphin1-5 (derived from β-casein) has the sequence YPFPG which contains the same bridged proline structure, P-X-P, found in enterostatin. The casomorphins are referred to as nutripoids due to their nutritional origin and to the fact that they activate the opioid receptor system in the intestines, specifically the μ-opioid receptors.
Experimental evidence indicates that the ability of enterostatin to regulate fat intake involves the process of thermogenesis (regulated energy consumption for heat generation) and the reward circuitry of the central nervous system. The reward pathways in the brain are complex and involve multiple pathways such as the mesolimbic dopaminergic system and the opioid system. Given that enterostatin binding to neural cell membranes can be competed by a peptide such as β-casomorphin which has high affinity for the μ-opioid receptors, it is not surprising that enterostatin may indeed effect regulation of opioid reward pathways. The μ- and the δ-opioid receptors are known to be involved in the regulation of rewards pathways related to feeding behavior. Agonists of these receptors stimulate feeding whereas, antagonists suppress feeding. Mu-opioid-specific agonists stimulate high-fat feeding and have been shown to block the inhibitory effects of enterostatin.
The role of enterostatin in the regulation of thermogenesis is suggested by the observation that the peptide binds to the β-subunit of F1F0-ATP synthase and this binding is also inhibited by β-casomorphin. However, binding of enterostatin to the ATP synthase subunit is not blocked or competed by addition of μ-opioid receptor-specific agonists. The binding of enterostatin to this protein was unexpected since F1F0-ATP synthase is the mitochondrial complex present in the inner mitochondrial membrane necessary for the generation of ATP during oxidative phosphorylation. In cell culture experiments it has been shown that the binding of enterostatin to the β-subunit of F1F0-ATP synthase results in decreased ATP production, increased thermogenesis, and increased oxygen consumption.
Of potential significance to diabetes is that these effects in pancreatic β-beta cells leads to decreased insulin secretion. Subsequent studies demonstrated that the β-subunit of the F1F0-ATP synthase complex can actually be found in the plasma membranes of hepatocytes associated with ATP hydrolase activity at the cell surface. The presence of this complex in the hepatocyte cell membrane has been shown to be involved in modulating lipoprotein uptake and given that enterostatin can bind to, and inhibit, the plasma membrane activity, the complex is likely also involved in modulating fat intake.
Enterostatin also regulates thermogenesis at the level of the family of uncoupling proteins (UCP). There are several UCP family members in humans and several of the encoding genes are induced under high-fat diet conditions. UCP1 is expressed in brown adipose tissue (BAT), UCP2 is expressed in a number of tissues such as the intestines and adipose tissue, while UCP3 is expressed in skeletal and heart muscle. Only UCP1 is involved in the process of adaptive thermogenesis. When animals are fed a high-fat diet the levels of UCP2 decline in the intestines but increase in adipose tissue. However, if enterostatin is added to the diet there is an observed increase in intestinal UCP2 expression. Although the role of UCP1 in the regulation of thermogenesis s well defined, the exact function of UCP2 is not. Evidence indicates that UCP2 is likely involved in the protection from reactive oxygen species (ROS) generated in conjunction with high-fat diets. Therefore, within the intestines, enterostatin may be playing a protective role by inducing increased levels of UCP2.
A current model for enterostatin function suggests that a high-fat diet leads to increased release of enterostatin into the intestinal lumen. Following its release, enterostatin can bind and activate μ-opioid receptors in the gut and/or a pathway involving F1F0-ATP synthase. Activation of the μ-opioid receptor signaling pathway likely influences the reward circuitry through vagal afferent connections that influence the central opioid signaling pathways. Simultaneously, or alternatively, the regulation of the F1F0-ATP synthase pathway could influences ATP production and overall energy utilization during high-fat feeding. Regardless of the pathway(s) affected by enterostatin, the evidence is clear that this pentapeptide influences fat intake by restricting an overconsumption of fat intake.
Fibroblast Growth Factor 19, FGF19
The fibroblast growth factor family of growth factors and hormones currently consists of 18 members in humans that function by binding to proteins of the FGF receptor (FGFR) family. The FGF proteins are divided into six subfamilies with subfamily 6 containing FGF19, FGF21 and FGF23 (commonly referred to as the FGF19 subfamily). All three members of the FGF19 subfamily function as endocrine hormones. FGF19 subfamily members have weak affinity for heparan sulfate proteoglycans (HSPG) which allows them to escape from the extracellular compartment into circulation enabling them to function as endocrine hormones.
Other members of the FGF family utilize their interactions with HSPG to form high-affinity interactions with FGF receptors (FGFR). However, since the FGF19 subfamily members have low affinity for HSPG they interact with the single-transmembrane containing Klotho proteins to facilitate their interactions with FGFR. There are two related Klotho proteins, αKlotho and βKlotho. FGF19 can interact with both Klotho co-receptor proteins. FGF19 shows preference for activation of FGF receptor 4 (FGFR4) but via its Klotho interactions can also activate FGFR1. FGF21 selectively uses βKlotho as its co-receptor, whereas, FGF23 uses αKlotho.
FGF19 Regulation of and by Bile Acids
FGF19 plays a critical role in the overall processes of the enterohepatic circulation. It carries out this role by regulation of bile acid synthesis in the liver, bile acid delivery to the gallbladder, and regulation of gallbladder function. The ability of intestinal FGF19 to regulate overall bile acid homeostasis is controlled by gut microbiota-derived bile acid derivatives. This process represents an intricate mechanism for feedback regulation of hepatic bile acid synthesis involving communication from the small intestine microbiota to the liver through the actions of FGF19.
When bile acids are secreted by the gallbladder, these compounds are acted upon by gut microbiota followed by re-uptake by intestinal enterocytes. Within the enterocytes the bile acids bind to, and activate, the nuclear receptor FXR. Bile acid activation of FXR leads to the enhanced expression of numerous genes in the enterocytes, with the FGF19 gene being one of these FXR targets. There are at least four FXR-binding sites in the promoter region of the FGF19 gene allowing it to be induced on the order of 300-fold via FXR activation.
Activation of the FGF19 gene results in the protein being secreted into the enterohepatic circulation. When the hormone reaches the liver, it interacts with the βKlotho/FGFR4 complex and triggers activation of a signal transduction cascade. The net result, in the liver, is transcriptional repression of the CYP7A1 gene, which encodes the rate-limiting enzyme in the “classic pathway” of bile acid synthesis. In addition to the regulation of hepatic bile acid synthesis, FGF19 enhances hepatic glycogen synthesis and reduces hepatic glucose output. Both of these latter consequences of FGF19 have significant clinical implications in the control of glucose homeostasis and insulin sensitivity in obesity and type 2 diabetes.
FGF19 Functions in the Hypothalamus
Emerging evidence has implicated FGF19 as a major hormone in the control of normal metabolic homeostasis, both via systemic actions and via actions within the CNS. In mouse models of obesity (diet-induced obesity: DIO), levels of FGF15 (the mouse ortholog of human FGF19) are reduced and in humans, serum levels of FGF19 are found to be significantly reduced in patients with type 2 diabetes. Systemic administration of FGF19 to DIO mice results in weight loss and reverses the associated diabetes.
The FGF receptors are known to be expressed in the brain. FGFR1 expression is abundant within the median eminence of the hypothalamus. Within the hypothalamus, expression of FGFR1 and FGFR4 is evident, particularly on NPY and AgRP expressing neurons. Experiments involving central administration of FGF19 result in improved glucose homeostasis primarily via suppression of NPY and AgRP release from hypothalamic neurons. Thus, although FGF19 clearly exerts a glucose lowering effect via its actions in the liver, functions of the hormone in the hypothalamus contribute to the overall glucose homeostatic functions of FGF19.
The primary central effects of FGF19 result in the normalization of glucose metabolism in obese subjects, enhancement in glucose tolerance and insulin sensitivity in the liver and skeletal muscle, and increased insulin secretion. The ability of FGF19 to suppress NPY and AgRP neuronal activity relies on the FGFR-mediated activation of the ERK1/2 signal transduction cascade. Of potential clinical significance is that the leptin-like effects of FGF19 on glucose homeostasis and insulin sensitivity are effective even in the DIO mouse where leptin effects are lost.
In addition to the metabolic regulation exerted by FGF19 within the brain, central administration of the hormone has been shown to result reductions in food intake. The reduced desire for food intake is what contributes to the weight loss observed in DIO mice following acute systemic administration of FGF19. However, the effects of FGF19 are not so clear-cut with respect to the control of feeding behavior and management of weight. This is likely due to the fact that normal functions of FGF19 include enhanced energy expenditure via actions within anorexigenic (α-MSH) neurons in the hypothalamus. Chronic systemic administration of FGF19 may result in increased food intake secondary to the resultant increased energy expenditure resulting from the actions of FGF19.
Secretin
Secretin is a 27 amino acid peptide that is secreted by enteroendocrine S cells of the duodenum. Secretin is a hormone that belongs to the vasoactive intestinal polypeptide (VIP)/secretin/glucagon/growth hormone-releasing hormone (GHRH) family of peptide hormones. Secretion of secretin is stimulated in response to the lowering of the pH of the lumen of the duodenum in response to the delivery of the gastric contents (gastric emptying) from the stomach.
Secretin is encoded by the SCT gene. The SCT gene is located on chromosome 11p15.5 and is composed of 4 exons that encode a 121 amino acid preproprotein. Secretin itself is encoded by amino acids 28-54 of the preproprotein. Although expression of the SCT gene is primarily found in the small intestines, it is also expressed in the pituitary and pineal glands and at lower levels in the hypothalamus, thalamus, and olfactory lobe.
The primary actions of secretin include induction of bile secretion from the gallbladder into the duodenum, enhanced secretion of bicarbonate and H2O into the duodenum from the pancreas, and increased gastric pepsin secretion. Secretin also inhibits gastric acid secretin and food-intake stimulated secretion of gastrin from the enteroendocrine G cells of the stomach. Secretin also inhibits gastric emptying by decreasing upper small intestinal motility. Another important effect of secretin is the stimulation of insulin secretion from β-cells of the pancreas in response to the ingestion of glucose.
Secretin exerts its effects by binding to a specific receptor, the secretin receptor (SR) which is a GPCR of the class B secretin-like family of receptors. The secretin receptor is coupled to a Gs-type G-protein such that binding of secretin leads to activation of membrane-localized adenylate cyclase which then leads to increased cAMP production.
The secretin receptor is encoded by the SCTR gene. The SCTR gene is located on chromosome 2q14.2 and is composed of 16 exons that encode a 440 amino acid precursor protein.
Within cholangiocytes (biliary epithelial cells that line the biliary tree) the activation of the SCTR results in increased secretion of Cl–, HCO3–, and H2O. These effects are due to PKA-mediated phosphorylation of the CFTR (cystic fibrosis transmembrane conductance regulator), AE2 (anion exchanger 2: encoded by the SLA4A2 gene), and AQP1 (aquaporin 1) transporters. CFTR transports Cl– across the apical membrane into the lumen of the bile ducts. AE2 transports bicarbonate into the biliary lumen in exchange for Cl– uptake. AQP1 transports H2O across the apical membrane into the lumen of the bile ducts. The effects of secretin receptor activation on Cl–, HCO3–, and H2O secretion from cholangiocytes is the process exerted by the hormone of pancreatic ductal cells.
In addition to its primary actions, secretin exerts effects on energy homeostasis, lipolysis in white adipose tissue (WAT), and thermogenesis in brown adipose tissue (BAT). Within WAT, the activation of the secretin receptor leads to increased PKA which phosphorylates numerous substrates involved in the regulation of adipocyte lipolysis. Within BAT, these effects on lipolysis result in increased thermogenesis resulting from increased mitochondrial fatty acid β-oxidation.