
Last Updated: July 3, 2026
Introduction to Fatty Acid Oxidation
Humans store fatty acids, without limit, primarily in adipose tissue, as a means to provide readily available energy from their oxidation during periods of fasting. Fatty acids, in most tissues, are stored the form of triglycerides (TG; or triacylglycerides, TAG). The process of lipolysis refers to the breakdown of triglycerides and the release of the stored fatty acids. Under conditions that promote lipolysis the released fatty acids are oxidized within the mitochondria and the process is coupled to the generation of ATP. Byproducts of fatty acid oxidation, important during periods of prolonged fasting, are the ketone bodies, acetoacetate and β-hydroxybutyrate. The ketones are important sources of energy for the brain in particular, but also the heart and skeletal muscle.
Lipid Digestion
Utilization of dietary lipids requires that they first be absorbed through the intestine. As these molecules are oils they would essentially be insoluble in the aqueous intestinal environment. Dietary lipid digestion and absorption are also covered in the Digestion and Digestive Processes page.
Solubilization (emulsification) of dietary lipid is accomplished, initially, via the agitation action as food passes through the stomach and then the emulsification process continues within the intestine via interaction with the bile salts that are synthesized in the liver and secreted from the gallbladder.
Dietary lipids, in the form of triglycerides, phospholipids, and cholesterol, are digested by various lipases. The bulk of dietary lipids in the human diet are in the form of triglycerides. The lipases found in the gastrointestinal tract include one originally identified as lingual lipase (secreted by acinar cells of von Ebner glands of the tongue), gastric lipase (encoded by the LIPF gene and secreted by Chief cells of the stomach), pancreatic lipase (PNLIP gene), and pancreatic lipase-related protein 2 (PNLIPRP2 gene). These enzymes generate free fatty acids and a mixture of mono- and diglycerides from dietary triglycerides.
Lingual lipase and gastric lipase are both derived from the lipase F, gastric (LIPF) gene and together constitute the acid lipases. The acidic lipases function essentially only in the acidic environment of the stomach. However, evidence suggests that lingual lipase functions within the mouth allowing for the ability to taste non-esterified fatty acids (NEFA). The acid lipases are distinct from pancreatic lipases in that they do not require a lipid-bile acid interface for activity nor do they require the presence of the accessory protein, colipase.
Pancreatic lipases, on the other hand, only function in the neutral pH environment generated in the small intestine by the secretion of pancreatic bicarbonate (HCO3–). Also, pancreatic lipases require the presence of colipase and a lipid-bile acid interface for their activity. The role of colipase in pancreatic lipase function is to anchor the lipase to the surface of an emulsified lipid droplet and to prevent it from being removed by bile salts. Pancreatic lipase degrades triglycerides at the sn-1 (C1) and sn-3 (C3) positions sequentially to generate 1,2-diacylglycerides (DAG) and 2-monoacylglycerides (MAG). Phospholipids are degraded at the sn-2 (C2) position by pancreatic phospholipase A2 releasing a free fatty acid and the lysophospholipid.
Table of the Major Lipases of Lipid Digestion and Plasma Lipid Metabolism
| Lipase Name | Gene and Structure | Primary Source | Functions / Comments |
| carboxy ester lipase, CEL | CEL: 9q34.13; 11 exons | pancreas | encoded precursor protein is 756 amino acids; hydrolyzes cholesteryl esters, tri-, di-, and monoglycerides, phospholipids, lysophospholipids, and ceramides; predominant site of expression is pancreas, also expressed, albeit at extremely low levels, in liver, intestine, and macrophages; participates in chylomicron assembly in intestinal enterocytes and their secretion |
| lysosomal acid lipase, LAL | LIPA: 10q23.31: 10 exons | lysosomes | enzyme is also called cholesterol ester hydrolase; three alternatively spliced mRNAs encode two distinct protein isoforms (399 and 283 amino acid precursor proteins); hydrolyzes cholesteryl esters and triglycerides |
| hepatic lipase | LIPC: 15q21.3; 15 exons | hepatocytes | encoded precursor protein is 499 amino acids; maturation requires activity of lipase maturation factor 1 (LMF1); protein is secreted from hepatocytes and binds to heparan-sulfate proteoglycans (HSPG) on surface of hepatocytes and endothelial cells; released to plasma via interactions involving apoA-II-enriched HDL; major substrates are IDL and triglyceride-rich HDL; activity is inhibited by angiopoietin-like protein 3 (ANGPTL3) |
| hormone sensitive lipase, HSL | LIPE: 19q13.2; 15 exons | adipocytes; testis | initially described as an epinephrine- and glucagon-responsive gene encoding an adipose tissue triglyceride lipase; alternative 5′ exon utilization results in a short form (775 amino acids) and a long form (1076 amino acids) of the enzyme; the short form is exclusive to adipose tissue, the long form was originally identified as a testis-specific isoform; pancreatic β-cells utilize an alternative translational start site generating a protein of 818 amino acids; primary substrates in adipose tissue are diglycerides; known to hydrolyze triglycerides and monoglycerides (albeit inefficiently), and cholesteryl esters, as well as other lipid and water soluble substrates; activities detailed in the next section |
| gastric/lingual lipase | LIPF: 10q23.31; 11 exons | gastric Chief cells | four alternatively spliced mRNAs; four protein isoforms; acidic lipase responsible for hydrolysis of dietary triglycerides within the stomach |
| endothelial lipase | LIPG: 18q21.1; 12 exons | endothelial cells | two alternatively spliced mRNAs encoding two protein isoforms (500 and 426 amino acid precursor proteins); remains associated with endothelial cells; maturation requires activity of lipase maturation factor 1 (LMF1); functions primarily as a phospholipase A1 (PLA1) type hydrolase with principal substrates being phosphatidylcholines; participates in HDL metabolism; activity inhibited by the hepatocyte-derived plasma protein, angiopoietin-like protein 3 (ANGPTL3) |
| lipoprotein lipase | LPL: 8p21.3; 10 exons | adipocytes, cardiac myocytes and skeletal muscle cells | 475 amino acid precursor protein; primarily expressed in adipose tissue, skeletal muscle, and cardiac capillary endothelial cells; functions as a homodimeric enzyme; synthesized in cells of the tissues via the ER secretory pathway; transferred across the subendothelial spaces to capillary endothelial cells through interaction with endothelial cell glycosylphosphatidylinositol-anchored HDL binding protein 1 (GPIHBP1); LPL remains attached to endothelial cell surfaces through its interaction with GPIHBP1; primary substrates are triglyceride-rich chylomicrons and VLDL; activity inhibited by the hepatocyte-derived plasma protein, angiopoietin-like protein 3 (ANGPTL3) and by ANGPTL4 |
| monoglyceride lipase | MGLL: 3q21.3; 12 exons | ten alternatively spliced mRNAs encoding nine distinct protein isoforms; in addition to hydrolyzing monoglycerides, this serine hydrolase also metabolizes the endocannabinoid, 2-arachidonoylglycerol (2-AG); activities detailed in the next section | |
| pancreatic lipase | PNLIP: 10q25.3; 13 exons | pancreas | 465 amino acid precursor protein; hydrolyzes dietary triglycerides contained in bile acid micelles within the small intestine; requires colipase, which is secreted by the pancreas, for activity |
| pancreatic lipase related protein 2 | PNLIPRP2: 10q25.3; 13 exons | pancreas | due to allelic variants there are coding and non-coding version of this gene; the coding version produces a 470 amino acid precursor protein; hydrolyzes dietary triglycerides, phospholipids, and galactolipids (from plants); human genome has two additional related genes PNLIPRP1 and PNLPRP3; protein encoded by PNLIPRP1 does not possess lipase activity |
Following absorption of the products of pancreatic lipase by the intestinal enterocytes, the re-synthesis of triglycerides occurs. The triglycerides are then solubilized in lipoprotein complexes (complexes of lipid and protein) called chylomicrons. A chylomicron contains lipid droplets surrounded by the more polar phospholipids and finally a layer of proteins. Triglycerides synthesized in the liver are packaged into VLDL and released into the blood directly. Chylomicrons from the intestine are first released into the lymphatic system and then into the blood at the left subclavian vein for delivery of dietary fatty acids to the various tissues for storage (adipose tissue) or production of energy through oxidation (primarily skeletal muscle and cardiac muscle).
There are exceptions to the packaging of dietary lipids into chylomicrons within the intestinal enterocytes. The exceptions are short-chain (less than 6 carbon atoms) and medium-chain (6 to 12 carbons) fatty acids, SCFA and MCFA, respectively. Although the bulk of dietary fatty acids are long-chain (14 to 22 carbon atoms) fatty acids in triglycerides (abbreviated LCT for long-chain triglycerides) some triglycerides contain MCFA, abbreviated MCT (medium-chain triglycerides). Naturally occurring dietary MCT are abundant in coconut oil and palm kernel oil. Coconut oil and palm kernel oil are both around 50%–60% MCFA with the bulk of the fatty acid being lauric acid, C12:0 (also identified as dodecanoic acid).
When MCT are hydrolyzed by pancreatic lipases the released fatty acids easily diffuse into the intestinal enterocytes and are passed directly to the portal circulation, bypassing the lymphatic system. Most, if not all, of the short-chain fatty acids (butyrate, propionate, and acetate) that are absorbed from the intestines are produced through the action of gut microbiota (bacteria) metabolism of soluble and insoluble fiber in the diet and these short-chain fatty acids also directly enter the blood from the intestinal enterocytes. MCFA freely enter hepatocytes and subsequently the mitochondria where they are oxidized to acetyl-CoA regardless of the level of ATP. The metabolic benefits of a diet rich in MCT is discussed in the Role of Medium-Chain Triglycerides (MCT) is Overall Metabolic Regulation section below.
The triglyceride components of VLDL and chylomicrons are hydrolyzed to free fatty acids and glycerol in the capillaries of tissues such as adipose tissue, heart, and skeletal muscle by the actions of lipoprotein lipase (LPL) and in the liver through the actions of hepatic triglyceride lipase [HTGL; encoded by the lipase C, hepatic (LIPC) gene]. The free fatty acids are then absorbed by the cells and the glycerol is returned, via the blood, to the liver (principal site) and kidneys. Within the liver the glycerol can then be converted to the glycolytic intermediate dihydroxyacetone phosphate (DHAP) and incorporated into the gluconeogenesis pathway or phosphorylated by glycerol kinase to glycerol-3-phosphate for reuse in triglyceride synthesis.
The classification of blood lipoprotein complexes is distinguished based upon the density of the different lipoproteins. As lipid is less dense than protein, the lower the density of lipoprotein the less protein there is in the particle.
Role of Medium-Chain Triglycerides (MCT) is Overall Metabolic Regulation
Mobilization of Fat Stores
The primary sources of fatty acids for oxidation are dietary and mobilization from cellular stores. Fatty acids from the diet are absorbed from the duodenum, packaged into lipoprotein particles called chylomicrons within intestinal enterocytes and then delivered to cells of the body via transport in the blood. Fatty acids are stored in the form of triglycerides within all cells but predominantly within adipose tissue.
In response to energy demands, the fatty acids of stored triglycerides can be mobilized for use by peripheral tissues. The release of metabolic energy, in the form of fatty acids, is controlled by a complex series of interrelated cascades that result in the activation of triglyceride hydrolysis.
The primary intracellular lipases are adipose triglyceride lipase (ATGL, also called desnutrin), hormone-sensitive lipase (HSL), and lipase A (LIPA; also called lysosomal acid lipase, LAL). LIPA is the most important lipase involved in lysosomal lipid metabolism as evidenced by the fact that LIPA deficiency results in the significant accumulation of cholesteryl esters in tissues such as the spleen and liver. LIPA deficiency is commonly called Wolman disease or cholesterol ester storage disease.
Adipose Triglyceride Lipase: ATGL
ATGL/desnutrin (one group that identified an activity that is ATGL originally called that enzyme iPLA2ζ) belongs to the family of patatin domain-containing proteins that consists of nine members in humans. The patatin domain was originally discovered in lipid hydrolases of certain plants and named after the most abundant protein of the potato tuber, patatin. Because some members of the family act as phospholipases, the proteins were originally called patatin-like phospholipase domain-containing protein A1 to A9 (PNPLA1–PNPLA9). ATGL (designated PNPLA2 in the patatin domain nomenclature) preferentially hydrolyzes long-chain fatty acids esterified to the sn-1 and sn-2 position of triglycerides, but does not hydrolyze fatty acids from the sn-3 position. ATGL also possesses PLA2 activity on the substrate: [1,2‑dilinoleoyl]‑phosphatidylcholine, and contributes to the mobilization of retinyl esters in hepatic stellate cells.
The human gene encoding ATGL is PNPLA2. The PNPLA2 gene is located on chromosome 11p15.5 and is composed of 10 exons encoding a 504 amino acids protein. Expression of the PNPLA2 gene is found in numerous tissues but the highest levels, on the order of 100 fold higher than any other tissue, are seen in adipose tissue.
In analogy with patatin, the active site of ATGL contains an unusual catalytic dyad (Ser47 and Asp166) within the patatin domain in the N-terminal half of the enzyme. The C-terminal half of the enzyme comprises the regulatory functions and also contains a predicted hydrophobic region involved in lipid droplet (LD) binding.
ATGL expression and enzyme activity are both under complex regulation. Expression of ATGL is induced by peroxisome proliferator-activated receptor (PPAR) agonists, glucocorticoids, and fasting. In addition, activation of the FoxO1 transcription factor by SIRT1-mediated deacetylation activates lipolysis by increasing ATGL expression. Conversely, silencing of SIRT1 has the opposite effect. Increased insulin release and food intake both result in decreased expression of ATGL. Reductions in ATGL expression have also been shown to be associated with mTOR complex 1 (mTORC1)-dependent signaling.
The level of lipase activity of ATGL (as well as HSL) does not always correlate to the level of expression of the gene. For example, TNFα activity reduces both ATGL and HSL expression at the level of the gene but induces increased lipase activity of pre-existing enzyme resulting in fatty acid and glycerol release from triglycerides. Similarly, the use of non-selective beta blockers (e.g. isoproterenol) results in the same type of gene inhibition with enzyme activation. The discrepancy between ATGL and HSL mRNA levels and enzyme activities is the result of extensive post-translational regulation of both ATGL and HSL.
There are two serine residues in ATGL that are subject to phosphorylation. Unlike phosphorylation of HSL (described below), ATGL phosphorylation is not PKA-dependent. In the mouse, it has been shown that AMPK phosphorylates ATGL resulting in increased lipase activity. However, it is important to point out that there remains a level of controversy as to the role of AMPK in the regulation of the activity of ATGL since induction, inhibition, and no effects results have been published.
Phosphorylation of ATGL is also carried out by Ca2+–calmodulin (CaM)-dependent protein kinase II, CaMKII. Within the liver this CAMKII mediated phosphorylation results in increased ATGL activity which, in turn, leads to increased rates of intrahepatic lipolysis and fatty acid oxidation. CaMKII is actually a family of four serine/threonine kinases, CaMKIIα, CaMKIIβ, CaMKIIγ, and CaMKIIδ, each of which is encoded by a separate gene.
The activity of CaMKII, which is regulated by cytosolic Ca2+, can be increased under conditions of abnormal mitochondrial calcium uptake. The major inner mitochondrial membrane (IMM) localized transporter responsible for mitochondrial uptake of calcium is the MCU (mitochondrial calcium uniporter). When mitochondrial calcium uptake is impaired in the liver there is increased cytosolic Ca2+ which results in increased activity of CaMKII. The increased phosphorylation of ATGL by CaMKII, under these conditions, leads to increased fatty acid oxidation and secondarily to increased gluconeogenesis.
In addition to phosphorylation regulating ATGL activity, the enzyme requires a coactivator protein for full lipase activity. The gene encoding this coactivator was originally identified as Comparative Gene Identification-58, CGI-58. The term comparative gene identification relates to the use of computational methods to identify protein sequences highly conserved across various species and CGI-58 was originally discovered in a screen comparing the proteomes of humans and C. elegans.
The official name for CGI-58 is α/β hydrolase domain-containing protein-5, lysophosphatidic acid acyltransferase (also identified as 1-acylglycerol-3-phosphate O-acyltransferase) owing to the presence of an α/β hydrolase domain commonly found in esterases, thioesterases, and lipases. The enzyme is encoded by the ABHD5 gene.
The ABHD5 gene is located on chromosome 3p21.33 and is composed of 10 exons that generate four alternatively spliced mRNAs that collectively encode three distinct protein isoforms.
It is unlikely that the ABHD5 protein possesses hydrolase activity because an asparagine residue is found in the catalytic domain where other hydrolases have a nucleophilic serine residue that is required for enzymatic activity. In addition to functioning as an ATGL coactivator, ABHD5 has also been shown to possess enzymatic activity as an acyl-CoA-dependent acylglycerol-3-phosphate acyltransferase (AGPAT). The physiological significance of this activity is currently unknown but it may be involved in the regulation of phosphatidic acid or lysophosphatidic acid signaling.
The activity of ATGL is also regulated by its state of acylation. In hepatocytes, ATGL undergoes S-acylation by the protein acyltransferase identified as ZDHHC11. ZDHHC11 belongs to the family of protein acyltransferases (PAT) that are members of the Asp-His-His-Cys-containing protein acyltransferase family, identified as DHHC-PAT. Due to the DHHC motif forming a zinc-finger domain the genes encoding these enzymes are termed zinc finger DHHC type containing (ZDHHC) with a number designating the specific gene. Currently 23 human ZDHHC genes have been identified and characterized, ZDHHC1–ZDHHC9, ZDHHC11–ZDHHC24 (there is no ZDHHC10 gene). The ZDHHC11-mediated S-palmitoylation of ATGL is required for its association with lipid droplets in order for triglyceride breakdown to take place. Indeed, the S-acylation of ATGL is required not only for lipid droplet localization but also to activate the lipase activity of the enzyme.
Additional proteins associated with lipid droplets (LD) in adipocytes participate in the ABHD5-mediated regulation of ATGL. In resting adipocytes of both WAT and BAT, the LD protein perilipin-1 interacts with ABHD5, preventing its binding to and, induction of ATGL. Following β-adrenergic stimulation of WAT, PKA phosphorylates perilipin-1 at multiple sites resulting in the release of ABHD5 which in turn, binds to and stimulates ATGL. This demonstrates that β-adrenergic stimulation of PKA induces ATGL activity, not by direct phosphorylation of ATGL itself, but through phosphorylation of perilipin-1. This model of ATGL regulation is evident from frameshift mutants that have been identified in the human perilipin-1 gene. These mutations are identified as V398fs and L404fs indicating that the frame shift occurs at valine 398 and leucine 404, respectively. Each of these mutant perilipin-1 proteins fails to bind ABHD5, resulting in unrestrained lipolysis, partial lipodystrophy, hypertriglyceridemia, and insulin resistance.
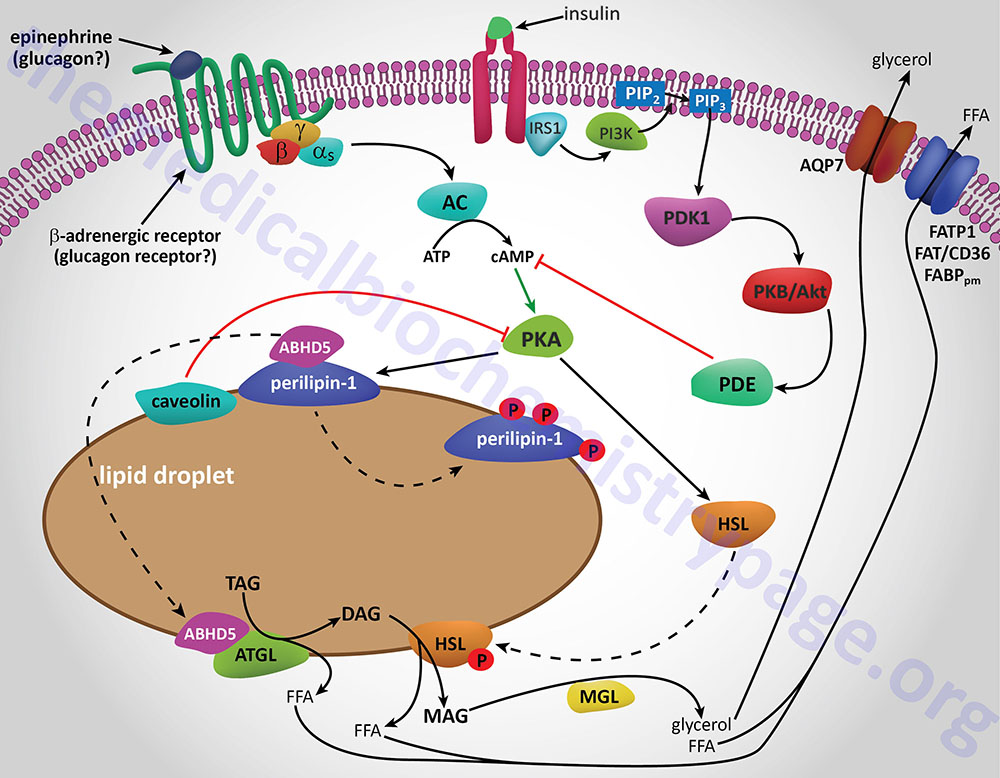
In non-adipose tissues with high rates of triglyceride hydrolysis, such as skeletal muscle and liver, regulation of ATGL activity occurs via a mechanism distinct from that in adipose tissue. In these tissues, perilipin-1 is replaced by perilipin-5. During fasting, perilipin-5 recruits both ATGL and ABHD5 to LD by direct binding of the enzyme and its coactivator. Data indicates that perilipin-5 is involved in the interaction of LD with mitochondria and thereby inhibits ATGL-mediated triglyceride hydrolysis.
Recently, a specific protein inhibitor for ATGL was isolated from white blood cells, specifically mononuclear cells. This protein was originally identified as being involved in the regulation of the G0 to G1 transition of the cell cycle. This protein was, therefore, called G0G1 switch protein 2 (encoded by the G0S2 gene). The protein is found in numerous tissues, with highest concentrations in adipose tissue and liver.
The G0S2 gene is located on chromosome 1q32.2 and is composed of 2 exons that encode a protein of 103 amino acids.
In adipose tissue G0S2 expression is very low during fasting but increases after feeding. Conversely, fasting or PPARα-agonists increase hepatic G0S2 expression. The protein has been shown to localize to LD, cytoplasm, ER, and mitochondria. These different subcellular localizations likely relate to multiple functions for G0S2 in regulating lipolysis, the cell cycle, and, possibly, apoptosis via its ability to interact with the mitochondrial antiapoptotic factor Bcl-2. With respect to ATGL regulation, the binding of the enzyme to LD and subsequent regulation is dependent on a physical interaction between the N-terminal region of G0S2 and the patatin domain of ATGL.
The delivery of ATGL to LD requires functional vesicular transport. When essential protein components of the transport machinery are defective or missing, such as ADP-ribosylation factor 1 (ARF1), small GTP-binding protein 1 (SAR1), the guanine-nucleotide exchange factor Golgi-Brefeldin A resistance factor (GBF1), or the coatamer proteins coat-complex I (COPI) and COPII, ATGL translocation to LD is blocked and the enzyme remains associated with the ER.
The association of ATGL with lipid droplets and the recruitment of these complexes to mitochondria can be altered under conditions of glucose deprivation, particularly in the liver. In glucose deficient hepatocytes, the glycolytic enzyme PFK-1 (encoded by the PFKL gene) is phosphorylated. When phosphorylated, PFK-1 interacts with the lipid droplet associated protein, perilipin-2 (PLIN2). The interaction of PFK-1 and perilipin-2 leads to PFK-1 functioning as a protein kinase where it phosphorylates perilipin-2. The phosphorylation of perilipin-2 promotes the interaction of perilipin-2 with carnitine palmitoyltransferase 1A (CPT1A) and the recruitment of ATGL. The consequence of these interactions is the tethering of lipid droplets to the outer mitochondrial membrane. The tethering of lipid droplets to the mitochondria enhances energy generation through the increased availability of fatty acids for mitochondrial β-oxidation.
Disorders Associated with ATGL and ABHD5
Mutations in the PNPLA2 gene result in the lipid storage myopathy (LSM) identified as neutral lipid storage disease with myopathy, NLSDM. Mutations in the ABHD5 gene result in the lipid storage myopathy (LSM) identified as neutral lipid storage disease with ichthyosis, NLSDI, also known as Chanarin-Dorfman syndrome. NLSDI is associated with massive triglyceride storage and defective long-chain fatty acid β-oxidation. The characteristic features of NLSDI are dry, scaly skin evident at birth as well as a progressive fatty infiltration of the liver.
Hormone-Sensitive Lipase: HSL
A landmark study published in 1964 demonstrated that a lipolytic activity present in adipose tissue was induced by hormonal stimulation. This work described the isolation and characterization of both HSL and monoglyceride lipase (MGL). This original study demonstrated that HSL had a higher level of activity as a diglyceride hydrolase than as a triglyceride hydrolase. Nevertheless, it became dogma that HSL was rate-limiting for the catabolism of fat stores in adipose and many non-adipose tissues.
However, when HSL-deficient mice were produced and shown to efficiently hydrolyze triglycerides the model began to emerge demonstrating ATGL, and not HSL, to be rate-limiting for adipose tissue triglyceride hydrolysis. HSL-deficient mice do not accumulate triglycerides in either adipose or non-adipose tissues, but they do accumulate large amounts of diglycerides in many tissues. This indicated for the first time that HSL was more important as a diglyceride hydrolase than a triglyceride hydrolase. It is now accepted that ATGL is responsible for the initial step of lipolysis in human adipocytes, and that HSL is rate-limiting for the catabolism of diglycerides. HSL not only hydrolyzes diglycerides but is also active at hydrolyzing ester bonds of many other lipids including triglycerides, monoglycerides, cholesteryl esters, retinyl esters, and short-chain carbonic acid esters.
The gene encoding HSL (official symbol: LIPE for lipase E, hormone sensitive) is located on chromosome 19q13.2 and is composed of 15 exons. Alternative exon usage results in tissue-specific differences in mRNA and protein size. In adipose tissue the HSL protein is composed of 775 amino acids, whereas the testicular form is composed of 1,076 amino acids. The larger HSL isoform found in testis is due to the inclusion of a novel exon located 16 kb upstream of the exons that encode the adipose tissue (and other tissues) form of HSL.
The expression profile of HSL, within adipocytes, essentially mirrors that of ATGL. Highest mRNA and protein concentrations are found in WAT and BAT with low levels of expression found in muscle, testis, steroidogenic tissues, and pancreatic islets as well as several other tissues.
Functional studies on the enzyme have identified an N-terminal lipid-binding region, the α/β hydrolase fold domain including the catalytic triad, and the regulatory module containing all known phosphorylation sites important for regulation of enzyme activity.
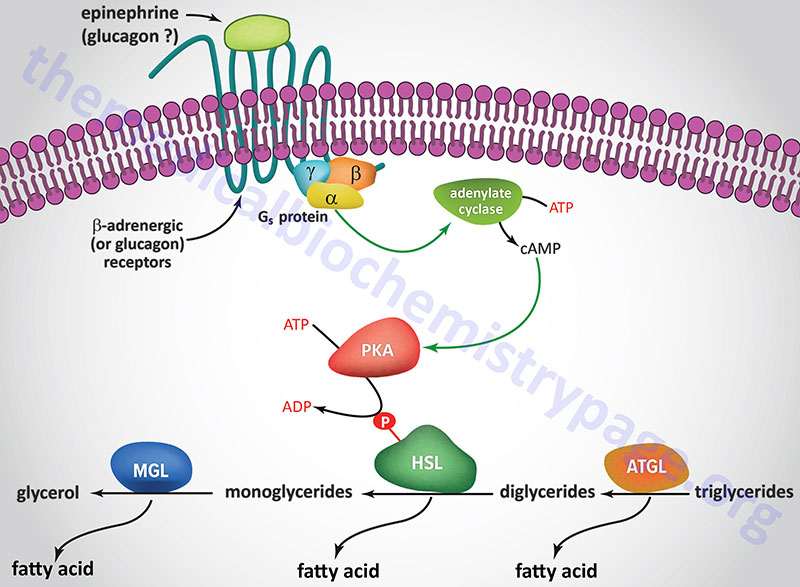
HSL and ATGL share many regulatory similarities yet the mechanisms of the regulatory processes differ markedly between the two enzymes. In adipose tissue, HSL enzyme activity is strongly induced by β-adrenergic stimulation, conversely insulin has a strong inhibitory effect. While β-adrenergic stimulation regulates ATGL primarily via recruitment of the coactivator ABHD5, HSL is a major target for PKA-mediated phosphorylation. Additional kinases, including AMPK, extracellular signal-regulated kinase (ERK), glycogen synthase kinase 3 (GSK3), and Ca2+/calmodulin-dependent kinase I (CaMKI), also phosphorylate HSL to modulate the activity of the enzyme. HSL has at least five potential phosphorylation sites, of which S660 and S663 appear to be particularly important for hydrolytic activity.
Phosphorylation of HSL affects enzyme activity only moderately resulting in an approximate 2-fold increase in hydrolytic activity. For full activation, HSL must gain access to LD, which, in adipose tissue, is mediated by perilipin-1. In addition to phosphorylating HSL, PKA also phosphorylates perilipin-1 on six consensus serine residues. The result of these phosphorylations is the binding of HSL to the N-terminal region of perilipin-1. This protein-protein interaction is the means by which HSL gains access to LD. The net effect, of HSL-phosphorylation and enzyme translocation to LD, coupled with ATGL activation by ABHD5, leads to a more than 100-fold increase in triglyceride hydrolysis in adipocytes.
Additional factors modulate the activation of HSL and ATGL. One such factor is nuclear receptor-interacting protein 140 (RIP140; encoded by the NRIP1 gene) which induces lipolysis by binding to perilipin-1, increasing HSL translocation to LD, and activating ATGL via ABHD5 dissociation from perilipin-1. In non-adipose tissues, such as skeletal muscle, HSL is activated by phosphorylation in response to epinephrine (β-adrenergic receptor-mediated activation of PKA) and muscle contraction (calcium release from sarcoplasmic reticulum). Since skeletal muscles lack perilipin-1 it has not yet been determined which alternative mechanisms regulate HSL access to LD.
Insulin-mediated deactivation of lipolysis is associated with transcriptional downregulation of ATGL and HSL expression. Additionally, insulin signaling results in phosphorylation and activation of various phosphodiesterase (PDE) isoforms (predominantly PDE3B) by PKB/AKT leading to PDE-catalyzed hydrolysis of cAMP which in turn results in reduced activation of PKA. These actions turn off lipolysis by preventing phosphorylation of both HSL and perilipin-1, activation and translocation of HSL, and activation of ATGL by ABHD5. In addition to its peripheral action, insulin also functions within the sympathetic nervous system to inhibit lipolysis in WAT. Increased insulin levels in the brain inhibit HSL and perilipin phosphorylation which results in reduced HSL and ATGL activities.
Nuclear-Localized HSL
In addition to its role as a lipase, localized to the cytosol, hormone-sensitive lipase (HSL) has been found to exert effects within the nucleus of adipocytes that are independent of its lipase activity. The nuclear localization of HSL occurs through its interaction with the TGF-β signal transduction protein, SMAD3. Indeed, the nuclear functions of HSL in adipocytes are related to normal TGF-β signaling which includes regulation of mitochondrial metabolism as well as remodeling of the extracellular matrix.
Within the nucleus HSL contributes to the regulation of gene expression. The mechanism by which HSL exerts effects on gene expression is by its interaction with numerous proteins involved in mRNA processing. HSL has been shown to interact with several RNA polymerase II subunits as well as the C-terminal domain (CTD) of RNA polymerase II itself. Additional proteins, to which HSL interacts, are involved in mRNA processing including splicing factor proline and glutamine rich (SFPQ) and non-POU domain-containing octamer binding protein (NONO). Both of these proteins are multifunctional regulators of gene expression. The nuclear effects of HSL have been shown to function independently of its lipase activity as demonstrated in experiments utilizing a catalytically inactive form of HSL.
One of the targets of HSL-mediated nuclear regulation of gene expression is PPARγ coactivator 1-α (PGC-1α) which is encoded by the PPARGC1A gene. When HSL is in the nucleus, the levels of PGC-1α mRNA, as well as the pre-mRNA, are reduced.
In order to regulate the nuclear activity of HSL, it is exported back out of the nucleus. The signal for nuclear export is phosphorylation and this is carried out by cAMP-dependent protein kinase, PKA. As discussed in greater detail, the catalytic subunit of PKA can be localized to the nucleus and this is the situation in adipocytes.
Monoglyceride lipase: MGL
MGL is considered to be the rate-limiting enzyme for the breakdown of monoglycerides that are the result of both extracellular and intracellular lipolysis pathways. The extracellular generation of monoglycerides is the result of the action of endothelial cell lipoprotein lipase (LPL) on lipoprotein particle-associated triglycerides. Intracellular hydrolysis of triglycerides by ATGL and HSL, as well as intracellular phospholipid hydrolysis by phospholipase C (PLC) and membrane-associated diglyceride lipase α and β results in the generation of MGL substrates.
The gene (MGLL) that encodes MGL is located on chromosome 3q21.3 and is composed of 12 exons that generate ten alternatively spliced mRNAs, that collectively encode nine distinct protein isoforms. MGL has been shown to localize to lipid droplets (LD), cell membranes, and the cytosol. The enzyme is ubiquitously expressed with highest levels of expression in adipose tissue. MGL shares homology with esterases, lysophospholipases, and haloperoxidases. The enzyme contains a consensus GXSXG motif within a catalytic triad that is typical of lipases and esterases.
MGL is critically important for efficient degradation of monoglycerides since it has been shown in mouse models that lack of MGL impairs lipolysis and is associated with increased MG levels in adipose and non-adipose tissues alike. MGL has received particular attention in recent years due to the discovery that the enzyme is responsible for the inactivation of 2-arachidonoylglycerol (2-AG) which is an endogenous cannabinoid monoglyceride (endocannabinoid).
For more information on the activities of ATGL, HSL and other lipases regulating triglyceride levels in adipocytes visit the Adipose Tissue: Not Just Fat page.
In contrast to the hormonal activation of adenylate cyclase and (subsequently) hormone-sensitive lipase in adipocytes, the mobilization of fat from adipose tissue is inhibited by numerous stimuli. The most significant inhibition is that exerted upon adenylate cyclase by insulin. When an individual is in the well fed state, insulin released from the pancreas prevents the inappropriate mobilization of stored fat. Instead, any excess fat and carbohydrate are incorporated into the triglyceride pool within adipose tissue.
Cellular Uptake of Fatty Acids
When fatty acids are released from adipose tissue stores they enter the circulation as free fatty acids (FFAs) and are bound to albumin for transport to peripheral tissues. When the fatty acid–albumin complexes interact with cell surfaces the dissociation of the fatty acid from albumin represents the first step of the cellular uptake process. Uptake of fatty acids by cells involves membrane proteins with high affinity for fatty acids. As opposed to long-chain fatty acids (LCFA) which are bound to albumin in the blood, medium-chain fatty acids (MCFA) generally do not interact with albumin with much affinity, except for lauric acid (C12:0) which binds with an affinity nearly equivalent to that of LCFA.
There are several members of the fatty acid receptor family including fatty acid translocase (FAT/CD36), plasma membrane-associated fatty acid-binding protein (FABPpm), and several fatty acid transport proteins (FATP). The FATP are a family of at least six fatty acid transport proteins (FATP1–FATP6) that are also members of the solute carrier family of transporters. The FATP facilitate the uptake of very long-chain (VLCFA) and long-chain fatty acids (LCFA).
FAT/CD36 is encoded by the CD36 gene. The CD36 encoded protein was originally identified a platelet receptor for thrombospondin and mutations in the CD36 gene are associated with platelet glycoprotein deficiency. The CD36 encoded protein is also known as scavenger receptor B3 (SCARB3 or SR-B3). With respect to fatty acid metabolism, FAT/CD36 is the major protein involved in the uptake of fatty acids by adipocytes, skeletal muscle myocytes, and heart cardiomyocytes. The localization of FAT/CD36 to the plasma membrane is facilitated by S-palmitoylation in the Golgi apparatus. The S-palmitoylation of FAT/CD36 is catalyzed by a member of the DHCC (Asp-His-Cys-Cys) domain-containing palmitoyltransferase encoded by the ZDHCC5 (zinc finger DHHC-type palmitoyltransferase 5) gene.
Within the plasma membrane FAT/CD36 is associated with the tyrosine kinase, LYN (LCK/YES-related novel tyrosine kinase). When fatty acids interact with FAT/CD36, the kinase activity of LYN is activated which then phosphorylates the ZDHHC5 encoded enzyme preventing palmitoylation of FAT/CD36. Under these conditions FAT/CD36 is depalmitoylated, a reaction catalyzed by the LYPLA1 (lysophospholipase 1) gene. The depalmitoylation of FAT/CD36 triggers endocytosis resulting in the internalization of the fatty acids.
Table of Mammalian Fatty Acid Transporters: Acyl-CoA Synthetases
| Fat Transporter | Comments |
| FAT/CD36 | fatty acid translocase; FAT is also known as CD36 which is a member of the scavenger receptor class (class B scavenger receptors) of receptors that bind lipids and lipoproteins of the LDL family; is a major regulator of ferroptosis via its interactions with the GPCR, GPR56 (encoded by the ADGRG1 gene); the CD36 gene is located on chromosome 7q21.11 and composed of 22 exons that generate 15 alternatively spliced mRNAs that encode six distinct isoforms of the protein |
| FABPpm | plasma membrane-associated fatty acid-binding protein; originally characterized as a plasma membrane-associated fatty acid transporter this protein was later demonstrated to be the mitochondrial isoform of glutamate-oxalate transaminase (gene symbol: GOT2); gene located on chromosome 16q21 and is composed of 10 exons that generate two alternatively spliced mRNAs encoding precursor proteins of 430 amino acids (isoform 1) and 387 amino acids (isoform 2) |
| FATP1 | FATP1 (fatty acid transport protein 1) encoded by SLC27A1 gene; FATP1 is also known as acyl-CoA synthetase very long-chain family, member 5 (ACSVL5); highest levels of expression in adipose tissue, skeletal and heart muscle; the SLC27A1 gene is located on chromosome 19p13.11 and is composed of 16 exons encoding a 646 amino acid protein |
| FATP2 | FATP2 (fatty acid transport protein 2) encoded by SLC27A2 gene; FATP2 is also known as acyl-CoA synthetase very long-chain family, member 1 (ACSVL1) as well as very long-chain acyl-CoA synthetase (VLCS); highest levels of expression in liver and kidney; present in peroxisome and microsomal membranes; the SLC27A2 gene is located on chromosome 15q21.2 composed of 10 exons that generate two alternatively spliced mRNAs that encode precursor proteins of 620 amino acids (isoform 1) and 567 amino acids (isoform 2) |
| FATP3 | FATP3 (fatty acid transport protein 3) encoded by SLC27A3 gene; FATP3 is also known as acyl-CoA synthetase very long-chain family, member 3 (ACSVL3); the SLC27A3 gene is located on chromosome 1q21.3 and is composed of 10 exons that generate two alternatively spliced mRNAs that encode precursor proteins of 683 amino acids (isoform 1) and 648 amino acids (isoform 2) |
| FATP4 | FATP4 (fatty acid transport protein 4) encoded by SLC27A4 gene; FATP4 is also known as acyl-CoA synthetase very long-chain family, member 4 (ACSVL4); is the major intestinal long-chain fatty acid transporter; the SLC27A4 gene is located on chromosome 9q34.11 and is composed of 14 exons that encode a 643 amino acid protein |
| FATP5 | FATP5 (fatty acid transport protein 5) encoded by SLC27A5 gene; FATP5 is also known as acyl-CoA synthetase very long-chain family, member 6 (ACSVL6), also as very long-chain acyl-CoA synthetase-related protein (VLACSR), also as very long-chain acyl-CoA synthetase homolog 2 (VLCSH2), and also as bile acid-CoA synthetase (BACS); ER-associated enzyme; highest levels of expression in the liver; capable of activating 24- and 26-carbon VLCFAs; catalyzes the activation of bile acids via formation of bile acid-CoA thioesters which then undergo conjugation with glycine and taurine (this activity is identified as BACS); the SLC27A5 gene is located on chromosome 19q13.43 and is composed of 11 exons that generate two alternatively spliced mRNAs encoding proteins of 690 amino acids (isoform 1) and 606 amino acids (isoform 2) |
| FATP6 | FATP6 (fatty acid transport protein 6) encoded by SLC27A6 gene; FATP6 is also known as acyl-CoA synthetase very long-chain family, member 2 (ACSVL2), very long-chain acyl-CoA synthetase homolog 1 (VLCSH1); expressed at highest levels in the heart; protein only detected in heart and testis; exhibits a preference for the transport of palmitic acid and linoleic acid, does not transport fatty acids less than 10 carbons long; the SLC27A6 gene is located on chromosome 5q23.3 and is composed of 112 exons that generate three alternatively spliced mRNAs all of which encode the same 619 amino acid protein |
The result of the interaction of fatty acids with plasma membrane receptors/binding proteins is a transmembrane concentration gradient. At the plasma membrane the apparent pKa of the fatty acid shifts from about 4.5 in aqueous solutions to about 7.6. This pKa change is independent of fatty acid type. As a consequence, about half of the fatty acids are present in the un-ionized form. This local environment effect promotes a transfer (flip-flop) of uncharged fatty acids from the outer leaflet across the phospholipid bilayer.
At the cytosolic surface of the plasma membrane, fatty acids can associate with the cytosolic fatty acid binding protein (FABPc) or with caveolin-1. Caveolin-1 is a constituent of caveolae (Latin for little caves) which are specialized “lipid rafts” present in flask-shaped indentations in the plasma membranes of many cells types that perform a number of signaling functions by serving as lipid delivery vehicles for subcellular organelles. In order that the fatty acids that are taken up to be directed to the various metabolic pathways (e.g. oxidation or triglyceride synthesis) they must be activated to acyl-CoA.
Members of the fatty acid transport protein (FATP) family have been shown to possess acyl-CoA synthetase (ACS) activity. Activation of fatty acids by FATPs occurs at the highly conserved cytosolic AMP-binding site of these proteins. The overall process of cellular fatty acid uptake and subsequent intracellular utilization represents a continuum of dissociation from albumin by interaction with the membrane-associated transport proteins, binding to FABPc and caveolin-1 at the cytosolic plasma membrane, activation to acyl-CoA (in many cases via FATP action) followed by intracellular trafficking via FABPc and/or caveolae to sites of metabolic disposition.
Roles of Intracellular Fatty Acid Binding Proteins: FABP
Fatty acid binding proteins (FABP) represent a family of intracellular lipid-binding proteins whose functions are to reversibly bind intracellular hydrophobic ligands and transport the bound ligand throughout the various cellular compartments, including the peroxisomes, mitochondria, endoplasmic reticulum, and nucleus.
FABP have broad binding characteristics which includes the ability to bind long-chain (C16–C20) fatty acids (LCFA), eicosanoids, bile salts, and peroxisome proliferators. There are currently nine well characterized FABP genes in the human genome, four of which (FABP4, FABP5, FABP9, and FABP12) are located in the same region of the q arm of chromosome 8 (8q21.13). The gene (PMP2) encoding peripheral myelin protein 2, also identified as FABP8, is also located on chromosome 8q21.13.
Each of these FABP was originally named for the tissue in which it was first isolated and characterized or in which it predominates. However, many of these FABP are expressed in numerous tissues. The nine FABP genes are members of the large fatty acid binding protein family of genes that included the genes encoding cellular retinoic acid binding proteins (CRABP) and those encoding retinol binding proteins (RBP).
Expression of a particular FABP gene directly reflects the lipid metabolic capacity of that tissue. In high lipid metabolizing tissues, such as the liver, adipose tissue, and the heart, the expressed FABP can account for 1%–5% of total soluble cytosolic proteins. The expression of FABP in the cell is essential for the binding of hydrophobic ligands, particularly free fatty acids, in order to reduce the detergent-like properties of high concentrations of fatty acids, thereby keeping them soluble. FABP are critical to the process of lipid trafficking within cells to the various cellular compartments where they will be stored, oxidized, utilized for membrane synthesis, and for their roles in the activation of nuclear receptors. With respect to the latter function, it has been shown that FABP are involved in the targeting of fatty acids to transcription factors of the peroxisome proliferator-activated receptor (PPAR) family.
In addition to their importance in intracellular lipid trafficking, many FABP interact with phospholipid-rich membranes and bind eicosanoid intermediates protecting these substrates against peroxidation strongly implicating these proteins in antioxidant-type behavior.
Table of Fatty Acid Binding Proteins
| FABP | Alternate Names | Tissue Location | Functions / Comments |
| FABP1 (L-FABP) | Z protein, hepatic FABP, heme-binding protein | liver, intestine, pancreas, kidney, lung, stomach | represents up to 5% of hepatocyte cytosolic protein; unique ability to bind multiple ligands at once; in addition to various free fatty acids FABP1 binds fatty acyl-carnitines, intermediates in glyceride synthesis, lysophospholipids, cholesterol, bile acids, prostaglandins, lipoxygenase products, retinoids, heme, and bilirubin; FABP1 also binds numerous xenobiotic drugs such as NSAIDs, fibrates, beta blockers, and benzodiazepines; the FABP1 gene is located on chromosome 2p11.2 and is composed of 4 exons encoding a 127 amino acid protein |
| FABP2 (I-FABP) | gut FABP, gFABP | intestine | mediates dietary fat absorption of free long-chain fatty acids (LCFAs); the FABP2 gene is located on chromosome 4q26 and is composed of 4 exons encoding a 132 amino acid protein; a polymorphism in codon 54 that causes a substitution of alanine for threonine (A54T) is associated with increased serum triglyceride accumulation, weight gain, and insulin resistance |
| FABP3 (H-FABP) | O-FABP, mammary-derived growth inhibitor, MDGI | skeletal and heart muscle, brain, kidney, lung, stomach, testes, placenta, ovary, brown adipose tissue (BAT), adrenal glands, mammary glands | makes up 4%–8% of cytosolic protein in the heart; major function is to traffic fatty acids to the mitochondria for oxidation; also binds non-prostanoid oxygenated fatty acids; measurement of protein in the blood is considered an early marker for myocardial infarct; may also be a marker for Creutzfeldt–Jakob disease (CJD) by measurement of levels in the cerebrospinal fluid; the FABP3 gene is located on chromosome 1p35.2 and is composed of 5 exons that generate two alternatively spliced mRNAs encoding proteins of 144 amino acids (isoform 1) and 133 amino acids (isoform 2) |
| FABP4 (A-FABP) | adipocyte protein 2, aP2 | adipocytes and macrophages of adipose tissue, dendritic cells | specific binding capacity for LCFAs; is a marker for adipocyte maturation; modulates the activity of HSL through direct interaction; macrophage FABP4 modulates inflammatory responses; recently demonstrated to be a secreted adipokine involved in regulating hepatic glucose production; the FABP4 gene is located on chromosome 8q21.13 and composed of 4 exons that encode a 132 amino acid protein |
| FABP5 (E-FABP) | psoriasis-associated FABP (PA-FABP); keratinocyte-type FABP (KFABP) | skin, brain, stomach, intestines, kidney, liver, lung, heart, skeletal muscle, tongue, adipocytes, macrophages, dendritic cells, testes, retina, placenta, spleen | physiological ligands not completely determined; in vitro the protein binds stearic acid with high affinity while having reduced affinity for unsaturated fatty acids; interacts with HSL like FABP4; the FABP5 gene is located on chromosome 8q21.13 and is composed of 4 exons that encode a 135 amino acid protein |
| FABP6 (Il-FABP) | ileal lipid-binding protein (ILBP); gastrotropin; intestinal bile acid-binding protein (I-BABP) | predominantly the ileum, also expressed at low levels in stomach, adrenal glands, ovary | involved in enterohepatic circulation of bile acids; binds bile acids with highest affinity then fatty acids; interacts with the ileal bile acid transporter protein; the FABP6 gene is located on chromosome 5q33.3 and is composed of 7 exons that generate three alternatively spliced mRNAs that encode proteins of 177 amino acids (isoform 1) and 128 amino acids (isoform 2) |
| FABP7 (B-FABP) | brain lipid-binding protein (BLBP) | brain, glial cells, mammary glands, retina | grey matter neurons do not express FABP7; highest affinity for long-chain omega-3 polyunsaturated fatty acids (PUFAs) particularly EPA and DHA; also binds oleic acid and arachidonic acid but does not bind palmitic acid or retinoic acid; the FABP7 gene is located on chromosome 6q22.31 and is composed of 5 exons that generate four alternatively spliced mRNAs, each of which encode a distinct protein isoform |
| FABP8 PMP2, M-FABP | peripheral myelin protein 2; myelin P2 protein | Schwann cells, peripheral nervous system | only member of the FABP family that is stably attached to the membrane; present on the cytoplasmic side of compact myelin membranes; binds LCFAs; thought to be involved in stabilizing myelin membranes; official gene symbol is PMP2; the PMP2 gene is located on chromosome 8q21.13 and is composed of 4 exons that generate two alternatively spliced mRNAs encoding proteins of 132 amino acids (isoform 1) and 60 amino acids (isoform 2) |
| FABP9 (T-FABP) | testes lipid-binding protein (TLBP); | testes, mammary glands, salivary glands | precise functions not clearly defined; thought to be involved in protection of fatty acids in sperm from oxidation; the FABP9 gene is located on chromosome 8q21.13 and is composed of 4 exons that encode a 132 amino acid protein |
| FABP12 | testes, lung, duodenum | the FABP12 gene is located on chromosome 8q21.13 and is composed of 7 exons that generate two alternatively spliced mRNAs |
Fatty Acid Activation
Oxidation of fatty acids occurs in the mitochondria and the peroxisomes. Fatty acids of between 2–6 carbon atoms (e.g. butyric acid, C4:0) and between 6–12 carbon atoms (e.g. octanoic acid, C8:0) in length, referred to as short- and medium-chain fatty acids (SCFA and MCFA), respectively, are oxidized exclusively in the mitochondria. Long-chain fatty acids (LCFA: 12–22 carbons long; e.g. palmitic acid, C16:0) are oxidized in both the mitochondria and the peroxisomes with the peroxisomes exhibiting preference for 14-carbon and longer LCFA. Very-long-chain fatty acids (VLCFA: C22–C28; often designated as any fatty acid of 22 carbons and longer; e.g. cerotic acid, C26:0) are preferentially oxidized in the peroxisomes. However, given that peroxisomes are unable to completely oxidize fatty acids, the chain shortened free fatty acids, or their carnitine esters, are released from peroxisomes and taken up by the mitochondria for complete oxidation to CO2 and H2O.
Long-chain fatty acids must be activated in the cytoplasm before being oxidized in the mitochondria. Medium-chain and short-chain fatty acids are activated within the matrix of the mitochondria. Fatty acid activation is catalyzed by fatty acyl-CoA synthetases (also called acyl-CoA ligases or thiokinases). The net result of this activation process is the consumption of two molar equivalents of ATP.
Humans express at least 26 acyl-CoA synthetases with several of these enzymes also being involved in fatty acid transport into cells (FATP1–FATP6) as indicated in the Table above in the Cellular Uptake of Fatty Acids section. The various acyl-CoA synthetases exhibit different substrate specificities, subcellular localization, and tissue distribution. Additional members of the family are the long-chain acyl-CoA synthetases, the medium-chain acyl-CoA synthetases, the short-chain acyl-CoA synthetases, and the bubblegum family acyl-CoA synthetases.
Fatty acid + ATP + CoA → Acyl-CoA + PPi + AMP
Table of the Mammalian Acyl-CoA Synthetase Family
| Gene Symbol | Aliases | Location | Enzyme name / Comments |
| AACS | 12q24.31 | acetoacetyl-CoA synthetase; primary enzyme for the conversion of acetoacetate to acetoacetyl-CoA; gene composed of 19 exons that encode a 672 amino acid protein | |
| AASDH | 4q12 | 2‑aminoadipic‑6‑semialdehyde dehydrogenase; also called acyl-CoA synthetase family member 4 isoform 2 (ACSF4); is also known as β-alanine activating enzyme; is one of only six human enzymes that is post-translationally modified by phosphopantetheinylation; gene composed of 17 exons that encode a protein of 992 amino acids | |
| ACSBG1 | 15q25.1 | acyl-CoA synthetase bubblegum family member 1; gene composed of 19 exons that generate two alternatively spliced mRNAs; involved in brain VLCFA metabolism and myelogenesis | |
| ACSBG2 | 19p13.3 | acyl-CoA synthetase bubblegum family member 2; gene composed of 16 exons that generate five alternatively spliced mRNAs encoding three distinct isoforms; expression restricted to the testis | |
| ACSF2 | 17q21.33 | acyl-CoA synthetase family member 2; gene composed of 19 exons that generate six alternatively spliced mRNAs encoding five distinct isoforms; has been shown to generate phenylacetyl-CoA from phenylacetate (phenylacetic acid, PAA) | |
| ACSF3 | 16q24.3 | acyl-CoA synthetase family member 3; mitochondrial protein, high affinity for methylmalonate and malonate; gene composed of 17 exons that generate four alternatively spliced mRNAs encoding two distinct isoforms; functions in the mitochondria to generate malonyl-CoA for mitochondrial fatty acid synthesis | |
| ACSS1 | 20p11.21 | acyl-CoA synthetase short-chain family member 1; mitochondrial protein primarily responsible for generation of mitochondrial acetyl-CoA from acetate; is also responsible for the generation of crotonyl-CoA from crotonate which is involved in the process of protein lysine crotonylation; gene composed of 15 exons that generate four alternatively spliced mRNAs that encode four distinct isoforms | |
| ACSS2 | 20q11.22 | acyl-CoA synthetase short-chain family member 2; cytosolic protein primarily responsible for generation of acetyl-CoA from acetate; is also responsible for the generation of lactoyl-CoA from lactate which is involved in the process of protein lysine lactylation which includes histone lactylation; gene expression controlled by SREBP; gene composed of 22 exons that generate three alternatively spliced mRNAs encoding three distinct isoforms | |
| ACSS3 | 12q21.31 | acyl-CoA synthetase short-chain family member 3; gene composed of 19 exons that encode a 686 amino acid precursor protein; involved in the conversion of propionic acid to propionyl-CoA; this enzyme is also known as propionyl-CoA synthetase; may also function in the synthesis of crotonyl-CoA, from dietary dietary crotonate, in the process of protein lysine crotonylation | |
| ACSM1 | 16p12.3 | acyl-CoA synthetase medium-chain family member 1; mitochondrial protein; gene composed of 18 exons that encode a 577 amino acid protein; functions in the activation of medium-chain fatty acids to medium-chain acyl-CoAs for oxidation in the mitochondrial matrix | |
| ACSM2A | 16p12.3 | acyl-CoA synthetase medium-chain family member 2A; mitochondrial protein; gene composed of 15 exons that generate three alternatively spliced mRNAs encoding two distinct isoforms; functions in the activation of medium-chain fatty acids to medium-chain acyl-CoAs for oxidation in the mitochondrial matrix | |
| ACSM2B | 16p12.3 | acyl-CoA synthetase medium-chain family member 2B; mitochondrial protein; gene composed of 16 exons that generate two alternatively spliced mRNAs encoding the same protein; functions in the activation of medium-chain fatty acids to medium-chain acyl-CoAs for oxidation in the mitochondrial matrix; also converts certain xenobiotics (e.g. benzoate and phenylacetate) to CoA derivatives, an activity referred to as a benzoyl-CoA ligase; the major biologically relevant phenylacetyl-CoA synthetase is encoded by the ACSF2 gene | |
| ACSM3 | 16p13.11 | acyl-CoA synthetase medium-chain family member 3; mitochondrial protein; gene composed of 15 exons that generate two alternatively spliced mRNAs that encode two distinct isoforms; functions in the activation of medium-chain fatty acids to medium-chain acyl-CoAs for oxidation in the mitochondrial matrix; primary substrate is lauric acid (C12:0; chemical name is dodecanoic acid) | |
| ACSM4 | 12p13.31 | acyl-CoA synthetase medium-chain family member 4; mitochondrial protein; gene composed of 13 exons that encode a 580 amino acid precursor protein; functions in the activation of medium-chain fatty acids to medium-chain acyl-CoAs for oxidation in the mitochondrial matrix | |
| ACSM5 | 16p12.3 | acyl-CoA synthetase medium-chain family member 5; mitochondrial protein; gene composed of 14 exons that encode a 579 amino acid precursor protein; functions in the activation of medium-chain fatty acids to medium-chain acyl-CoAs for oxidation in the mitochondrial matrix | |
| ACSL1 | 4q35.1 | acyl-CoA synthetase long-chain family member 1; substrate preference for fatty acids of 16–20 carbons; expression highest in heart, liver, and adipose tissue; involved in activation of inflammasomes in neutrophils during sepsis; activity reduces the level of lipid oxidation and increases the resistance of cells to ferroptosis; gene composed of 28 exons that generate five alternatively spliced mRNAs encoding four distinct isoforms | |
| ACSL3 | 2q36.1 | acyl-CoA synthetase long-chain family member 3; involved in the production of monounsaturated fatty acids (MUFA); substrate preference for myristic (C14:0), arachidonic (C20:4), and eicosapentaenoic, EPA (C20:5) acids; highly expressed in the brain, testes, and skeletal muscle; localized in the Golgi apparatus, endoplasmic reticulum (ER), peroxisomes, and mitochondrial outer membrane; may be involved in resistance to ferroptosis; gene composed of 17 exons that generate two alternatively spliced mRNAs encoding the same protein | |
| ACSL4 | Xq23 | acyl-CoA synthetase long-chain family member 4; involved in the production of polyunsaturated fatty acids (PUFA); expression highest in adrenal gland, ovary, testis, and brain; localized in the endoplasmic reticulum (ER), mitochondria, plasma membrane, and peroxisomes; substrate preference for fatty acids of 16–20 carbons; enhances possibility for plasma membrane lipid peroxidation and therefore to an increase in the potential for ferroptosis; involved in activation of inflammasomes in neutrophils during sepsis; gene composed of 17 exons that generate two alternatively spliced mRNAs encoding two distinct isoforms; preferentially recognizes arachidonic acid as substrate | |
| ACSL5 | 10q25.2 | acyl-CoA synthetase long-chain family member 5; expressed in liver, small intestine, adipose tissue, skeletal muscle, spleen, lung, and uterus; localized to the endoplasmic reticulum (ER) and mitochondrial outer membrane; substrate preference for palmitic (C16:0), palmitoleic (C16:1), oleic (C18:1), and linoleic (C18:2) acids; gene composed of 23 exons that generate three alternatively spliced mRNAs encoding two distinct isoforms; functions as tumor suppressor in a cytotoxic T-cell (CTL)-dependent manner | |
| ACSL6 | 5q31 | acyl-CoA synthetase long-chain family member 6; near exclusive expression in the brain; substrate preference for docosahexaenoic acid, DHA (C22:6); required for the enrichment of DHA in the brain; gene composed of 23 exons that generate six alternatively spliced mRNAs encoding six distinct isoforms | |
| SLC27A1 | FATP1, ACSVL5 | 19p13.11 | solute carrier family 27 (fatty acid transporter), member 1; also known as fatty acid transport protein 1 and acyl-CoA synthetase very long-chain family, member 5; see details above |
| SLC27A2 | FATP2, ACSVL1 | 15q21.2 | solute carrier family 27 (fatty acid transporter), member 2; also known as fatty acid transport protein 2 and acyl-CoA synthetase very long-chain family, member 1; see details above |
| SLC27A3 | FATP3, ACSVL3 | 1q21.3 | solute carrier family 27 (fatty acid transporter), member 3; also known as fatty acid transport protein 3 and acyl-CoA synthetase very long-chain family, member 3; see details above |
| SLC27A4 | FATP4, ACSVL4 | 9q34.11 | solute carrier family 27 (fatty acid transporter), member 4; also known as fatty acid transport protein 4 and acyl-CoA synthetase very long-chain family, member 4; see details above |
| SLC27A5 | FATP5, ACSVL6 | 19q13.43 | solute carrier family 27 (fatty acid transporter), member 5; also known as fatty acid transport protein 5 and acyl-CoA synthetase very long-chain family, member 6; see details above |
| SLC27A6 | FATP6, ACSVL2 | 5q23.3 | solute carrier family 27 (fatty acid transporter), member 6; also known as fatty acid transport protein 6 and acyl-CoA synthetase very long-chain family, member 2; see details above |
Fatty Acid Deactivation
Once a fatty acid is esterified to coenzyme A forming an acyl-CoA, its fate is not destined for oxidation in the mitochondria or the peroxisomes. Fatty acyl-CoA molecules are substrates for a family of enzymes called acyl-CoA thioesterases (ACOT). The products of ACOT activity are free fatty acids and CoASH. The action of ACOT enzymes serves to deactivate fatty acids.
There are two classes of ACOT enzymes, identified as type I and type II. The type I acyl-CoA thioesterases are characterized by a molecular mass of approximately 40 kDa and they exhibit positive responses to peroxisome proliferator treatment. Type I ACOT enzymes belong to the large α/β-hydrolase family of enzymes that includes lipases and esterases. The type II acyl-CoA thioesterases are characterized by a molecular mass of approximately 110–150 kDa and they exhibit variable responses peroxisome proliferator treatment. The type II ACOT enzymes, despite possessing limited amino acid sequence similarity, do have structural features in common. One distinctive feature is termed the hot-dog domain. The hot-dog domain consists of a seven-stranded antiparallel β-sheet (the “bun”) that wraps around a hydrophobic five-turn α-helical region (the “hot dog”), and a layer comprising loops over the helix referred to as the “condiments”.
Humans express 12 genes that encode enzymes that possess acyl-CoA thioesterase activity. There are 10 genes identified by the ACOT nomenclature, ACOT1, ACOT2, ACOT4, ACOT6, ACOT7, ACOT8, ACOT9, ACOT11, ACOT12, and ACOT13 and two genes that are designated by the thioesterase superfamily nomenclature, THEM4 and THEM5. The THEM5 gene has also been identified as ACOT15. Several ACOT genes are also identified with the THEM nomenclature with ACOT11 being identified as THEM1 and ACOT13 being identified as THEM2.
The ACOT11 and ACOT12 encoded proteins possess multiple functional domains with one domain being the thioesterase domain and the other being a START (STeroidogenic Acute Regulatory protein-related lipid Transfer) domain. As such ACOT11 has also been identified as STARD14 and ACOT12 has been identified as STARD15.
The ACOT1, ACOT2, and ACOT4 encoded enzymes are type I acyl-CoA thioesterases while the ACOT7, ACOT8, ACOT9, ACOT11, ACOT12, ACOT13, THEM4, and THEM5 encoded enzymes are type II acyl-CoA thioesterases.
Four genes, ACOT1, ACOT2, ACOT4, and ACOT6 are clustered on chromosome 14q24.3. the ACOT6 encoded protein possesses a catalytic site but it is unclear whether the protein has a functional enzymatic activity.
Expression of the ACOT1, ACOT2, and ACOT4 genes is highest in adipose tissue, liver, heart, and kidney. The ACOT1 encoded enzyme is localized to the cytosol and exhibits substrate specificity for long-chain (C12-C20) saturated- and monounsaturated acyl-CoAs. The ACOT2 encoded enzyme is targeted to the mitochondrial matrix where it exhibits substrate specificity for long-chain fatty acyl-CoAs. The ACOT4 encoded enzyme possesses a type I peroxisomal targeting sequence and exhibits substrate specificity for short-chain dicarboxylic acyl-CoA esters, particularly succinyl-CoA, and medium- to long-chain acyl-CoAs.
Mitochondrial (beta) β-Oxidation Reactions
Fatty Acid Transport into Mitochondria
Short-chain and medium-chain fatty acids require no specific transport mechanism to enter the mitochondria for oxidation and, as indicated earlier, are activated by CoA attachment within the mitochondrial matrix. Also, because dietary short-chain and medium-chain fatty acids directly enter the portal circulation (they are not packaged into chylomicrons) they are rapidly oxidized within the liver.
Long-chain fatty acids, present in the triglycerides of chylomicrons, or VLDL, or as free fatty acids released from adipose tissue, require a specific mitochondrial transport mechanism to be oxidized. The transport of long-chain fatty acyl-CoAs into the mitochondria is accomplished via an acyl-carnitine intermediate, which itself is generated by the action of carnitine palmitoyltransferase 1 (CPT1 or CPT-I), an enzyme that resides in the outer mitochondrial membrane.
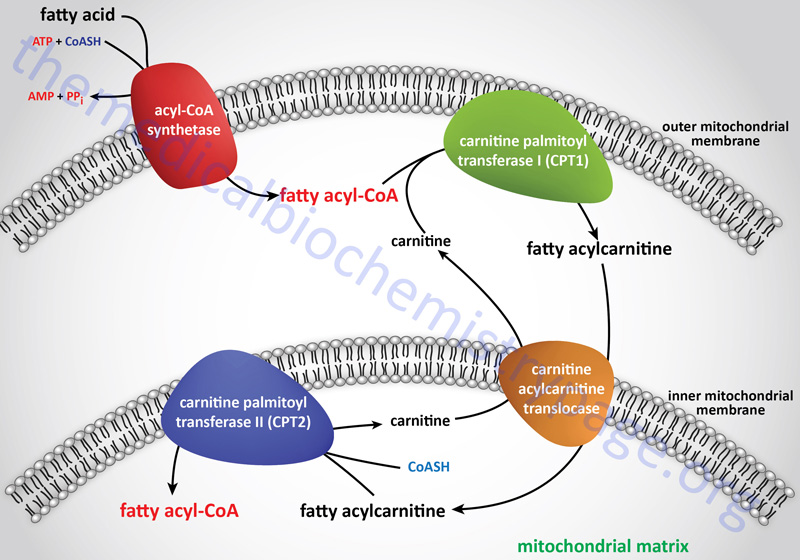
Carnitine Transport
The carnitine that is used by carnitine palmitoyltransferase 1 (CPT1) can be derived from the diet or it can be synthesized de novo in cells from the modified amino acid trimethylysine. The significance of de novo carnitine synthesis to overall metabolism was demonstrated in SLC25A45 knock-out mice. The SLC25A45-mediated transport of trimethyllysine (TML) into the mitochondria was shown to contribute to overall fuel choice adaptation. In SLC25A45 negative mice there is an observable increase in glucose oxidation with a concomitant decrease in mitochondrial fatty acid β-oxidation. In addition, the loss of SLC25A45 results in suppression of fatty acid β-oxidation in brown adipose tissue (BAT) resulting in decreased adaptive thermogenesis.
Whether dietary or synthesized de novo, carnitine is transported into cells by the plasma membrane localized transporter encoded by the SLC22A5 gene. The SLC22A5 encoded transporter is commonly identified as organic zwitterion/cation transporter 2 (OCTN2). OCTN2 is the only carnitine transporter localized to the plasma membrane and as such represents the major transporter for cellular uptake of carnitine.
The SLC22A5 gene is located on chromosome 5q31.1 and is composed of 11 exons that generate two alternatively spliced mRNAs that encode proteins of 581 amino acids (isoform a) and 557 amino acids (isoform b). Expression of the SLC22A5 gene is ubiquitous but the highest levels are seen in the small intestines and kidneys.
Free carnitine is transported out of the mitochondria, to the cytosol, via the action of the carnitine-acylcarnitine translocase, CACT. The CACT transporter is also capable of transporting acyl-carnitine molecules from the cytosol into the mitochondria, as described in the next section.
Carnitine Palmitoyltransferase 1 (CPT1)
There are three CPT1 genes in humans identified as CPT1A, CPT1B, and CPT1C. Expression of CPT1A predominates in the liver and is thus, referred to as the liver isoform. CPT1B expression predominates in skeletal muscle and is thus, referred to as the muscle isoform. CPT1C expression is exclusive to the brain and testes.
The CPT1A gene is located on chromosome 11q13.2 and consists of 22 exons that generate two alternatively spliced mRNAs encoding isoform 1 (773 amino acids) and isoform 2 (756 amino acids).
The CPT1B gene is located on chromosome 22q13.33 and consists of 21 exons that generate six alternatively spliced mRNAs, all of which yield two distinct isoforms of the enzyme. Isoform a is 772 amino acids and isoform c is 738 amino acids.
The CPT1C gene is located on chromosome 19q13.33 and consists of 24 exons that generate eleven alternatively spliced mRNAs that encode five different protein isoforms.
The activity of CPT1C is distinct from those of CPT1A and CPT1B in that it does not act on the same types of fatty acyl-CoAs that are substrates for the latter two enzymes, nor does it participate in the mobilization of fatty acids into the mitochondria. CPT1C does exhibit high-affinity for malonyl-CoA binding. Within the hypothalamus, brain CPT1C serves as a sensor of nutrient availability by binding malonyl-CoA which triggers a reduction in the release of the appetite promoting neuropeptides, neuropeptide Y (NPY) and Agouti-related peptide (AgRP) and an increase the release of the appetite suppressing neuropeptides, α-melanocyte stimulating hormone (α-MSH) and cocaine and amphetamine regulated transcript (CART). The net effect of increased hypothalamic malonyl-CoA binding to CPT1C is, therefore, satiety and appetite suppression.
Once a fatty acylcarnitine is generated at the outer mitochondrial membrane it is transported into the mitochondria through the action of carnitine-acylcarnitine translocase, CACT. The CACT transporter is a member of the SLC family of transporters and as such is encoded by the SLC25A20 gene. The carnitine acylcarnitine translocase is located in the inner mitochondrial membrane where it facilitates acylcarnitine transport across the outer and inner mitochondrial membranes in exchange for free carnitine.
The SLC25A20 gene is located on chromosome 3p21.31 and is composed of 9 exons that encode a 301 amino acid protein.
Carnitine Palmitoyltransferase 2 (CPT2)
Following CACT-mediated transfer of the CPT1-generated fatty acylcarnitines across the inner mitochondrial membrane, the fatty acyl-carnitine molecules are acted on by the inner mitochondrial membrane-associated carnitine palmitoyltransferase 2 (CPT2 or CPT-II) regenerating the fatty acyl-CoA molecules.
The CPT2 gene is located on chromosome 1p32.3 and consists of 5 exons that generate two alternatively spliced mRNAs. These mRNAs encode precursor proteins of 658 amino acids (isoform 1) and 635 amino acids (isoform 2).
Additional Carnitine Acyltransferases
In addition to the genes of the CPT family of carnitine acyltransferases, humans express two additional carnitine acyltransferases. The two additional enzymes are carnitine O-acetyltransferase encoded by the CRAT gene and carnitine octanoyltransferase encoded by the CROT gene.
The CRAT gene is located on chromosome 9q34.11 and is composed of 18 exons that generate seven alternatively spliced mRNAs that collectively encode six distinct protein isoforms. The different CRAT mRNAs encode enzymes that are found in the mitochondrial matrix, the peroxisomes, and the nucleus. The substrate specificity of CRAT is for short-chain acyl-CoA esters.
The CROT gene is located on chromosome 7q21.12 and is composed of 21 exons that generate three alternatively spliced mRNAs that encode three distinct protein isoforms.
The CROT encoded enzyme is found in the peroxisomes where it has substrate preference for medium-chain acyl-CoAs. The major substrate for the CROT enzyme is 4,8-dimethylnonanoyl-CoA. The function of the CROT enzyme is to participate in the transport of medium- and long-chain fatty acyl-CoA molecules out of the peroxisomes to the cytosol where they can then be transported into the mitochondria.
Processes of Mitochondrial Fatty Acid β-Oxidation
The process of mitochondrial fatty acid oxidation is termed β-oxidation since it occurs through the sequential removal of 2-carbon units by oxidation at the β-carbon position, relative to the carboxylic acid group, of the fatty acyl-CoA molecule. The oxidation of fatty acids and lipids in the peroxisomes (see below) also occurs via a process of β-oxidation but the enzymes are distinct from those used within the mitochondria.
Each round of mitochondrial fatty acid β-oxidation involves four steps that, in order, are oxidation, hydration, oxidation, and cleavage. The first oxidation step in mitochondrial β-oxidation involves a family of FAD-dependent acyl-CoA dehydrogenases that act on saturated fatty acids. Each of these dehydrogenases has a range of substrate specificities determined by the length of the fatty acid.
Each round of fatty acid β-oxidation produces one mole of FADH2, one mole of NADH, and one mole of acetyl-CoA. The acetyl-CoA, the end product of each round of β-oxidation, then enters the TCA cycle, where it is further oxidized to CO2 with the concomitant generation of three moles of NADH, one mole of FADH2 and one mole of ATP. The NADH and FADH2 generated during the fat oxidation and acetyl-CoA oxidation in the TCA cycle then can enter the respiratory pathway for the production of ATP via oxidative phosphorylation.
Short-Chain Acyl-CoA dehydrogenase: SCAD
Short-chain acyl-CoA dehydrogenase (SCAD, also called butyryl-CoA dehydrogenase) prefers fats of 4–6 carbons in length. The SCAD enzyme (also known as ACAD3) is encoded by the ACADS (acyl-CoA dehydrogenase, short chain) gene.
The ACADS gene is located on chromosome 12q24.31 and is composed of 11 exons that generate two alternatively spliced mRNAs encoding proteins of 412 amino acids and 408 amino acids.
Medium-Chain Acyl-CoA Dehydrogeanse: MCAD
Medium-chain acyl-CoA dehydrogenase (MCAD; also known as ACAD1) prefers fats of 4–16 carbons in length with maximal activity for C10 acyl-CoAs. The MCAD enzyme is encoded by the ACADM gene.
The ACADM gene is located on chromosome 1p31.1 and is composed of 13 exons generate five alternatively spliced mRNAs each encoding a unique protein isoform.
Long-Chain Acyl-CoA Dehydrogenase
Long-chain acyl-CoA dehydrogenase (LCAD; also known as ACAD4) prefers fats of 10-18 carbons in length with maximal activity for C12 acyl-CoAs. The LCAD enzyme is encoded by the ACADL gene. The ACADL gene is located on chromosome 2q34 and is composed of 12 exons that encode a 430 amino acid precursor protein. However, due to low level expression of the ACADL gene in humans, the encoded acyl-CoA dehydrogenase plays a limited, if at all, role in mitochondrial fatty acid β-oxidation.
Expression of the ACADL gene in humans primarily occurs in alveolar type II pneumocytes which are the specialized cells of the alveolar epithelium that synthesize and secrete pulmonary surfactant. Pulmonary surfactant is a complex of lipid (90%) and protein (10%) where the primary lipid is dipalmitoylphosphatidylcholine (DPPC; also called dipalmitoyllecithin).
Very Long-Chain Acyl-CoA Dehydrogenase
Very long-chain acyl-CoA dehydrogenase (VLCAD; also known as ACAD6) prefers fats of 16-24 carbons and is inactive on any fatty acid less than 12 carbons. Unlike the localization of SCAD and MCAD to the mitochondrial matrix, VLCAD is an inner mitochondrial membrane-localized enzyme.
The VLCAD enzyme is encoded by the ACADVL gene. The ACADVL gene is located on chromosome 17p13.1 and is composed of 22 exons that generate four alternatively spliced mRNAs each of which encode distinct protein isoforms. The isoform 4 encoding ACADVL mRNA does not initiate translation efficiently and is, therefore, a likely mRNA candidate for nonsense mediated decay, NMD.
Additional Acyl-CoA Dehydrogenase Family Members
In addition to the fatty acyl-CoA dehydrogenases, SCAD, MCAD, LCAD, and VLCAD, humans express seven additional acyl-CoA dehydrogenases, not all of which are involved in lipid oxidation. These additional enzymes are isovaleryl-CoA dehydrogenase (also known as ACAD2; encoded by the IVD gene), glutaryl-CoA dehydrogenase (also known as ACAD5; encoded by the GCDH gene), short/branched-chain acyl-CoA dehydrogenase (also known as ACAD7; encoded by the ACADSB gene), ACAD8, ACAD9, ACAD10, and ACAD11.
The IVD and ACAD8 encoded proteins are involved in the catabolism of leucine and valine, respectively. The GCDH encoded enzyme is involved in the conversion of glutaryl-CoA to crotonyl-CoA derived through the catabolism of tryptophan and the catabolism of lysine.
The ACADSB encoded enzyme prefers the branched-chain acyl-CoA, 2-methylbutryl-CoA, which is a product of the catabolism of isoleucine, but is also functional on the short straight-chain acyl-CoAs, butyryl-CoA and hexanoyl-CoA.
Although ACAD9 has dehydrogenase activity towards palmitoyl-CoA and stearoyl-CoA in vitro, the enzyme appears to be involved in the assembly of complex I of mitochondrial oxidative phosphorylation.
The ACAD10 encoded enzyme is functional only on the CoA derivative of 2-methyl substituted pentadecanoic (C15:0) acid.
The ACAD11 encoded enzyme appears to be specific for the CoA derivative of the saturated fatty acid, behenic (C22:0) acid. The major source of behenic acid is from the drumstick tree (moringa tree: Moringa oleifera) but is also present in the oils of rapeseed and peanut. Although behenic acid is poorly absorbed in humans it has been purported to result in significant increases in serum cholesterol.
Similarities and Differences in Oxidation of Different Length Fatty Acids
Although the oxidation of long-chain saturated fatty acids occurs with a chemistry that is the same as that for the oxidation of medium-chain and short-chain saturated fatty acids, there are different sets of enzymes involved. Following the FAD-dependent acyl-CoA dehydrogenase step, the next three steps in mitochondrial β-oxidation involve a hydration step, another oxidation step, and finally a hydrolytic reaction that requires CoA and releases acetyl-CoA and a fatty acyl-CoA two carbon atoms shorter than the initial substrate.
The enzymes of long-chain fatty acid oxidation are localized to the inner mitochondrial membrane. In the oxidation of these substrates, until they become medium-chain length, the water addition is catalyzed by an enoyl-CoA hydratase activity, the second oxidation step is catalyzed by an NAD+-dependent long-chain hydroxacyl-CoA dehydrogenase activity [long-chain 3-hydroxyacyl-CoA dehydrogenase (LCHAD) activity], and finally the cleavage into an acyl-CoA and an acetyl-CoA is catalyzed by a thiolase activity. For the oxidation of long-chain saturated fatty acids these three activities are encoded in a multifunctional enzyme called the mitochondrial trifunctional protein, MTP (also known simply as trifunctional protein: TFP).
MTP is a heterooctameric complex composed of four α-subunits encoded by the HADHA gene and four β-subunits encoded by the HADHB gene. The α-subunits contain the enoyl-CoA hydratase (also called long-chain enoyl-CoA hydratase, LCEH) and long-chain hydroxyacyl-CoA dehydrogenase (LCHAD) activities, while the β-subunits possess the long-chain 3-ketoacyl-CoA thiolase (LCKAT; also called β-ketothiolase or just thiolase) activity.
It is important to point out that there is a β-ketothiolase involved in isoleucine catabolism. As indicated in the Table below, this enzyme is encoded by the ACAT1 gene. The significance of this information relates to the fact that there is an inborn error in metabolism referred to as β-ketothiolase deficiency and this disorder is not related to fatty acid metabolism.
The HADHA gene is located on chromosome 2p23.3 and is composed of 20 exons that encode a 763 amino acid precursor protein.
The HADHB gene is also located on chromosome 2p23.3 in a head-to-head orientation with the HADHA gene. The HADHB gene is composed of 17 exons that generate three alternatively spliced mRNAs, each of which encode a distinct protein isoform.
Medium-chain and short-chain saturated fatty acids are oxidized by soluble mitochondrial matrix enzymes. Following the action of MCAD and SCAD the resulting enoyl-CoAs are substrates for the enoyl-CoA hydratase encoded by the ECHS1 (enoyl-CoA hydratase, short chain 1; also identified as SCEH) gene. The ECHS1 encoded enzyme is often referred to as crotonase. The ECHS1 gene is located on chromosome 10q26.3 and is composed of 8 exons that encode a 290 amino acid protein.
The next step in medium- and short-chain fatty acid β-oxidation is catalyzed by hydroxyacyl-CoA dehydrogenase (also called short-chain L-3-hydroxyacyl-CoA dehydrogenase, SCHAD) which is encoded by the HADH gene. This dehydrogenase exhibits highest specificity for medium-chain saturated fatty acids. The HADH gene is located on chromosome 4q25 and is composed of 11 exons that generate three alternatively spliced mRNAs, each of which encoded a distinct protein isoform. Mutations in the HADH gene are associated with familial hyperinsulinemic hypoglycemia.
The thiolase reaction is catalyzed by the ACAA2 encoded enzyme (see the Table below) which is also referred to as medium-chain 3-ketoacyl-CoA thiolase, MCKAT.
The mammalian genome actually encodes five distinct enzymes with thiolase activity as outlined in the following Table.
Table of Mammalian Thiolase Genes
| Thiolase Gene Symbol | Comments |
| ACAA1 | acetyl-CoA acyltransferase 1; also called peroxisomal 3-oxoacyl-CoA thiolase; involved in peroxisomal fatty acid β-oxidation; located on chromosome 3p22.2 spanning 11 kb composed of 12 exons that generate two alternatively spliced mRNAs encoding two isoforms of the enzyme (isoform a is 424 amino acids, isoform b is 331 amino acids) |
| ACAA2 | acetyl-CoA acyltransferase 2; also called mitochondrial 3-oxoacyl-CoA thiolase or medium-chain 3-ketoacyl-CoA thiolase (MCKAT); catalyzes the terminal reaction of mitochondrial fatty acid β-oxidation of medium- and short-chain fatty acids and the breakdown of the ketone, acetoacetyl-CoA, to two moles of acetyl-CoA; activity similar to that catalyzed by HADHB of the MTP; located on chromosome 18q21.1 and composed of 10 exons that encode a 397 amino acid protein |
| ACAT1 | acetyl-CoA acetyltransferase 1; also called mitochondrial acetoacetyl-CoA thiolase; involved in ketone body synthesis (see below) in the liver; located on chromosome 11q22.3 spanning 27 kb composed of 17 exons that generate 13 alternatively spliced mRNAs that collectively encode five distinct protein isoforms; 9 of the 13 alternatively spliced mRNAs encode the same 337 amino acid protein (isoform e); the protein encoded by the SOAT1 gene (sterol O-acyltransferase 1) was originally referred to as acyl-CoA: cholesterol acyltransferase 1, ACAT1 but due to the nomenclature conflict is no longer designated with this acronym |
| ACAT2 | acetyl-CoA acetyltransferase 2; also called cytosolic acetoacetyl-CoA thiolase; involved in cholesterol biosynthesis and in the utilization of ketone bodies by the brain, cardiac muscle, and several other tissues; located on chromosome 6q25.3 and is composed of 10 exons that generate two alternatively spliced mRNAs encoding two isoforms of the enzyme (isoform 1 is amino 397 acids, isoform 2 is 426 amino acids); the protein encoded by the SOAT2 gene (sterol O-acyltransferase 2) was originally referred to as acyl-CoA: cholesterol acyltransferase 2, ACAT2 but due to the nomenclature conflict is no longer designated with this acronym |
| HADHB | hydroxyacyl-CoA dehydrogenase/3-ketoacyl-CoA thiolase/enoyl-CoA hydratase, beta subunit; 3-ketoacyl-CoA thiolase; β-ketothiolase; HADHB encodes the β-subunit of mitochondrial trifunctional protein (MTP); located on chromosome 2p23.3 composed of 17 exons that generate three alternatively spliced mRNAs each of which encode a distinct protein isoform |
Pathway of Mitochondrial β-Oxidation of Long-Chain Fatty Acids
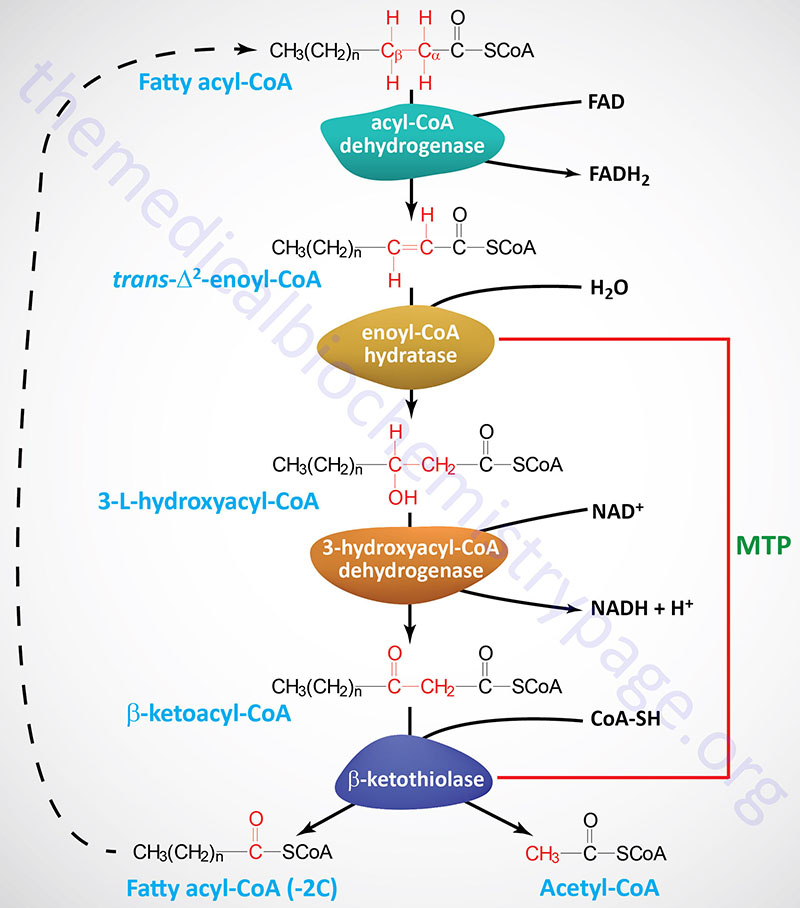
Pathway of Mitochondrial β-Oxidation of Medium- and Short-Chain Fatty Acids
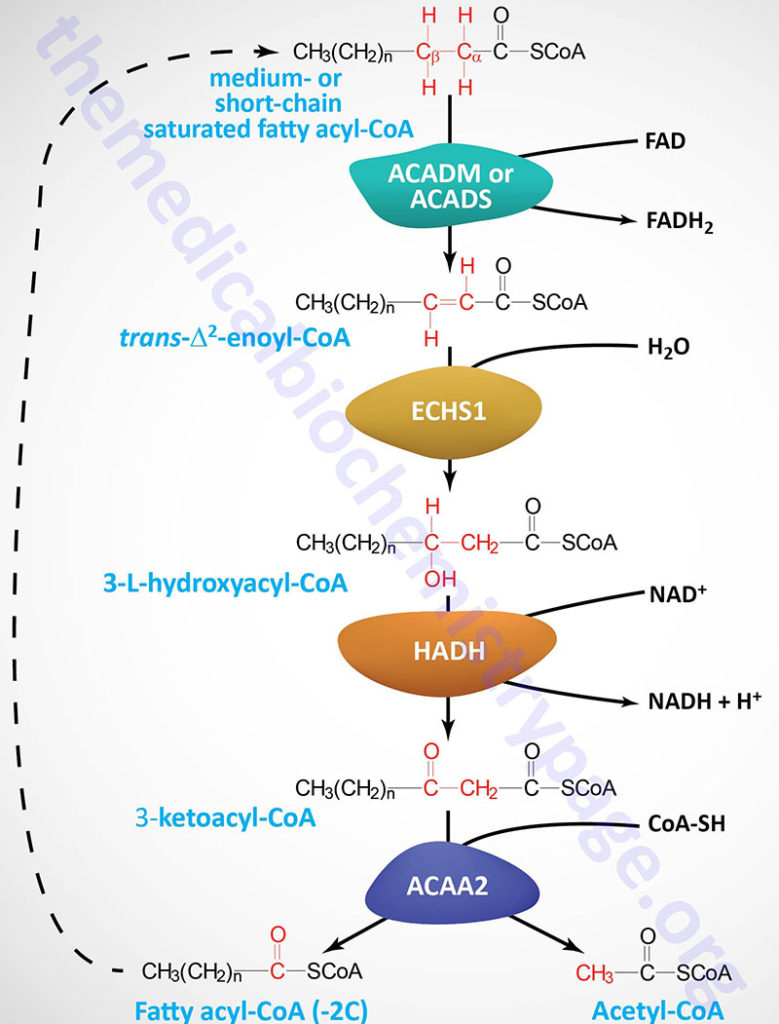
Role of Medium-Chain Triglycerides (MCT) is Overall Metabolic Regulation
As noted above, dietary medium-chain fatty acids (MCFA), primarily those found in dietary triglycerides and thus referred to as medium-chain triglycerides (MCT), are released by pancreatic lipases and the MCFA are rapidly absorbed from the lumen of the small intestinal and pass through intestinal enterocytes to the portal circulation. MCFA freely enter hepatocytes and are oxidized to acetyl-CoA, regardless of the ATP demand of the cell. The acetyl-CoA then contributes to ketone production yielding levels in the blood that are similar to those found after two days of fasting. Diets high in MCT are associated with health benefits that includes weight loss and cardiovascular functioning, much of which is attributed to the effects of ketones. The greatest benefits of a diet rich in MCT is associated with MCT predominantly containing octanoic acid (C8:0; also commonly called caprylic acid).
One of the major metabolic benefits of consuming MCT (enriched in C8:0 and C10:0 fatty acids) is decreased postprandial blood glucose along with increased insulin levels and decreased peripheral insulin resistance. Consuming MCT is also associated with decreased (albeit not dramatically so) circulating levels of LDL cholesterol (LDL-C). Overall energy homeostasis benefits from MCT consumption due to increased postprandial energy expenditure coupled with decreased desire for food intake.
Energy Yield from Mitochondrial β-Oxidation of Fatty Acids
The oxidation of fatty acids yields significantly more energy per carbon atom than does the oxidation of carbohydrates. The net result of the oxidation of one mole of oleic acid (an 18-carbon fatty acid) will be 146 moles of ATP (2 mole equivalents are used during the activation of the fatty acid), as compared with 114 moles from an equivalent number of glucose carbon atoms.
β-Oxidation of Fatty Acids with Odd Numbers of Carbon Atoms: Propionate Metabolism
The majority of natural animal derived fatty acids contain an even number of carbon atoms. Fatty acids with an odd number of carbon atoms (odd-chain fatty acids, OCFA) are primarily found in the fat and milk of ruminant animals with a small proportion of plant derived fatty acids being OCFA. Upon complete β-oxidation of OCFA there results a single mole of propionyl-CoA. The propionyl-CoA is converted, via a mitochondrially-localized three reaction ATP-dependent pathway, to succinyl-CoA. The succinyl-CoA can then enter the TCA cycle for further oxidation or can be used for the synthesis of heme.
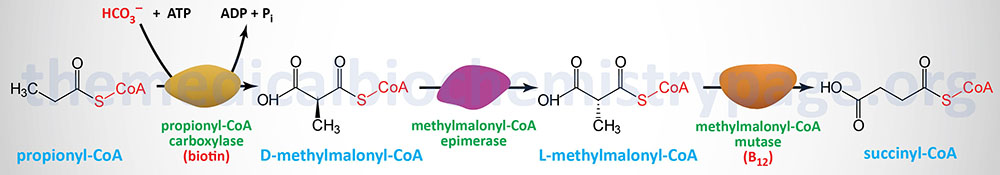
Propionyl-CoA carboxylase is called an ABC enzyme due to the requirements for ATP, Biotin, and CO2 for the reaction. The metabolic significance of methylmalonyl-CoA mutase in this pathway is that it is one of only two enzymes that requires a vitamin B12-derived co-factor for activity. The other B12-requiring enzyme is methionine synthase (see the Amino Acid Biosynthesis page). This same propionyl-CoA conversion pathway is required for the metabolism of the amino acids valine, methionine, isoleucine, and threonine. For this reason this three-step reaction pathway is often remembered by the mnemonic as the VOMIT pathway, where V stands for valine, O for odd-chain fatty acids, M for methionine, I for isoleucine, and T for threonine. In addition to these five compounds, there is another source of propionyl-CoA that is exclusive to the liver which is the pathway of bile acid synthesis from cholesterol.
Propionyl-CoA carboxylase functions as a heterododecameric enzyme (subunit composition: α6β6) and the two different subunits are encoded by the PCCA and PCCB genes, respectively. The biotin-dependent carboxylating enzymes in mammals are multifunctional and contain three distinct enzymatic activities that may be contained in a single protein or in different subunits of the multisubunit enzymes. These three enzymatic activities are the biotin carboxylase (BC), the carboxyltransferase (CT), and the biotin carboxyl carrier protein (BCCP) activities. The α-subunit of propionyl-CoA carboxylase contains the these activities of a typical biotin-dependent carboxylase. The BC domain of the α-subunit resides in the N-terminal region and the BCCP domain resides in the C-terminal region. Propionyl-CoA carboxylase has a domain, called the biotin transfer (BT) domain, that is not found in the other biotin-dependent carboxylase family enzymes. The BT domain is found in the middle of the α-subunit and is required for interactions between the α- and β-subunits.
The PCCA gene is located on chromosome 13q32.3 and is composed of 32 exons that generate eleven alternatively spliced mRNAs, each of which encode a unique protein isoform.
The PCCB gene is located on chromosome 3q22.3 and is composed of 17 exons that generate two alternatively spliced mRNAs encoding precursor proteins of 539 amino acids (isoform 1) and 559 amino acids (isoform 2).
Methylmalonyl-CoA epimerase is encoded by the MCEE gene located on chromosome 2p13.3 and is composed of 4 exons that encode a 176 amino acid precursor protein.
Methylmalonyl-CoA mutase is encoded by the MMUT gene located on chromosome 6p12.3 and is composed of 13 exons that encode a precursor protein of 750 amino acids. Mutations in the MMUT gene are one cause of the methylmalonic acidemias.
Mutations in either the PCCA or PCCB gene are associated with propionic acidemia associated with severe ketoacidosis. The original identification of a child suffering from propionyl-CoA deficiency was in 1961. This child suffered frequent episodes of severe ketoacidosis, all of which were precipitated by protein ingestion. Blood and urine analysis demonstrated marked elevations in glycine levels. These initial laboratory studies lead to the disorder being called ketotic hyperglycinemia. However, there is no defect in glycine metabolism with inherited mutations in PCCA or PCCB. The clinical hallmark of the disease is severe ketoacidosis of an episodic nature precipitated primarily due to intake of protein.
Unsaturated Fatty Acid β-Oxidation
The mitochondrial oxidation of unsaturated fatty acids is essentially the same process as for saturated fats, except when a double bond is encountered. In such a case, the bond is isomerized by one of two specific enoyl-CoA isomerases and oxidation continues. These isomerases are encoded by the enoyl-CoA delta isomerase 1 (ECI1) and ECI2 genes. The ECI1 and ECI2 encoded enzymes catalyze the transformation of 3-cis and 3-trans-enoyl-CoA esters that are the result of the mitochondrial β-oxidation of cis-, mono-, and polyunsaturated fatty acids. The products of the isomerases are the 2-trans-enoyl-CoA intermediates.
The ECI1 gene is located on chromosome 16p13.3 and is composed of 7 exons that generate two alternatively spliced mRNAs, both of which encode distinct precursor proteins.
The ECI2 gene is located on chromosome 6p25.2 and is composed of 12 exons that generate three alternatively spliced mRNAs, that collectively encode two distinct protein isoforms.
In the case of polyunsaturated fatty acids (PUFA), such as linoleate, the presence of the Δ12 unsaturation results in the formation of a dienoyl-CoA during oxidation. These dienoyl-CoA molecules are substrates for an additional oxidizing enzyme, the NADPH requiring 2,4-dienoyl-CoA reductase which is encoded by the DECR1 gene. The DECR1 gene is located on chromosome 8q21.3 and is composed of 13 exons that generate two alternatively spliced mRNAs, both of which encode distinct precursor proteins.
Because the DECR1 encoded enzyme is localized to the mitochondria and requires NADPH as a co-factor. The activity of DECR1 is, therefore, dependent upon the enzymatic conversion of NAD+ to NADP+. The phosphorylation of NAD+ in the mitochondria is catalyzed by one of the two NAD kinases, specifically the kinase encoded by the NADK2 gene.
The NADK2 gene is located on chromosome 5p13.2 and is composed of 17 exons that generate four alternatively spliced mRNAs that collectively encode three distinct protein isoforms. Expression of the NADK2 gene is highest in the liver.
2,4-Dienoyl-CoA Reductase Deficiency
Loss of function of the NADK2 gene results in loss of mitochondrial NADPH production and as a consequence reduced activity of 2,4-dienoyl-CoA reductase. This rare autosomal disorder is aptly called 2,4-dienoyl-CoA reductase deficiency. Affected individuals exhibit a variable phenotype dependent upon the type of mutation in the NADK2 gene.
Symptoms of 2,4-dienoyl-CoA reductase deficiency range from severe neurologic and metabolic dysfunction beginning in early infancy to more subtle symptoms such as childhood-onset optic atrophy or intermittent muscle weakness. In the most severe cases patients have elevated serum levels of C10:2-carnitine. Biochemical analysis in 2,4-dienoyl-CoA reductase deficiency patients will show hyperlysinemia due to the requirement for NADPH of the mitochondrial lysine catabolic enzyme, α-aminoadipate-6-semialdehyde synthase that is encoded by the AASS gene.
Peroxisomal (beta) β-Oxidation Reactions
In addition to mitochondrial oxidation of fatty acids, the peroxisomes play an important role in overall fatty acid metabolism. Very-long-chain fatty acids (VLCFAs: C18–C26) are preferentially oxidized in the peroxisomes with cerotic acid (a 26:0 fatty acid) being solely oxidized in this organelle. Peroxisomal fatty acid oxidation does not proceed to CO2 and H2O as in the mitochondria. The chain-shortened fatty acyl-CoAs, or carnitine esters, are transported out of the peroxisomes and taken up by the mitochondria where their complete oxidation takes place.
The peroxisomes also metabolize di– and trihydroxycholestanoic acids (DHCA and THCA; bile acid intermediates); long-chain dicarboxylic acids that are produced by ω-oxidation of long-chain monocarboxylic acids; pristanic acid via the α-oxidation pathway (see below); certain polyunsaturated fatty acids (PUFA) such as tetracosahexaenoic acid (24:6), which by β-oxidation yields the important PUFA docosahexaenoic acid (DHA); and certain prostaglandins and leukotrienes. The di- and trihydroxycholestanoic acids are also known as di and trihydroxycoprostanoic acids.
In order to be metabolized, the various lipids that are oxidized in the peroxisomes are activated by CoA addition and then transported into the peroxisomes by members of the ABC family of transporters. Very long-chain fatty acyl-CoAs are transported by ABCD1. Tetracosahexaenoic acid is transported by ABCD2. Di- and trihydroxycholestanoic acids are transported by ABCD3. ABCD3 also transports phytanoyl-CoA and pristanoyl-CoA.
The enzymatic processes of peroxisomal β-oxidation are very similar to those of mitochondrial fatty acid β-oxidation with one major difference. During mitochondrial fatty acid β-oxidation the first oxidation step, catalyzed by various acyl-CoA dehydrogenases, results in the generation of the reduced electron carrier, FADH2, that then delivers its’ electrons directly to the electron transport chain for synthesis of ATP. In the peroxisome the first oxidation step is catalyzed by acyl-CoA oxidases which are coupled to the reduction of O2 to hydrogen peroxide (H2O2). Thus, the reaction is not coupled to energy production but instead yields a significant reactive oxygen species (ROS). Peroxisomes also contain the enzyme catalase that degrades the hydrogen peroxide back to O2.
Humans contain three peroxisomal acyl-CoA oxidases, ACOX1, ACOX2, and ACOX3. The ACOX1 gene is located on chromosome 17q25.1 and contains 15 exons that generate three alternatively spliced mRNAs, each of which encode a distinct protein.
The ACOX2 gene is located on chromosome 3p14.3 and contains 16 exons that encode a 681 amino acid protein.
The ACOX3 gene is located on chromosome 4p16.1 and contains 23 exons that generate 10 different alternatively spliced mRNA that collectively encode seven distinct protein isoforms.
Human and rodent ACOX1 (also referred to as palmitoyl-CoA oxidase) is the major peroxisomal acyl-CoA oxidase in human peroxisomes. ACOX1 is responsible for the oxidation of straight-chain mono- and dicarboxylic fatty acids, very long-chain fatty acids, prostaglandins, and xenobiotics.
In humans, in contrast to rodents, 2-methyl (α-methyl) branched-chain fatty acids (primarily pristanoic acid) and the bile acid intermediates, di- and trihydroxycholestanoic acids, are desaturated in the peroxisomes by a single enzyme ACOX2 (also called branched-chain acyl-CoA oxidase). Expression of the human ACOX3 gene and the encoded enzyme (also referred to as pristanoyl-CoA oxidase) are detected in normal tissue only at extremely low levels. Due to the pattern of expression that has been determined thus far, the function of ACOX3 may be restricted to certain developmental stages or to tissues that have yet to be tested for expression.
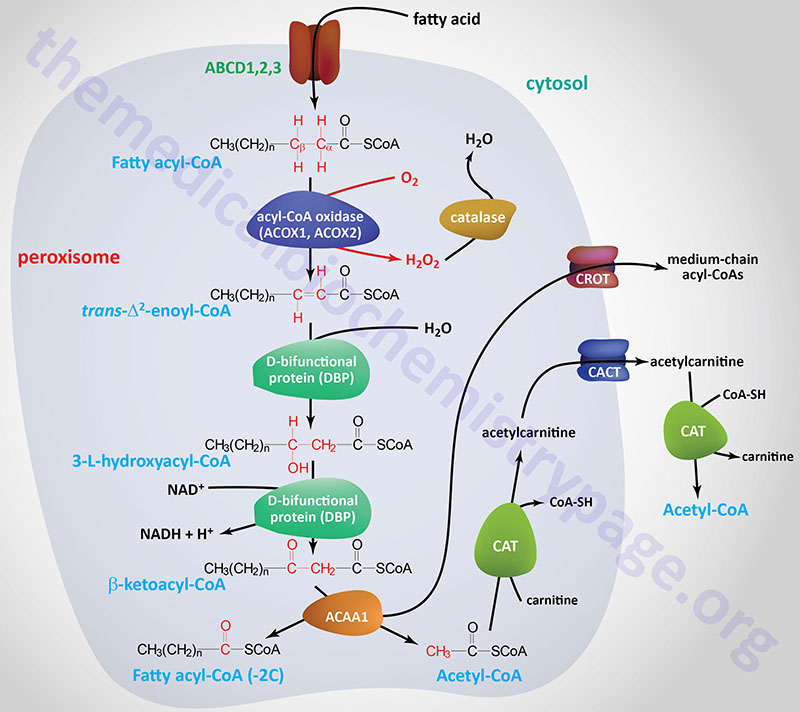
Mutations in the ACOX1 gene result in a very rare autosomal recessive disorder referred to as pseudo-neonatal adrenoleukodystrophy (pseudo-NALD). ACOX1 deficiency presents in the neonatal period with failure to thrive, hypotonia, and seizures. As the disease progresses there is developmental delay and neurological regression beginning some time between 1 and 3 years of age. Biochemical studies will find elevated plasma levels of the saturated very long-chain fatty acids (VLCFA), lignoceric acid (C24:0, systematic name is tetracosanoic acid), pentacosylic acid (C25:0, systematic name is pentacosanoic acid), and cerotic acid (C26:0, systematic name is hexacosanoic acid).
The hydration step and second oxidation step in peroxisomal β-oxidation is carried out by a single bifunctional enzyme as opposed to two separate enzymes. The two enzymatic activities are a hydratase and a dehydrogenase. There are two distinct bifunctional enzymes identified as L-bifunctional protein (LBP) and D-bifunctional protein (DBP). LBP is specific for L-3-hydroxyacyl-CoAs and DBP is specific for D-3-hydroxyacyl-CoAs. These bifunctional enzymes are also referred to as multifunctional proteins 1 and 2 (MFP-1 and -2) or L- and D-peroxisomal bifunctional enzymes (L-PBE and D-PBE).
The DBP protein is encoded by the hydroxysteroid 17-β dehydrogenase 4 (HSD17B4) gene which is located on chromosome 5q23.1 and is composed of 27 exons that generate 12 alternatively spliced mRNAs that collectively encode ten distinct protein isoforms. Mutations in the HSD17B4 gene are the cause of the autosomal recessive disorder referred to as D-bifunctional protein deficiency.
The LBP protein is encoded by the enoyl-CoA hydratase and 3-hydroxyacyl CoA dehydrogenase gene (EHHADH) which is located on chromosome 3q27.2 and is composed of 8 exons that generate two alternatively spliced mRNAs encoding two distinct protein isoforms.
DBP is the primary, if not exclusive enzyme involved in the oxidation of VLCFA, pristanic acid, and di- and trihydroxycholestanoic acids. The precise role of LBP in human peroxisomal lipid oxidation is unclear. Human peroxisomes contain the thiolase, acetyl-CoA C-acyltransferase 1 (ACAA1), that catalyzes the terminal step in the peroxisomal β-oxidation pathway.
The clinical significance of the activity of the acyl-CoA oxidases of peroxisomal β-oxidation is related to tissue specific oxidation processes. In the pancreatic β-cell there is little, if any, catalase expressed so that peroxisomal oxidation of VLCFA results in an increased release of ROS that can damage the β-cell contributing to the progressive insulin deficiency seen in obesity.
Peroxisomal Branched-Chain Fatty Acid Metabolism
Metabolism of branched-chain fatty acids and related lipids begins predominantly in peroxisomes due to the lack of substrate recognition by the mitochondrial β-oxidation enzymes. Within the mitochondria, oxidation of 2-methyl (α-methyl) branched-chain lipids requires conversion (racemization) of the 2R stereoisomers to the 2S configuration.
In addition to branched-chain fatty acid oxidation, the peroxisomes metabolize branched-chain drugs, e.g. ibuprofen. In humans, branched-chain lipids are primarily derived from the catabolism of isoprenoids and from chlorophyll in the form of phytanic acid.
The 2R to 2S racemization reactions are catalyzed by 2-methyl (α-methyl) acyl-CoA racemase which is encoded by the AMACR gene. The AMACR gene is located on chromosome 5p13.2 and is composed of 5 exons that generate three alternatively spliced mRNAs, each of which encode a distinct protein isoform.
The substrates for the AMACR encoded enzyme include (2R)/(2S)-pristanoyl-CoA and the bile acid precursor molecule (25R)/(25S)-trihydroxy cholestanoyl-CoA. Pristanoyl-CoA is a byproduct of the peroxisomal α-oxidation of phytanic acid (see next Section). The 2-methyl acyl-CoA racemase is localized both the peroxisomes and the mitochondria with 80%-90% of the total cellualar activity localized to the peroxisomes. Within the mitochondria 2-methyl acyl-CoA racemase induces β-oxidation of branched chain fatty acids and fatty acid derivatives by catalyzing the conversion of several (2R) methyl-branched chain fatty acid acyl-CoA molecules to the (S) stereoisomer.
Mutations in the AMACR gene result in two types of related disorders. The first is a late-onset disorder associated with sensory-motor neuropathy, cognitive decline, migraines, seizures, and liver abnormalities. The neuropathy symptoms of late-onset AMACR deficiency are similar to those of Refsum disease. The other form of AMACR deficiency is an early onset disorder characterized by severe liver abnormalities.
Peroxisomal (alpha) α-Oxidation Reactions
Phytanic acid [(7R,11R)-3,7,11,15-tetramethylhexadecanoic acid] is a 3-methyl branched-chain fatty acid derived from the chlorophyll component, phytol. Phytanic acid is present in the tissues of ruminants, certain fishes, and in dairy products and is, therefore, an important dietary component of fatty acid intake. Because phytanic acid is a 3-methyl branched-chain fatty acid, it cannot act as a substrate for any of the first enzymes of the mitochondrial β-oxidation pathway (the acyl-CoA dehydrogenases).
Phytanic acid is first converted to its CoA-ester and then phytanoyl-CoA serves as a substrate in an α-oxidation process. The α-oxidation reaction (as well as the remainder of the reactions of phytanic acid oxidation) occurs within the peroxisomes and requires a specific α-hydroxylase (specifically phytanoyl-CoA hydroxylase, PhyH), which adds a hydroxyl group to the α-carbon of phytanic acid. PhyH is also called phytanoyl-CoA 2-hydroxylase or phytanoyl-CoA dioxygenase. Phytanoyl-CoA hydroxylase is a member of the large family of 2-oxoglutarate and Fe2+-dependent dioxygenases (2-OGDD).
Phytanoyl-CoA hydroxylase is one of two human enzymes that are classified as fatty acid 2-hydroxylases. The other enzyme in this family is fatty acid 2-hydroxylase which is encoded by the FA2H gene. The FA2H encoded enzyme is involved in the generation of a specific family of sphingolipids and ceramides. This pathway is described in the Sphingolipid Metabolism and the Ceramides page.
The PhyH enzyme is derived from the PHYH gene that is located on chromosome 10p13 and is composed of 10 exons that generate six alternatively spliced mRNAs that encode a total of five distinct protein isoforms. One of the mRNAs encodes the larger protein of 338 amino acids that contains a cleavable peroxisomal targeting signal type 2 (PTS2). The PTS2 signal is found at the N-terminus of the PhyH protein.
The product of the PhyH reaction is 2-hydroxyphytanoyl-CoA (2-OH phytanoyl-CoA) which is a substrate for 2-hydroxyphytanoyl-CoA lyase. The activity of 2-hydroxyphytanoyl-CoA lyase is dependent on thiamine pyrophosphate (TPP) as a cofactor.
2-Hydroxyphytanoyl-CoA lyase is encoded by the HACL1 gene. The HACL1 gene is located on chromosome 3p25.1 and is composed of 17 exons that generate four alternatively spliced mRNAs, each of which encode a distinct protein isoform.
The action of 2-hydroxyphytanoyl-CoA lyase yields formyl-CoA and pristanal. Pristanal is converted to pristanic acid via the action of the aldehyde dehydrogenase encoded by the ALDH3A2 (aldehyde dehydrogenase 3 family member A2) gene. The ALDH3A2 encoded enzyme is also known as fatty aldehyde dehydrogenase (FALDH).
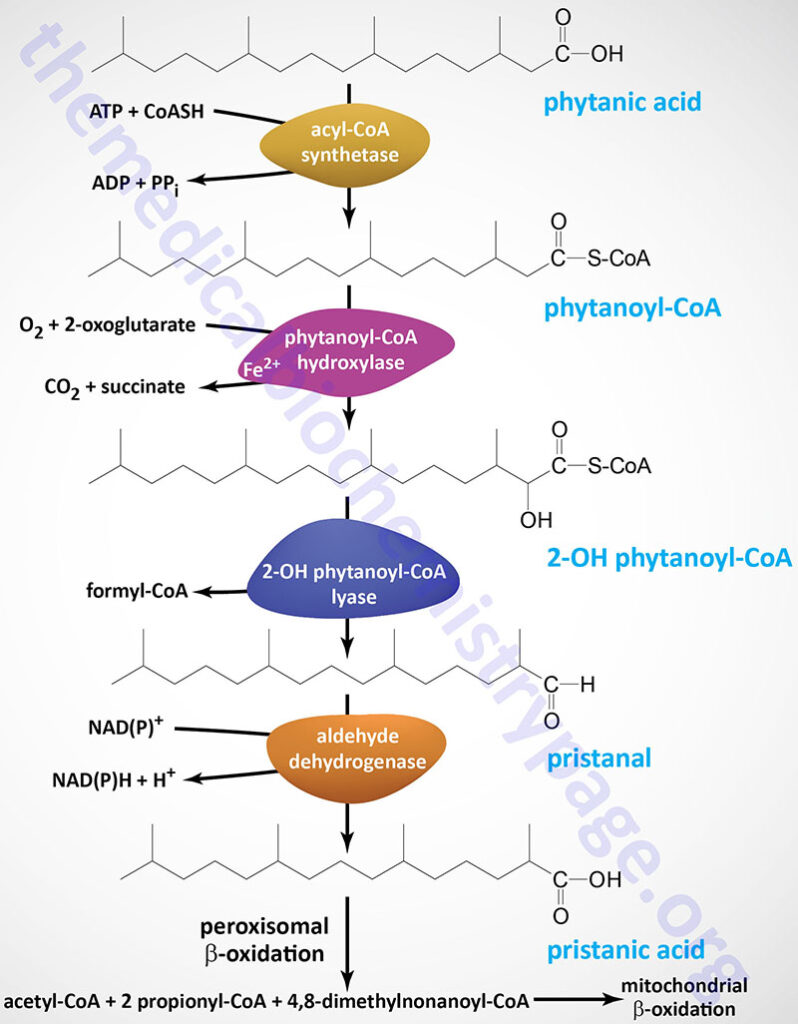
Pristanic acid (2,6,10,14-tetramethylpentadecanoic acid) then serves as a substrate for the peroxisomal process of β-oxidation. Pristanic acid is activated to pristanoyl-CoA which then serves as a substrate for D-bifunctional protein of the peroxisomal β-oxidation pathway.
After three steps of peroxisomal β-oxidation, the medium-chain fatty acid product (4,8-dimethylnonanoic acid) is esterified to carnitine and transported out of the peroxisome for uptake by the mitochondria where it is further oxidized via the mitochondrial β-oxidation pathway.
The propionyl-CoAs are transported out of the peroxisomes for metabolism in the mitochondria to the TCA cycle intermediate, succinyl-CoA. Within the liver the acetyl-CoA is transported out of the peroxisomes and can be taken up by the mitochondria for synthesis of the ketones, acetoacetate and β-hydroxybutyrate.
Microsomal (omega) ω-Oxidation: Dicarboxylic Acid Synthesis
The microsomal (endoplasmic reticulum, ER) pathway of fatty acid ω-oxidation represents a minor pathway of overall fatty acid oxidation. However, in certain pathophysiological states, such as diabetes, chronic alcohol consumption, and starvation, the ω-oxidation pathway may provide an effective means for the elimination of toxic levels of free fatty acids. The pathway refers to the fact that fatty acids first undergo a hydroxylation step at the terminal (omega, ω) carbon. Human ω-hydroxylases are all members of the cytochrome P450 (CYP) family of enzymes. These enzymes are abundant in the liver and kidneys. Specifically, the human ω-hydroxylase enzymes are members of the CYP4A and CYP4F families that preferentially hydroxylate the terminal methyl group of C10–C26 length fatty acids.
CYP4A11 is the human homolog of the rat liver CYP4A1 gene whose encoded enzyme was the first ω-hydroxylase characterized. CYP4A11 utilizes NADPH and O2 to introduce an alcohol to the ω-CH3– of several fatty acids including lauric (12:0), myristic (14:0), palmitic (16:0), oleic (18:1), and arachidonic acid (20:4).
Following addition of the ω-hydroxyl the fatty acid is a substrate for an alcohol dehydrogenase (ADH) which generates an oxo-fatty acid, followed by generation of the corresponding dicarboxylic acid via the action of an aldehyde dehydrogenase (ALDH) family member enzyme. Further metabolism then takes place via the β-oxidation pathway in peroxisomes.
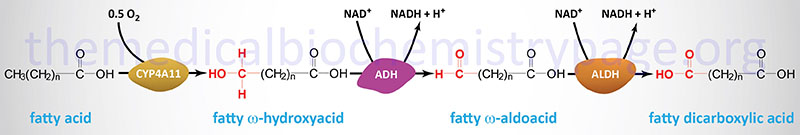
The process of generating ω-hydroxy fatty acids is not solely for the eventual conversion to a dicarboxylic acid. Several ω-hydroxy fatty acids are substrates for the synthesis of a specialized class of sphingolipids identified as the ω-hydroxyceramides and the ω-O-acylceramides. These ω-hydroxy fatty acids are generated by the CYP4F family member ω-hydroxylase, CYP4F22, acting on a class of fatty acids referred to as ultra-long-chain fatty acids (ULCFA) because they are greater than 28 carbons in length. The ω-O-acylceramides are particularly important in the skin where they function in the skin barrier formation.
The formation of ω-hydroxylated arachidonic acid, identified as 20-hydroxyeicosatetraenoic acid (20-HETE), by CYP4A11 plays an important role in various pathologies. 20-HETE is involved in the activation of pro-inflammatory pathways, and the signaling pathways leading to hypertension. Both of these pathologic states contribute to the development of obesity, type 2 diabetes, and cardiovascular disease. In addition to CYP4A11 the CYP4F family member ω-hydroxylase, CYP4F22, can also generate 20-HETE.
The function of 20-HETE is exerted through its binding to a specific GPCR identified as GPR75. GPR75 is coupled to a Gq-type G-protein and thus, its activation results in increased IP3 and DAG production. The increased production of IP3 results in release of ER stored Ca2+ and activation of various Ca2+-mediated signaling cascades.
With respect to the vasculature, the activation of GPR75 by 20-HETE in vascular smooth muscle cells leads to inhibition of the large conductance calcium- and voltage-activated potassium channel (designated KCa1.1 and encoded by the KCNMA1 gene). The KCNMA1 encoded potassium channel is also known as MaxiK or BK (big potassium). The inhibition of the KCNMA1 channel underlies the hypertension-inducing effects of 20-HETE. Indeed, polymorphisms in the CYP4A11 gene are associated with hypertension in certain populations, particular Asian populations.
In addition to ω-hydroxylation of arachidonic acid and LTB4, CYP4F2 has been shown to be responsible for the ω-hydroxylation of the phytyl tail of the tocopherols and tocotrienols (collectively known as vitamin E). Metabolism of vitamin E requires an initial ω-hydroxylation step followed by subsequent β-oxidation.
Another human CYP4A family member has been identified and designated CYP4A22. The CYP4A22 protein is highly homologous with CYP4A11 and has been shown to exhibit lauric acid ω-hydroxylase activity. Expression of CYP4A22 is low in all tissues in which it is found.
The CYP4A family is not the only CYP4 family of enzymes that have been found to possess ω-hydroxylase activity. The CYP4F family enzyme CYP4F3A, which is expressed in leukocytes, is necessary for the ω-hydroxylation and subsequent degradation of leukotriene B4 (LTB4). LTB4 plays an important role in the modulation of inflammatory processes.
The CYP4F3 gene is subject to alternative promoter usage and tissue-specific alternative mRNA splicing, which results in two different proteins being produced. These two enzymes are designated CYP4F3A and CYP4F3B, with the latter enzyme being expressed in the liver. CYP4F3B has higher affinity for arachidonic acid.
Another CYP4F family member, identified as CYP4F2, has also been shown to exhibit LTB4-hydroxylating activity. The CYP4F2 protein has a high degree of homology to the CYP4F3B protein and is expressed in the liver and kidneys. CYP4F2 has been shown to be the major arachidonic acid ω-hydroxylase in human liver and kidney. Indeed, the substrate specificity of CYP4F2 for arachidonic acid is much higher than that of CYP4A11, which was originally described as a significant arachidonic acid ω-hydroxylase. In addition to ω-hydroxylation of arachidonic acid and LTB4, CYP4F2 has been shown to be responsible for the ω-hydroxylation of the phytyl tail of the tocopherols and tocotrienols (collectively known as vitamin E). Metabolism of vitamin E requires an initial ω-hydroxylation step followed by subsequent β-oxidation.
Additional members of the CYP4F subfamily have been identified in humans. These genes are designated CYP4F8, CYP4F11, and CYP4F12. CYP4F8 is present in epithelial linings and catalyzes the (ω1)-hydroxylation of prostaglandin H2 (PGH2). CYP4F11 is primarily expressed in liver, but also found in kidney, heart, brain and skeletal muscle. The primary endogenous substrates for CYP4F11 are long-chain 3-hydroxydicarboxylic acids (3-OHDCA) and the enzyme is also very active at hydroxylating various xenobiotics. CYP4F12 is expressed liver, heart, gastrointestinal and urogenital epithelia and its primary substrates are eicosanoids and xenobiotics.
Peroxisomal Oxalate (Dicarboxylic Acid) Metabolism
Generally, dicarboxylic acids generated via the ω-oxidation pathway, are degraded in the mitochondria as well as the peroxisomes by their respective fatty acid β-oxidation pathways. Of clinical significance is the short-chain dicarboxylic acid, oxalic acid (oxalate). Oxalate is a dicarboxylic acid composed of two carbon atoms and exits in solution as a dianion of the formula C2O42– (–OOC—COO–).
Oxalate is present in many foods such as spinach, chard, nuts, black pepper, poppy seeds, and also occurs as a result of the metabolism of ascorbic acid, and the amino acids glycine, serine, and 4-hydroxyproline as well as metabolism of ethanolamine. The majority (40%) of daily endogenous oxalate production comes from the metabolism of dehydroascorbate. Only around 0.1% of endogenous oxalate arises from glycine catabolism, whereas approximately 15% of the daily endogenous production of glyoxylate derives from 4-hydroxyproline. The origin of 4-hydroxyproline is either the diet or the degradation of collagen proteins.
Glyoxylate and L-glycerate glycolate are immediate precursors of oxalate. Glyoxylate is a byproduct of the metabolism of serine and glycine. The majority of serine catabolism involves its conversion to glycine, followed by glycine decarboxylase-mediated metabolism. Alternatively, glycine can be converted to serine and serine can be converted to 3-hydroxypyruvate via transamination of pyruvate. The 3-hydroxypyruvate can then be converted to glycoaldehyde via the action of glyoxylate and hydroxypyruvate reductase (encoded by the GRHPR gene). The glycoaldehyde is ultimately metabolized to glyoxylate.
The modified amino acid, 4-hydroxyproline (released from collagen breakdown), is ultimately metabolized to glyoxylate and pyruvate in the pathway outlined in the Figure below where the formation of glyoxylate and pyruvate is catalyzed via the action of 4-hydroxy-2-oxoglutarate aldolase 1 (encoded by the HOGA1 gene).
Glyoxylate can be metabolized to oxalate by hydroxyacid oxidase 1 (encoded by the HAO1 gene; also called glycolate oxidase) as well as by lactate dehydrogenase. Glyoxylate and alanine can also be converted to glycine and pyruvate, respectively, via the action of the vitamin B6-dependent enzyme, alanine–glyoxylate and serine–pyruvate aminotransferase (encoded by the AGXT gene). The AGXT gene is expressed exclusively in hepatocytes and is localized to the peroxisomes.
Glycolate, produced from glyoxylate in the mitochondria of non-hepatic tissues via the activity of the HAO1 encoded enzyme, can delivered to the blood and transferred to the liver where it can be used for glycine synthesis in the peroxisomes. This pathways involves conversion of glycolate back to glyoxylate and then the AGXT encoded enzyme converts glyoxylate to glycine and pyruvate as detailed in the Figure above.
Excess glyoxylate will enhance the metabolism of glyoxylate to oxalate and can lead to hyperoxaluria. There are several forms of inherited hyperoxaluria. Oxalate is excreted in the urine and approximately 50% – 80% is derived from the diet. If levels of oxalate in the glomerular filtrate increase (hyperoxaluria) it can chelate Ca2+ ions forming the insoluble compound, calcium oxalate. Indeed, calcium oxalate crystals are the most common type of compound contributing to renal calculi (kidney stones).
Hyperoxaluria
The hyperoxalurias are divided into two categories: the primary hyperoxalurias (PH) and the secondary hyperoxalurias. Primary hyperoxalurias are rare autosomal recessive disorders of glyoxylate metabolism that are associated with high oxalate production, primarily within the liver. There are three distinct forms of PH identified as PH1, PH2, and PH3 (or PH I, PH II, and PH III). The primary hyperoxalurias are associated with overproduction and excessive urinary excretion of oxalate. The excess urinary oxalate leads to recurrent urolithiasis and nephrocalcinosis. Glomerular filtration rates decline in PH due to progressive renal involvement leading to oxalate accumulation and systemic oxalosis that manifests with metabolic bone disease, anemia, and skin ulcerations.
Primary hyperoxaluria type 1 is the most common inherited form of hyperoxaluria, representing 70%-80% of all cases. PH 1 results from mutations in the peroxisomal specific enzyme alanine–glyoxylate and serine–pyruvate aminotransferase encoded by the AGXT gene. The AGXT gene is located on chromosome 2q37.3 and is composed of 11 exons that encode a 392 amino acid protein.
Primary hyperoxaluria type 2 accounts for around 10% of all cases. PH 2 results from mutations in glyoxylate and hydroxypyruvate reductase encoded by the GRHPR gene. The GRHPR gene is located on chromosome 9p13.2 and is composed of 12 exons that encode a 328 amino acid protein. Expression of the GRHPR gene is seen in most tissues with the highest levels in the liver and then the kidney.
Primary hyperoxaluria type 3 results from mutations in 4-hydroxy-2-oxoglutarate aldolase 1 encoded by the HOGA1 gene. The HOGA1 gene is located on chromosome 10q24.2 and is composed of 7 exons that generate two alternatively spliced mRNAs that encode proteins of 327 (isoform 1) and 164 (isoform 2) amino acids. The HOGA1 gene is predominantly expressed in the kidney and the enzyme is localized to the mitochondria.
Fatty Acid Oxidation and Thermogenesis in Brown Adipose Tissue
Brown adipose tissue (BAT) is so-called due to the presence of high concentrations of mitochondria in the individual adipose cells. The primary function of BAT is the regulation body temperature, particularly in response to cold exposure, as well as in the control of energy expenditure. The ability of BAT to regulate body temperature (thermogenesis) involves the uncoupling of proton flow in mitochondria. This uncoupling process releases the energy of the electrochemical proton gradient as heat. This process, referred to as adaptive thermogenesis, is a normal physiological function of brown adipose tissue. Newborn babies contain brown fat in their neck and upper back that serves the function of non-shivering thermogenesis.
The muscle contractions that take place in the process of shivering not only generates ATP but also produces heat, referred to as shivering thermogenesis. Non-shivering thermogenesis is a hormonal stimulus for heat generation without the associated muscle contractions of shivering. The process of thermogenesis in brown fat is initiated by the release of free fatty acids from the triglycerides stored in the adipose cells. The hormonal release of fatty acids in brown fat is the same as that described for the mobilization of stored fat in the Mobilization of Fat Stores section above.
The most studied regulator of the metabolic and thermogenic properties of BAT is the catecholamine, norepinephrine. Within BAT, norepinephrine interacts with all three types of adrenergic receptors (α1, α2, and β) each of which activates distinct signaling pathways in the brown adipocyte. With respect to the β-adrenoreceptors, the β3 subtype is the most significant, β1 receptors are expressed in brown preadipocytes but not mature adipocytes and β2 receptors are expressed in BAT but not brown adipocytes themselves.
Of significance to the role of BAT in thermogenesis and the major role of β3-adrenoreceptors is the fact that these receptors are not subject to desensitization as are the β1 and β2 receptors. However, this is not to say that continuous adrenergic stimulation has no negative effect in BAT. Indeed, the level of β3 receptor expression is downregulated during continuous adrenergic stimulation. However, this effect is transient and the mRNA rapidly re-accumulates after termination of the stimulatory signal.
Signal transduction events triggered by adrenergic stimulation of BAT are effected via the activation of Gs type G-proteins associated with the β-adrenergic receptors. Gs-type G-proteins couple to the activation of adenylate cyclase resulting in increased cAMP production and activation of PKA. PKA then phosphorylates and activates HSL, the consequences of which are increased release of free fatty acids from triglycerides. These effects are identical to the effects of catecholamines in white adipose tissue (WAT).
The free fatty acids are taken up by the mitochondria where they can undergo β-oxidation resulting in increased energy consumption. However, in BAT, with respect to the process of thermogenesis, the fatty acids can also interact with and activate the proton gradient uncoupling activity of uncoupling protein 1 (UCP1, also known as thermogenin).
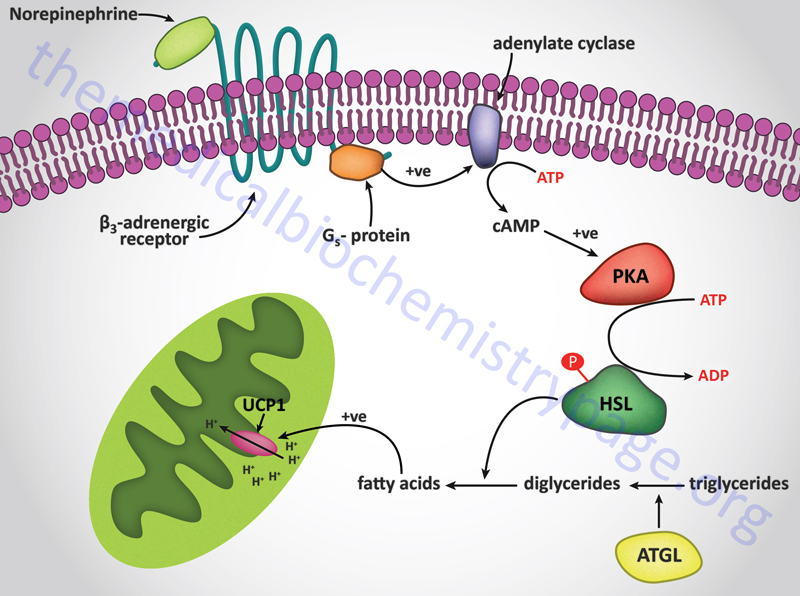
There are three uncoupling protein genes in humans identified as UCP1, UCP2, and UCP3. UCP1 is expressed in brown adipose tissue (BAT), UCP2 is expressed in a number of tissues, and UCP3 is expressed in skeletal and heart muscle. Only UCP1 is involved in the process of adaptive thermogenesis. UCP1 acts as a channel in the inner mitochondrial membrane to control the permeability of the membrane to protons. The binding of free fatty acids to UCP1 triggers an uncoupling of the proton gradient and the release of the energy of the gradient as heat.
In addition to norepinephrine acting through the adrenergic receptors, adenosine and secretin stimulate lipolysis and thermogenesis in BAT. Adenosine binds to the adenosine 2A (A2A) receptor (encoded by the ADORA2A gene) which is coupled to a Gs-type G-protein. Secretin binds to the secretin receptor (encoded by the SCTR gene) which is also coupled to a Gs-type G-protein. Thus, the effects of adenosine and secretin are similar to those of norepinephrine in BAT.
Another Gs-type GPCR that functions in BAT to regulate lipolysis and thermogenesis is GPR3. GPR3 is a constitutively active receptor meaning that it activates the production of cAMP in the absence of a ligand. In addition to being expressed in BAT, GPR3 is also expressed in the central nervous system and the ovaries.
Receptors on brown adipocytes that counter the actions of norepinephrine, adenosine, and secretin include the insulin receptor and hydroxycarboxylic acid receptor 2 (HCA2; encoded by the HCAR2 gene). The HCA2 receptor was originally identified as GPR109A. Activation of the insulin receptor in BAT results in the activation of the phosphodiesterase, PDE3B. Activation of PDE3B results in the breakdown of cAMP, thus terminating the activation of lipolysis and thermogenesis in BAT. HCA2 is coupled to a Gi-type G-protein and is activated by the ketone, β-hydroxybutyrate (BHB). The activation of Gi-type G-proteins leads to the inhibition of adenylate cycles and, thus, reduced production of cAMP, resulting in the same effects as those initiated by activation of the insulin receptor..
Regulation of Fatty Acid Metabolism
In order to understand how the synthesis and degradation of fats needs to be exquisitely regulated, one must consider the energy requirements of the organism as a whole. The blood is the carrier of triglycerides in the form of VLDLs and chylomicrons, fatty acids bound to albumin, amino acids, lactate, ketone bodies and glucose. The pancreas is the primary organ involved in sensing the organism’s dietary and energetic states by monitoring glucose concentrations in the blood. Low blood glucose stimulates the secretion of glucagon, whereas, elevated blood glucose calls for the secretion of insulin.
The metabolism of fat is regulated by two distinct mechanisms. One is short-term regulation, which can come about through events such as substrate availability, allosteric effectors and/or enzyme modification. The other mechanism, long-term regulation, is achieved by alteration of the rate of enzyme synthesis and turn-over.
Fat metabolism can also be regulated by malonyl-CoA-mediated inhibition of CPT1. Such regulation serves to prevent de novo synthesized fatty acids from entering the mitochondria and being oxidized. The predominant source of the malonyl-CoA that inhibits CPT1 is from the ACC2 enzyme.
Regulation of ACC1 and ACC2
ACC1 catalyzes the rate-limiting (committed) step in fatty acid synthesis. There are two major isoforms of ACC in mammalian tissues. These are identified as ACC1 and ACC2. ACC1 is strictly cytosolic and is enriched in liver, adipose tissue, and lactating mammary tissue. ACC2 was originally discovered in rat heart but is also expressed in liver and skeletal muscle. ACC2 has an N-terminal extension that contains a mitochondrial targeting motif and is found associated with CPT1 allowing for rapid regulation of CPT1 by the malonyl-CoA produced by ACC2. The localization of ACC2, and the inhibition of CPT1 by the malonyl-CoA generated by the enzyme, indicate that ACC2 is primarily responsible for the regulation of fatty acid oxidation.
Both isoforms of ACC are allosterically activated by citrate and inhibited by palmitoyl-CoA and other short- and long-chain fatty acyl-CoAs. Citrate triggers the polymerization of ACC1 which leads to significant increases in its activity. Although ACC2 does oligomerization similar to that of ACC1, it does not appear to occur to the extent of that of ACC presumably due to its mitochondrial association. However, ACC2 is also allosterically activated by citrate. Glutamate and other dicarboxylic acids can also allosterically activate both ACC isoforms.
ACC activity can also be affected by phosphorylation. Both ACC1 and ACC2 contain at least eight sites that undergo phosphorylation. The sites of phosphorylation in ACC2 have not been as extensively studied as those in ACC1. Phosphorylation of ACC1 at three serine residues (S79, S1200, and S1215) by AMPK leads to inhibition of the enzyme. AMPK-mediated phosphorylation of Ser79 appears to be the most significant for the regulation of ACC1 activity. Glucagon-stimulated increases in cAMP, and subsequently to increased PKA activity, also lead to phosphorylation of ACC. ACC2 is a better substrate for PKA than is ACC1.
The activating effects of insulin on ACC are complex and not completely resolved. It is known that insulin leads to the dephosphorylation of the serines in ACC1 that are AMPK targets in the heart enzyme. This insulin-mediated effect has not been observed in hepatocytes or adipose tissues cells. At least a portion of the activating effects of insulin are related to changes in cAMP levels. Activation of α1-adrenergic receptors in liver and skeletal muscle cells inhibits ACC activity as a result of phosphorylation by an as yet undetermined kinase.
Insulin, a product of the well-fed state, stimulates ACC1 and FAS synthesis, whereas starvation leads to a decrease in the synthesis of these enzymes. Adipose tissue levels of lipoprotein lipase also are increased by insulin and decreased by starvation. However, the effects of insulin and starvation on lipoprotein lipase in the heart are just the inverse of those in adipose tissue. This sensitivity allows the heart to absorb any available fatty acids in the blood in order to oxidize them for energy production. Starvation also leads to increases in the levels of cardiac enzymes of fatty acid oxidation, and to decreases in FAS and related enzymes of fatty acid synthesis.
The activity of ACC2 is also regulated by hydroxylation, a modification that enhances its activity. One of the prolyl hydroxylase domain (PHD) family enzymes has been shown to modify the activity of ACC2. The PHD enzymes are 2-oxoglutarate and Fe2+ dependent enzymes whose roles in the regulation of cellular responses to hypoxia have been well studied. With respect to ACC2, the PHD3 enzyme has been shown to be responsible for hydroxylation of the enzyme. In contrast to the phosphorylation of ACC2 by AMPK during energy-deficient states, hydroxylation of ACC2 increases under conditions of high glucose content, in particular in skeletal muscle. When ACC2 is phosphorylated the interaction with PHD3 is impaired.
Regulation of Hormone Sensitive Lipase
Adipose tissue contains hormone-sensitive lipase (HSL), which is activated by PKA-dependent phosphorylation; this activation increases the release of fatty acids into the blood. This in turn leads to the increased oxidation of fatty acids in other tissues such as muscle and liver. In the liver, the net result (due to increased acetyl-CoA levels) is the production of ketone bodies (see below). This would occur under conditions in which the carbohydrate stores and gluconeogenic precursors available in the liver are not sufficient to allow increased glucose production. The increased levels of fatty acid that become available in response to epinephrine are assured of being completely oxidized, because PKA also phosphorylates and inhibits ACC1; the synthesis of fatty acid is thereby inhibited.
The activity of HSL is also affected via phosphorylation by AMPK. In this case the phosphorylation inhibits the enzyme. Inhibition of HSL by AMPK may seem paradoxical since the release of fatty acids stored in triglycerides would seem necessary to promote the production of ATP via fatty acid oxidation and the major function of AMPK is to shift cells to ATP production from ATP consumption. This paradigm can be explained if one considers that if the fatty acids that are released from triglycerides are not consumed they will be recycled back into triglycerides at the expense of ATP consumption. Thus, it has been proposed that inhibition of HSL by AMPK mediated-phosphorylation is a mechanism to ensure that the rate of fatty acid release does not exceed the rate at which they are utilized either by export or oxidation.
Insulin has the opposite effect to epinephrine in adipose tissue: it increases the synthesis of triglycerides. One of the many effects of insulin is to lower cAMP levels, which leads to increased dephosphorylation through the enhanced activity of protein phosphatases such as PP-1. With respect to fatty acid metabolism, this yields dephosphorylated and inactive hormone-sensitive lipase. Insulin also stimulates certain phosphorylation events. This occurs through activation of several cAMP-independent kinases.
Exercising Muscle and Regulation of Fatty Acid Oxidation
During exercise, skeletal muscle cells produce and secrete numerous factors referred to as myokines and also as exerkines. One such exerkine, that exerts effects on both fatty acid and glucose metabolism, is β-aminoisobutyrate (BAIBA). BAIBA is a non-protein amino acid that exists in two enantiomeric forms, D-BAIBA and L-BAIBA.
The synthesis of D-BAIBA involves the metabolism of thymine. Thymine is first converted to dihydrothymine through the action of dihydropyrimidine dehydrogenase (encoded by the DPYD gene). Dihydropyrimidine dehydrogenase represents the rate-limiting enzyme in the catabolism of both thymine and uracil. Dihydrothymine is then converted to N-carbamoyl-BAIBA via the action of dihydropyrimidase (encoded by the DPYS gene). Finally, N-carbamoyl-BAIBA is converted to D-BAIBA via the action of beta-ureidopropionase 1 (encoded by the UPB1 gene). These same three enzymes catabolize uracil to β-alanine.
The synthesis of L-BAIBA involves the catabolism of valine. One of the last products of valine catabolism is L-methylmalonic acid semialdehyde which can be reversibly converted to L-BAIBA via the actions of 4-aminobutyrate aminotransferase (encoded by the ABAT gene). The ABAT encoded enzyme is primarily expressed in the brain, skeletal muscle, liver, and kidney. Within the brain the function of 4-aminobutyrate aminotransferase is the metabolism of the neurotransmitter, GABA.
The catabolism of both D-BAIBA and L-BAIBA involves conversion to D-methylmalonic acid semialdehyde and L-methylmalonic acid semialdehyde, respectively. Conversion of D-BAIBA to D-methylmalonic acid semialdehyde is catalyzed by pyridoxal phosphate-dependent enzyme, alanine–glyoxylate aminotransferase 2 (encoded by the AGXT2 gene). Conversion of L-BAIBA to L-methylmalonic acid semialdehyde involves the ABAT encoded enzyme. Both methylmalonic acid semialdehydes are converted to propionyl-CoA via the actions of aldehyde dehydrogenase 6 family member A1 (encoded by the ALDH6A1 gene). The ALDH6A1 encoded enzyme is more commonly referred to as methylmalonate semialdehyde dehydrogenase (MMSDH). Mutations in the AGXT2 gene are associated with benign β-aminoisobutyric aciduria.
BAIBA exerts numerous effects that modulate fatty acid metabolism in the liver that includes increased fatty acid β-oxidation and ketone body production. These effects are, in part, the result of increased expression of the gene encoding carnitine palmitoyltransferase 1 (CPT1) in hepatocytes. BAIBA has been shown to activate AMPK as well as the transcription factors of the peroxisome proliferator-activated receptor family, PPARα and PPARβ/δ, while simultaneously inhibiting PPARγ. Via the activation of AMPK, BAIBA induces inhibition of the rate-limiting enzyme of fatty acid synthesis, acetyl-CoA carboxylase 1 (ACC1) as well as the activity of the metabolic regulatory transcription factor, SREBP-1c. The activation of AMPK and the transcription factors of the PPAR family are the direct contributors to the increased hepatic fatty acid β-oxidation observed in response to BAIBA. The increase in hepatic fatty acid oxidation contributes to increased insulin sensitivity and glucose homeostasis. The effects of BAIBA also contribute to reductions in inflammation since both AMPK and PPARβ/δ decrease the activity of the potent inflammatory gene inducing transcription factor, NF-κB.
BAIBA also exerts effects in adipose tissue that result in increased “browning” of adipose tissue leading to increased fatty acid storage in triglycerides. These effects of BAIBA are, in part, due to the activation of PPARα in adipocytes. The activation of PPARα leads to increased expression of the transcriptional co-activator, PGC-1α as well as the genes encoding uncoupling protein-1 (UCP1) and components of mitochondrial oxidative phosphorylation such as cytochrome c (CYCS).
The mechanism by which BAIBA exerts these effects on lipid metabolism in the liver and adipose tissue is not fully elucidated but is likely to involve its binding to, and activating of, one or more receptors. Candidate receptors of BAIBA include the free fatty acid receptor 3 (FFAR3 which was originally identified as GPR41) and MAS related GPR family member D (MRGPRD), both of which are members of the GPCR family. Additional potential receptors for BAIBA are the receptor for glycine (GlyR) and the GABA-A receptor, GABAAR. Both of these latter two receptors are members of the family of ligand-gated ion channels.
The Glucose-Fatty Acid Cycle
The glucose-fatty acid cycle describes interrelationships of glucose and fatty acid oxidation as defined by fuel flux and fuel selection by various organs. This cycle is not a metabolic cycle such as can be defined by the TCA cycle as an example, but defines the dynamic interactions between these two major energy substrate pools. The glucose-fatty acid cycle was first proposed by Philip Randle and co-workers in 1963 and is, therefore, sometimes referred to as the Randle cycle or Randle hypothesis. The cycle describes how nutrients in the diet can fine-tune metabolic processes on top of the more coarse control exerted by various peptide and steroid hormones. The underlying theme of the glucose-fatty acid cycle is that the utilization of one nutrient (e.g. glucose) directly inhibits the use of the other (in this case fatty acids) without hormonal mediation. The general interrelationships between glucose and fatty acid utilization in skeletal muscle and adipose tissue that constitutes the glucose-fatty acid cycle are diagrammed in the Figure below.
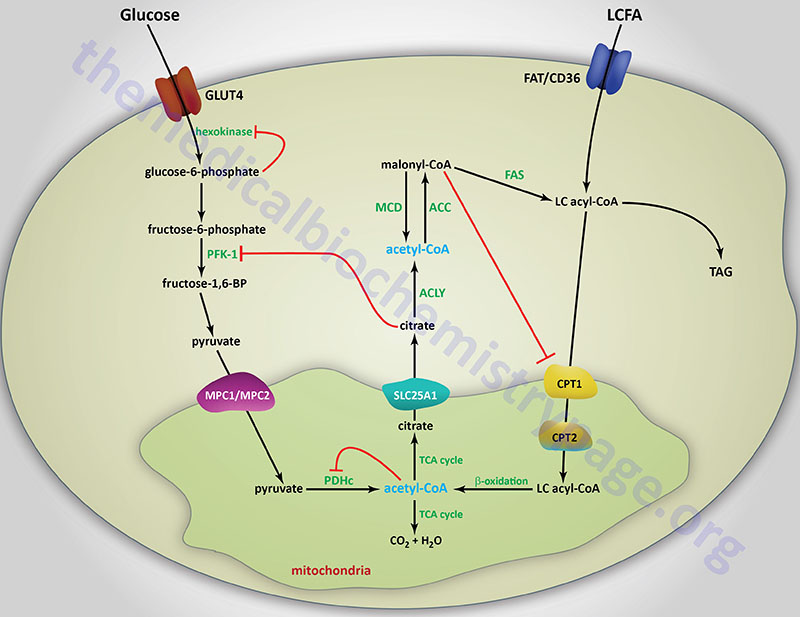
How do the dynamics of the glucose-fatty acid cycle play out under various physiological conditions and changing fuel substrate pools? In the fasted state it is imperative that glucose be spared so that the brain can have adequate access to this vital fuel. Under these conditions, epinephrine (and possibly glucagon), stimulate adipose tissue lipolysis releasing free fatty acids (FFAs) to the blood for use as a fuel by other peripheral tissues. When the released FFAs enter the liver they oxidized and also serve as substrates for ketogenesis. The oxidation of fatty acids inhibits glucose oxidation as outlined in the above figure. In addition to sparing glucose for the brain, fatty acid oxidation also preserves pyruvate and lactate which are important gluconeogenesis substrates. The effects of fatty acids on glucose utilization can also be observed in the well fed state after a high fat meal and during periods of exercise.
As outlined in the above Figure, the inhibition of glucose utilization by fatty acid oxidation is mediated by short-term effects on several steps of overall glycolysis that include glucose uptake, glucose phosphorylation and pyruvate oxidation. During fatty acid oxidation the resultant acetyl-CoA allosterically activates PDKs that phosphorylate and inhibit the PDHc. PDKs are also activated by increasing levels of NADH that will be the result of increased fatty acid oxidation. Thus, two products of fat oxidation result in inhibition of the PDHc. In addition, excess acetyl-CoA is transported to the cytosol either as citrate (as diagrammed in the Synthesis of Fatty Acids page) or as acetylcarnitine. Mitochondrial acetylcarnitine is formed through the action of carnitine O-acetyltransferase (CAT: encoded by the CRAT gene).
Acetyl-carnitine is transported out of the the mitochondria via the action of carnitine-acylcarnitine translocase (CACT: encoded by the SLC25A20 gene). Once in the cytosol acetyl-carnitine is converted to acetyl-CoA via the action of cytosolic CAT. In the cytosol, citrate serves as an allosteric inhibitor of PFK1 thus limiting entry of glucose into glycolysis. The increase in glucose-6-phosphate that results from inhibition of PFK1 leads to feed-back inhibition of hexokinase which in turn limits glucose uptake via GLUT4. Additional mechanisms of fatty acid metabolism that lead to interference in glucose uptake and utilization are the result of impaired insulin receptor signaling. These latter processes are discussed in detail in the Insulin Function, Insulin Resistance, and Food Intake Control of Secretion page.
Mechanisms by which glucose utilization inhibits fatty acid oxidation are tissue specific due primarily to the differences in Km of hepatic glucokinase and skeletal muscle and adipose tissue hexokinase. In addition, hepatic CPT1 is approximately 100-fold less sensitive to inhibition by malonyl-CoA than are the skeletal muscle and cardiac isoforms. When glucose is oxidized in glycolysis the resultant pyruvate enters the mitochondria via the pyruvate symporter. Increasing mitochondrial pyruvate inhibits the PDKs allowing for rapid decarboxylation of pyruvate by the PDHc ensuring continued entry of glucose into the glycolytic stream. Some of the acetyl-CoA derived from pyruvate oxidation will be diverted from the TCA cycle as citrate and transported to the cytosol by the tricarboxylic acid transporter (TCAT). The citrate is converted to acetyl-CoA and oxaloacetate by ATP-citrate lyase (ACLY) and can now serve as a substrate for ACC. The resultant malonyl-CoA will inhibit CPT1 thus, restricting mitochondrial uptake and oxidation of fatty acyl-CoAs. The inhibition of fatty acid oxidation in the liver re-routes LCFAs into triglycerides (TAGs). Long term effects of excess glucose are reflected in hepatic steatosis resulting from the diversion of fats into TAGs instead of being oxidized.
In addition to being regulated by intermediates of glucose and fat oxidation, several enzymes in these two pathways are regulated at the level of post-translational modification and/or gene expression. Most of these regulatory schemes have been covered in the above sections.
Clinical Significance of Defects in Fatty Acid Metabolism
The majority of clinical problems related to fatty acid metabolism are associated with the processes of mitochondrial fatty acid β-oxidation but also include disorders in peroxisomal lipid metabolism. Disorders that result from defects in mitochondrial fatty acid β-oxidation fall into three main groups:
Deficiencies in Carnitine
Deficiencies in carnitine can be either primary or secondary. Primary carnitine deficiencies are due to defects in carnitine transport into cells or due to defects in synthesis from lysine. Mutations in the major carnitine transporter, encoded by the SLC22A5 gene (also known as organic zwitterion/cation transporter 2 (OCTN2), are the predominant causes of primary carnitine deficiency (correctly identified as carnitine deficiency, systemic primary).
Secondary carnitine deficiencies are most commonly the result of trapping in fatty acylcarnitines due to defects in mitochondrial fatty acid oxidation enzymes.
Carnitine deficiencies lead to an inability to transport long-chain fatty acids into the mitochondria for oxidation. Carnitine deficiencies can occur in newborns and particularly in pre-term infants. Carnitine deficiencies also are found in patients undergoing hemodialysis or those affected by any one of various organic acidemias/acidurias (e.g. propionic acidemia or isovaleric acidemia).
Carnitine deficiencies may manifest with systemic symptomology or may be limited to only muscles. Symptoms can range from mild occasional muscle cramping to severe weakness or even death. Treatment is by oral carnitine administration.
Carnitine Palmitoyltransferase Deficiencies
Deficiencies in CPT1 are relatively rare and affect primarily the liver and lead to reduced fatty acid oxidation and ketogenesis. The most common symptom associated with CPT1 deficiency is hypoketotic hypoglycemia associated with hepatic pathology. There is also an elevation in blood levels of carnitine. The liver involvement results in hepatomegaly and the muscle involvement results in weakness.
CPT2 deficiencies can be classified into three main forms. The adult form is the most common and affects primarily the skeletal muscles and is called the adult myopathic form. This form of the disease causes muscle pain and fatigue and myoglobinuria following prolonged exercise. This form of CPT2 deficiency manifests with symptoms that are very similar to those exhibited by patients with the glycogen storage disease, McArdle disease. However, with McArdle disease the muscle pain and cramping is associated with short bursts of aerobic exercise, such as following sprinting, as opposed to following sustained exercise as in the case of CPT2 deficiency.
The severe infantile multisystem form manifests in the first 6–24 months of life with most afflicted infants demonstrating significant involvement before 1 year. The primary symptom of this form of CPT2 deficiency is hypoketotic hypoglycemia. Symptoms will progress to severe hepatomegaly and cardiomyopathy. Often times death from CPT2 deficiency may be misdiagnosed as sudden infant death syndrome, SIDS.
The rarest form of CPT2 deficiency is referred to as the neonatal lethal form. Symptoms of this form appear within hours to four days after birth and include respiratory failure, hepatomegaly, seizures, hypoglycemia, and cardiomegaly. The cardiomegaly will lead to fatal arrhythmias.
Carnitine acyltransferases may also be inhibited by sulfonylurea drugs, such as tolbutamide and glyburide, that are used in the treatment of the hyperglycemia of type 2 diabetes.
Carnitine Acylcarnitine Translocase Deficiency
Mutations in the SLC25A20 gene that encodes the carnitine-acylcarnitine translocase (CACT) enzyme are associated with a rare autosomal recessive disorder. Carnitine-acylcarnitine translocase deficiency manifests in the neonatal period (within hours of birth) with breathing difficulty, seizures, cardiomyopathy, and arrhythmia leading to cardiac arrest.
Metabolic consequences of CACT deficiency are hypoketotic hypoglycemia, dicarboxylic aciduria, elevated serum levels of long-chain acylcarnitines, reduced serum carnitine, and hyperammonemia.
Deficiencies in Acyl-CoA Dehydrogenases
The acyl-CoA dehydrogenase deficiency disorders represent a group of inherited disorders that are associated with impairment of mitochondrial fatty acid β-oxidation. As the name implies, these disorders result from deficiencies in the family of acyl-CoA dehydrogenases that catalyze the first reaction in the mitochondrial β-oxidation of long-chain fatty acids. The enzymes affected belong to one of three categories:
- Very long-chain acyl-CoA dehydrogenase (VLCAD): The symptoms that result from deficiencies in VLCAD were originally ascribed to loss of long-chain acyl-CoA dehydrogenase (LCAD) activity. However, expression of the gene encoding LCAD (ACADL) is very low and restricted in humans and, therefore, LCAD does not contribute, to any significant extent, to long-chain fatty acid oxidation. Mitochondrial oxidation of long-chain fatty acids begins through the activity of VLCAD.
- Medium-chain acyl-CoA dehydrogenase (MCAD): MCAD deficiency (MCADD) is the most common form of acyl-CoA dehydrogenase deficiency. In the first years of life this deficiency will become apparent following a prolonged fasting period. Symptoms include vomiting, lethargy and frequently coma. Excessive urinary excretion of medium-chain dicarboxylic acids as well as their glycine and carnitine esters is diagnostic of this condition. In the case of this enzyme deficiency taking care to avoid prolonged fasting is sufficient to prevent clinical problems.
- Short-chain acyl-CoA dehydrogenase (SCAD)
Mitochondrial Trifunctional Protein (MTP) Deficiency
In addition to deficiencies in the fatty acyl-CoA dehydrogenases, that catalyze the first reaction of mitochondrial long-chain fatty acid β-oxidation, deficiencies in the activities of the mitochondrial trifunctional protein (MTP), the enzyme complex that catalyzes the last three reactions of mitochondrial long-chain fatty acid β-oxidation, have also been identified. The MTP is composed of α-subunits and β-subunits encoded by the HADHA and HADHB genes, respectively.
Mutations in the gene encoding the α-subunits (HADHA) represent one form of the MTP deficiency syndromes and complete loss of MTP activity represents the other. Because the HADHA encoded enzyme possesses long-chain L-3-hydroxyacyl-CoA dehydrogenase (LCHAD) activity the disorder is also commonly referred to as long-chain 3-hydroxyacyl-CoA dehydrogenase deficiency (LCAHDD).
Missense mutations in the HADHB gene have been identified that result in the loss of the long-chain 3-keto-acyl-CoA thiolase activity. These HADHB mutations are collectively part of the MTP deficiency spectrum. Females harboring HADHB mutations are at risk of developing a disorder termed acute fatty liver of pregnancy (AFLP) as well as the disorder referred to as hemolysis, elevated liver enzymes, and low platelet count (HELLP) syndrome during pregnancy.
MTP deficiency is an autosomal recessive disorder with a frequency of 1 in 100,000. The clinical symptoms associated with MTP deficiency include hypoketotic hypoglycemia, metabolic acidosis, metabolic encephalopathy, liver dysfunction, cardiomyopathy, exercise-induced myoglobinuria, and rhabdomyolysis. MTP deficiency can sometimes be mistaken as Reye syndrome.
Total loss of MTP activity is often associated with a very high rate of early lethality. However, some patients with total MTP deficiency have been reported that exhibit a much less severe phenotype, with peripheral neuropathy being the only prominent symptom. To date, a total of 72 mutations have been identified in the HADHA gene and 67 mutations in the HADHB gene that are associated with the symptoms of MTP deficiency.
Lipid Storage Myopathies (LSM)
The lipid storage myopathies (LSM) represent a heterogeneous group of inherited disorders that, in addition to other pathologies, are all characterized by abnormal lipid accumulation in muscle fiber. The LSM are classified based upon pathological findings which result in large part due to the accumulation of lipid droplets (LD) in muscle but also in other tissues.
Four discernable LSM have been categorized and includes the above indicated primary carnitine deficiency (PCD), multiple acyl-CoA dehydrogenase deficiency (MADD; also known as glutaric acidemia type II, GAII), and the neutral lipid storage diseases (NLSD) which includes NLSD with myopathy (NLSDM) and NLSD with ichthyosis (NLSDI).
MADD is a disorder that results from mutations in either of the two genes that encode the protein components of the heterodimeric complex identified as electron transfer flavoprotein (ETF) or the gene encoding electron transfer flavoprotein dehydrogenase (encoded by the ETFDH gene). The ETF is composed of an α-subunit encoded by the ETFA gene and a β-subunit encoded by the ETFB gene. The ETF is localized to the inner mitochondrial membrane where it accepts electrons from the FAD-dependent dehydrogenases of mitochondrial fatty acid β-oxidation as well as several dehydrogenases involved in amino acid metabolism. The function of ETFDH is to transfer the electrons from the ETF to ubiquinone (CoQ10) of the electron transport chain.
The lipid myopathies also include several of the diseases indicated above such as VLCAD, MCAD, and SCAD deficiencies, CPT2 deficiency, and the mitochondrial trifunctional protein (MTP) deficiencies.
Other lipid myopathies have been shown to be the result of lipin 1 (phosphatidic acid phosphatase) deficiency, acyl-CoA dehydrogenase 9 (ACAD9) deficiency, and acetyl-CoA acyltransferase 2 (ACAA2) deficiency (also known as medium-chain 3-ketoacyl-CoA thiolase, MCKAT deficiency).
Peroxisomal Fatty Acid Oxidation Defects
Disorders related to the peroxisomes are divided into two categories. One category, exemplified by Zellweger syndrome, contains the disorders that result from the defects in the biogenesis of the peroxisomes. The other category, exemplified by Refsum disease, contains the disorders that are due to mutations in a single peroxisomal enzyme.
Refsum disease is a rare autosomal recessive disorder in which patients harbor a defect in the peroxisomal α-oxidizing enzyme, phytanoyl-CoA hydroxylase (PhyH). Although mutations in PhyH are the primary cause of Refsum disease, the syndrome can also result from defects in the peroxisomal protein (PEX7) responsible for the import of PhyH into the peroxisome. Patients accumulate large quantities of phytanic acid in their tissues and serum. This leads to severe symptoms, including cerebellar ataxia, retinitis pigmentosa, nerve deafness and peripheral neuropathy. As expected, the restriction of dairy products and ruminant meat from the diet can ameliorate the symptoms of this disease. It should be noted that accumulation of phytanic acid is not solely the result of defects in PhyH.
Phytanic acid accumulation is also seen when there are inherited defects in peroxisome function leading to Zellweger syndrome, neonatal adrenoleukodystrophy and infantile Refsum disease. In addition, rhizomelic chondrodysplasia punctata, type 1 (RCDP1) results in phytanic acid accumulation. Refsum disease due to deficiency in PhyH is properly referred to as classical Refsum disease to distinguish it from infantile Refsum due to peroxisome dysfunction.
Mutations in the HSD17B4 gene are the cause of the autosomal recessive disorder referred to as D-bifunctional protein deficiency.
Synthesis and Utilization of the Ketones
During high rates of fatty acid oxidation, primarily in the liver, large amounts of acetyl-CoA are generated. Indeed, the major source of acetyl-CoA for hepatic ketone synthesis is from the mitochondrial β-oxidation of fatty acids. However, amino acid oxidation can and does contribute to the pool of available acetyl-CoA. Catabolism of amino acids, where leucine is a major contributor, contributes up to 5% of the acetyl-CoA diverted into ketone synthesis in the liver. The excess acetyl-CoA exceeds the capacity of the TCA cycle, and one result is what has been referred to as a spillover pathway, the synthesis of ketone bodies.
The synthesis of the ketone bodies (ketogenesis) occurs in the mitochondria allowing this process to be intimately coupled to the rate of hepatic fatty acid oxidation. Conversely, the utilization of the ketones (ketolysis) occurs in the cytosol. The ketone bodies are acetoacetate, β-hydroxybutyrate, (BHB) and acetone. The pathways of ketone synthesis and utilization are classically viewed as being required for energy generation during periods of carbohydrate restriction, during fasting, and during a prolonged fast (starvation). However, more recent studies have discovered that ketones are important mediators of metabolic and signal transduction processes when carbohydrates are in abundance.
Ketone Synthesis
The formation of acetoacetyl-CoA occurs by condensation of two moles of acetyl-CoA. This reaction is essentially a reversal of the thiolase (HADHB or ACAA2) catalyzed reaction of fatty acid β-oxidation but is in fact catalyzed by the mitochondrial enzyme acetoacetyl-CoA thiolase (encoded by the ACAT1 gene). Acetoacetyl-CoA and an additional acetyl-CoA are converted to β-hydroxy-β-methylglutaryl-CoA (HMG-CoA: 3-hydroxy-3-methylglutaryl-CoA) by mitochondrial HMG-CoA synthase (encoded by the HMGCS2 gene), an enzyme found in large amounts only in the liver and intestinal epithelial cells. The reaction catalyzed by the HMGCS2 encoded enzyme represents the regulated and rate-limiting step in ketone synthesis.
Humans express two HMG-CoA synthase genes, the mitochondrial HMGCS2 gene just described, and the HMGCS1 gene which encodes a cytosolic enzyme that is involved in cholesterol biosynthesis.
The HMGCS2 gene is located on chromosome 1p12 and is composed of 10 exons that generate two alternatively spliced mRNAs, both of which encode distinct protein isoforms (isoform 1 is 508 amino acids and isoform 2 is 466 amino acids). Expression of the HMGCS2 gene is positively regulated via glucagon signal transduction events and negatively regulated by insulin signal transduction events (see Regulation of Ketogenesis below).
Mutations in the HMGCS2 gene result in a severely impaired ability to carry out ketogenesis. Mitochondrial HMG-CoA synthase deficiency is a rare autosomal disorder. Most patient will manifest with an initial episode of metabolic decompensation following gastroenteritis or gastroenteritis-like symptoms. Additional symptoms that are commonly found in these patients include dyspnea, seizures, and hepatomegaly. Most patients will present with acute metabolic decompensation accompanied with hypoglycemia, dicarboxylic aciduria, and little to no detectable urinary ketones. One prominent abnormal metabolite detected in the urine of mitochondrial HMG-CoA synthase deficient patients is 4-hydroxy-6-methyl-2-pyrone (4HMP). Indeed, measurement of urinary 4HMP levels significantly facilitates a diagnosis mitochondrial HMG-CoA synthase deficiency.
HMG-CoA (3-hydroxy-3-methylglutaryl-CoA) in the mitochondria is converted to acetoacetate by the action of HMG-CoA lyase. HMG-CoA lyase is also involved in catabolism of the amino acid leucine, catalyzing the final step in that pathway.
The HMG-CoA lyase gene (HMGCL) is located on chromosome 1p36.11 and is composed of 9 exons that generate two alternatively spliced mRNAs which encode two distinct precursor proteins (isoform 1 is 325 amino acids and isoform 2 is 254 amino acids). Expression of the HMGCL gene is highest in the liver and kidneys.
Mutations in the HMGCL gene result in HMG-CoA lyase deficiency, a disorder referred to as 3-hydroxy-3-methylglutaric aciduria. Inheritance of 3-hydroxy-3-methylglutaric aciduria occurs as an autosomal recessive disorder. Since the enzyme encoded by the HMGCL gene is also involved in leucine catabolism, mutations in HMGCL also result in defects in metabolism of this amino acid and consequently leads to accumulation of leucine metabolites, 3-hydroxy-3-methylglutaric acid, 3-methylglutaconic acid, 3-methylglutaric acid, and 3-hydroxyisovaleric acid.
The reduced capacity to synthesize ketone bodies results in inefficient energy production, particularly by the brain, during periods of fasting. Reduced HMG-CoA lyase activity is associated with episodes of hypoglycemia and metabolic acidosis. HMG-CoA lyase deficiency is treatable by diet, particularly leucine restriction, and avoidance of prolonged fasting. Supplementary glucose is administered to prevent hypoglycemia. Without prompt and proper therapeutic intervention, death occurs early in life.
Acetoacetate can undergo spontaneous decarboxylation to acetone, or be enzymatically converted to β-hydroxybutyrate (BHB) through the action of mitochondrial β-hydroxybutyrate dehydrogenase which is encoded by the BDH1 gene. The BDH1 gene is located on chromosome 3q29 and is composed of 16 exons that generate three alternatively spliced mRNAs, each of which encode the same 343 amino acid precursor protein. Functional β-hydroxybutyrate dehydrogenase is a homotetrameric complex.
Humans express a second BDH gene, BDH2, whose encoded protein is only 20% identical to the BDH1 encoded protein. The BDH2 encoded protein appears to play a role in iron homeostasis.
The precise mechanism by which the ketone bodies cross the inner mitochondrial membrane is unclear but may occur by simple diffusion out of the mitochondria of hepatocytes. Once out of the mitochondria the ketone bodies are transported into the blood via the action of the plasma membrane localized monocarboxylate transporter 1 (MCT1) or MCT2 which are encoded by the SLC16A1 and SLC16A7 genes, respectively.
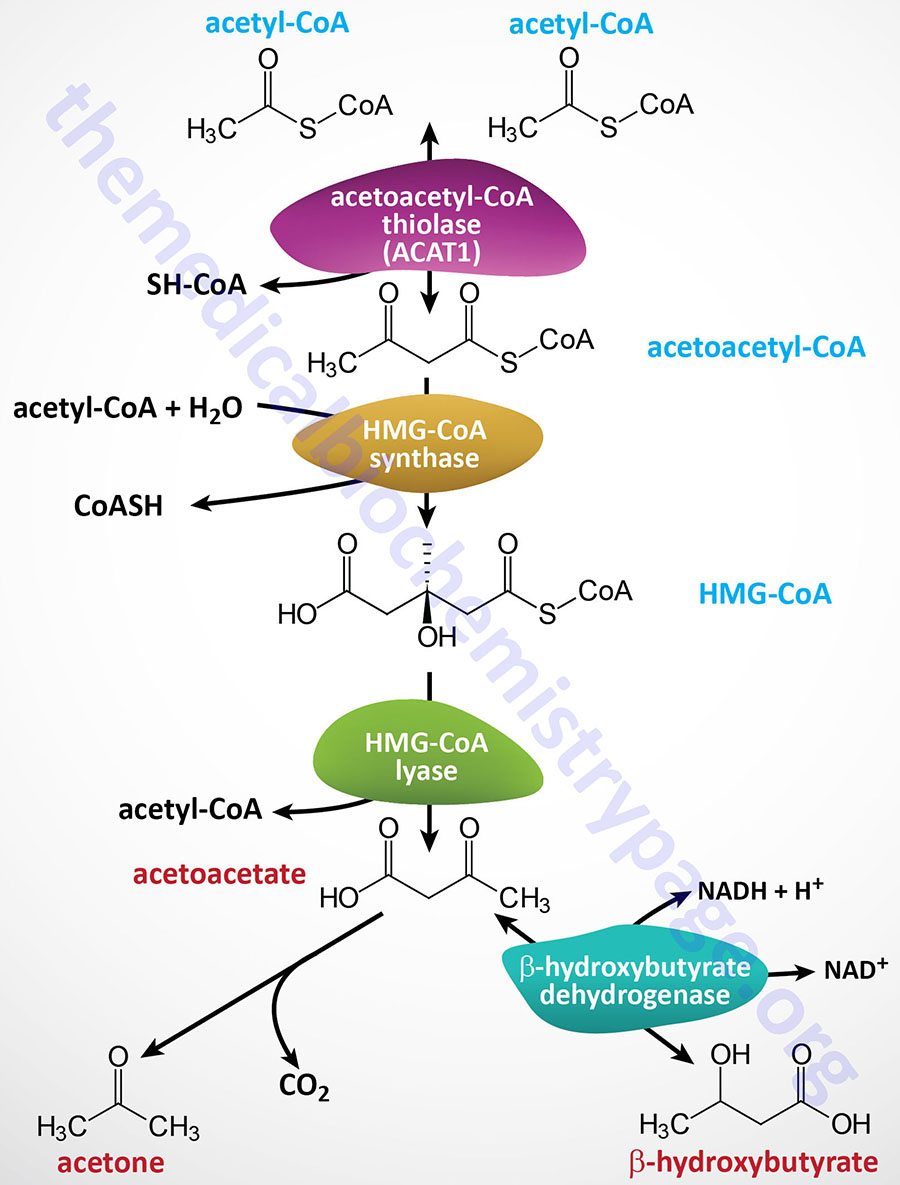
Ketone Utilization
When the level of glycogen in the liver is high the production of β-hydroxybutyrate (BHB) increases. When carbohydrate utilization is low or deficient, the level of oxaloacetate will also be low, resulting in a reduced flux through the TCA cycle. This in turn leads to increased release of ketone bodies from the liver for use as fuel by other tissues. In early stages of starvation (beyond one week without food intake), when the last remnants of readily releasable fat are oxidized, heart and skeletal muscle will consume primarily ketone bodies to preserve glucose for use by the brain and erythrocytes.
Eventually the brain will adapt to ketone oxidation for energy in order to preserve what little glucose can be made via gluconeogenesis in a starvation scenario for use by erythrocytes as these cells can get the necessary ATP from no other source.
Acetoacetate and predominantly β-hydroxybutyrate (BHB) also serve as major substrates for the biosynthesis of neonatal cerebral lipids.
Once in the circulation the ketones are taken up by non-hepatic tissues most likely by the plasma membrane localized monocarboxylate transporter 1 (MCT1). Evidence for the role of MCT1 in ketone uptake came from the observations that loss-of-function mutations in the SLC16A1 gene (which encodes the MCT1 protein) are associated with spontaneous ketoacidosis.
Both β-hydroxybutyrate and acetoacetate are taken up by the mitochondria of non-hepatic tissues for their reconversion back to acetyl-CoA. Non-hepatic ketone body utilization occurs via a series of mitochondrial reactions (shown in the Figure below) that are essentially a reversal of ketone body synthesis.
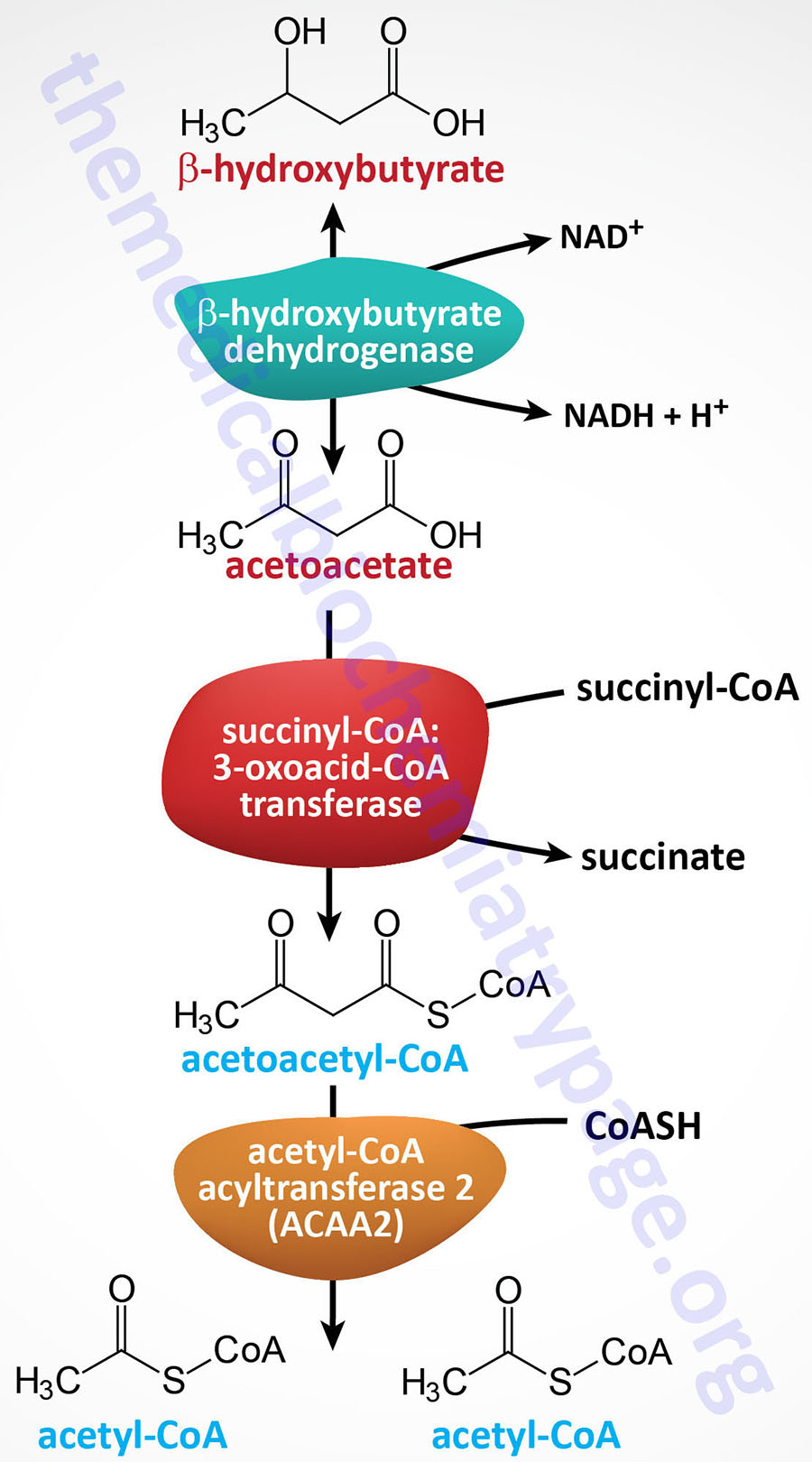
The initial steps involve the conversion of β-hydroxybutyrate to acetoacetate and of acetoacetate to acetoacetyl-CoA. The first step in ketone utilization involves the reversal of the β-hydroxybutyrate dehydrogenase reaction. It is important to appreciate that under conditions where tissues are utilizing ketones for energy production their NAD+/NADH ratios are going to be relatively high thus, driving the β-hydroxybutyrate dehydrogenase catalyzed reaction in the direction of acetoacetate synthesis.
The second reaction of ketone utilization involves the action of 3-oxoacid-CoA transferase 1 (encoded by the OXCT1 gene) which is also known as succinyl-CoA:3-oxoacid-CoA transferase (SCOT). The latter enzyme is expressed in all human cells that contain a mitochondria except for hepatocytes. Importantly, the lack of OXCT1 expression in the liver allows the liver to produce ketone bodies but not to utilize them. This ensures that non-hepatic tissues have access to ketone bodies as a fuel source during prolonged fasting and starvation.
The OXCT1 gene is located on chromosome 5p13.1 and is composed of 19 exons that generate six alternatively spliced mRNAs, each of which encode a distinct precursor protein. Expression of the OXCT1 gene is essentially ubiquitous, except for the liver, with highest levels in the heart, brain, and adipose tissue.
Mitochondrial acetoacetyl-CoA is hydrolyzed into two molecules of acetyl-CoA primarily by the ACAA2 encoded mitochondrial thiolase (see Table above in the section covering mitochondrial β-oxidation). The other two mitochondrial thiolases, encoded by the ACAT1 and HADHB genes, can also hydrolyze acetoacetyl-CoA.
Although ketones are not actively utilized by the liver, due to low level expression of the OXCT1 gene, cytosolic acetoacetate can contribute to de novo lipogenesis in the liver. Cytosolic acetoacetate is converted to acetoacetyl-CoA which is then hydrolyzed into two moles of acetyl-CoA as described in the Nonoxidative Metabolic Fates of Ketones section below.
Mechanisms Regulating Ketogenesis
The fate of the acetyl-CoA derived from the metabolism of fatty acids, pyruvate, and numerous amino acids is determined by an individual’s dietary, physiological, and hormonal status. Although, as indicated, acetyl-CoA can be derived through oxidative metabolism of lipids, carbohydrates, and amino acids, the predominant source of the acetyl-CoA diverted into ketone synthesis in the liver is from fatty acid oxidation. As indicated above, amino acid catabolism, in particular leucine catabolism, contributes around 5% of the total pool of acetyl-CoA for hepatic ketone synthesis. Although carbohydrate and pyruvate catabolism yield acetyl-CoA these reactions are negligible with respect to substrate generation for ketogenesis. The overall rate of hepatic ketogenesis may by affected by several factors that includes substrate availability and the transcriptional and post-transcriptional regulation of ketogenesis enzymes.
Control in the release of free fatty acids from adipose tissue directly affects the level of ketogenesis in the liver. This is, of course, substrate-level regulation. Fatty acid release from adipose tissue is controlled via the activity of hormone-sensitive lipase (HSL). When glucose levels fall, pancreatic glucagon secretion increases resulting in phosphorylation of adipose tissue HSL, thus resulting in increased hepatic ketogenesis due to increased substrate (free fatty acids) delivery from adipose tissue. Conversely, insulin, released in the well-fed state, inhibits ketogenesis via the triggering of dephosphorylation and inactivation of adipose tissue HSL.
Once fats enter the liver, they have two distinct fates. They may be activated to acyl-CoAs and oxidized, or esterified to glycerol in the production of triglycerides. If the liver has sufficient supplies of glycerol-3-phosphate, most of the fats will be turned to the production of triglycerides.
The acetyl-CoA generated by the oxidation of fats and amino acids can be completely oxidized in the TCA cycle or it can be diverted into lipid biosynthesis. If the hepatic demand for ATP is high the fate of acetyl-CoA is likely to be further oxidation to CO2. This is especially true under conditions of hepatic stimulation by glucagon which results in increased gluconeogenesis and the energy for this process is derived primarily from the oxidation of fatty acids supplied from adipose tissue.
The regulation of the HMGCS2 gene and the encoded enzyme itself are subject to various regulatory processes. Glucagon also directly enhances hepatic ketone synthesis at the level of gene expression. The HMGCS2 gene is transcriptionally activated by the forkhead transcription factor, FOXA2 whose activity is enhanced in response to elevated cAMP levels as occurs via glucagon activation of its receptor. Conversely, the activity of FOXA2 is inhibited by insulin via the insulin receptor-mediated activation of the kinases, phosphatidylinositol-3-kinase (PI3K) and AKT/PKB.
The transcription factor PPARα induces the expression of the HMGCS2 gene in the liver in response to the consumption of a ketogenic diet (e.g. high-fat diet) as well as in response to the physiological changes occurring during starvation.
The enzymatic activity of HMGCS2 is regulated by both allosteric effectors and by post-translational modifications. Succinyl-CoA allosterically inhibits HMGCS2 and it also serves as a substrate for succinylation of lysine residues in the enzyme. In fact, in addition to HMGCS2, HMG-CoA lyase, and β-hydroxybutyrate dehydrogenase (BDH1) are also subjected to lysine succinylation. When these enzymes are succinylated they are targets for the sirtuin 5 (SIRT5) deacetylase. Humans express seven sirtuin genes that each encode NAD+-dependent enzymes. In contrast to SIRT5-induced inhibition of activity, the action of the SIRT3 deacetylase on HMGCS2 results in enhanced enzyme activity.
HMGCS2 and succinyl-CoA:3-oxoacid-CoA transferase (SCOT) also undergo modification via lysine acetylation. Hypersuccinnylation and hyperacetylation of HMGCS2 is correlated with reduced rates of hepatic ketogensis. Hypersuccinnylation and hyperacetylation of SCOT, particularly in the brain, is associated with diminished metabolism of ketones.
In addition, glucagon results in phosphorylation and inhibition of acetyl-CoA carboxylase (ACC), the rate limiting enzyme of de novo fatty acid synthesis. Conversely, under conditions of insulin release, hepatic ACC is activated and the excess acetyl-CoA will be converted into malonyl-CoA and then free fatty acids. The increased malonyl-CoA results in inhibition of fatty acid transport into the mitochondria resulting in reduced fat oxidation and reduced production of excess acetyl-CoA.
Metabolic and Clinical Significance of Ketones
The production and utilization of the ketone bodies are interconnected with most aspects of metabolic homeostasis including that of fatty acid oxidation, the TCA cycle, gluconeogenesis, and endogenous lipid and sterol biosynthesis. Synthesis of ketones occurs at a relatively low rate during normal feeding and under conditions of normal physiological status.
The normal concentration of total ketone bodies in the circulation of healthy adult humans will exhibit circadian oscillations of between approximately 100μM and 250μM. The level of circulating ketones can rise as high as 1mM after prolonged exercise or following a 24 hr period of fasting.
Normal physiological responses to carbohydrate shortages cause the liver to increase the production of ketone bodies from the acetyl-CoA generated from fatty acid oxidation. The human liver produces up to 300 grams of ketone bodies per day and these molecules will contribute to between 5% and 20% of the total body energy expenditure depending upon the fed, fasted, or starved states.
During ketotic states both acetoacetate and β-hydroxybutyrate will be excreted in the urine and their levels in the urine will be proportionate to those in the circulation. However, it should be noted that most clinical measurements of urine ketones are only detecting acetoacetate and not β-hydroxybutyrate.
Ketone body oxidation is a significant contributor to overall energy metabolism in humans within extrahepatic tissues, particularly in the brain, under many different physiological states. The principal normal physiological conditions that result in increased ketone utilization are fasting and post-exercise recovery as well as in response to the consumption low carbohydrate diets. During pregnancy and in the neonate the utilization of ketones is also elevated.
The most significant disruption in the level of ketosis, leading to profound clinical manifestations, occurs in untreated insulin-dependent diabetes mellitus, type 1 diabetes. This physiological state, diabetic ketoacidosis (DKA), results from a reduced supply of glucose, due to lack of circulating insulin in the absence of therapeutic injection, and a concomitant increase in fatty acid oxidation due, in large part, to an unregulated increase in circulating glucagon. The DKA state can result in circulating ketone bodies being as high as 20mM. The increased production of acetyl-CoA leads to ketone body production that exceeds the ability of peripheral tissues to oxidize them. Ketone bodies are relatively strong acids (pKa around 3.5), and their increase lowers the pH of the blood resulting in significant metabolic acidosis. This acidification of the blood is dangerous for many reasons but in particular because it impairs the ability of hemoglobin to bind oxygen.
Nonoxidative Metabolic Fates of Ketones
Although the primary function of ketones is for energy generation by non-hepatic tissues during periods of fasting (particularly the brain), they can be directed into lipid metabolic pathways within the liver. Acetoacetate can be exported out of the mitochondria for conversion to acetoacetyl-CoA by a cytoplasmic acetoacetyl-CoA synthetase encoded by the AACS gene. The cytoplasmic acetoacetyl-CoA can be hydrolyzed to two molecules of acetyl-CoA by the action of the cytosolic thiolase (acetyl-CoA acetyltransferase 2) encoded by the ACAT2 gene.
The two moles of cytosolic acetyl-CoA generated by the action of ACAT2 on acetoacetyl-CoA can be utilized for the synthesis of fatty acids.
Cytosolic acetoacetyl-CoA and acetyl-CoA can also be directed into the cholesterol biosynthesis pathway via the action of cytosolic HMG-CoA synthase encoded by the HMGCS1 gene.
β-Hydroxybutyrate-Mediated Signaling
In addition to hepatocyte synthesis of the ketone, β-hydroxybutyrate (BHB), colonic bacteria also contribute to the generation of BHB. Colonic bacteria generate short-chain fatty acids (SCFA) through fermentation of soluble fiber. These SCFA include acetate, propionate, and butyrate which are absorbed by colonocytes. Metabolically the gut bacteria-derived SCFA can be used for oxidation or diverted into the ketogenesis pathway. In addition to hepatocytes, gut epithelial cells are the only other cell to express the HMGCS2 gene allowing them to contribute to ketone synthesis. However, gut-derived SCFA also exert other important cell signaling effects.
Although the beneficial effects of these SCFA can be attributed to all three, the most extensively studied effects are those exerted by butyrate. Butyrate promotes colonocyte cell differentiation, suppresses colonic inflammation, and of clinical significance it induces cell cycle arrest and apoptosis in colon cancer cells. These beneficial effects of butyrate (and also shown for propionate), within the colon are mediated, in part, by its ability to inhibit the activity of histone deacetylases (HDAC). Like butyrate, the ketone β-hydroxybutyrate (BHB) has also been shown to inhibit the activity of HDAC. The effects of β-hydroxybutyrate-mediated HDAC inhibition are enhanced expression of genes that reduce the level of oxidative stress.
In addition to altering the patterns of gene expression through modification of HDAC activity, β-hydroxybutyrate can alter gene expression patterns by serving as a direct modifier of lysine residues in histones resulting in lysine β-hydroxybutyrylation. The level of histone β-hydroxybutyrylation increases in hepatocytes in response to prolonged fasting. The effects histone β-hydroxybutyrylation on gene expression represents a novel form of epigenetic control. The level of histone β-hydroxybutyrylation is similar to the level of the more well studied epigenetic histone modification, acetylation.
The consequences of histone lysine β-hydroxybutyrylation are changes in expression of numerous genes such as the gene for the transcriptional co-activator, PGC-1β (encoded by the PPARGC1B gene) which is itself involved in the regulation of expression of numerous genes involved in energy homeostasis, the insulin receptor substrate 2 (IRS2) gene whose encoded protein is involved in insulin signaling, and the carnitine palmitoyltransferase 1A (CPT1) gene whose encoded protein regulates the ability of the mitochondria to oxidize fatty acids.
β-Hydroxybutyrate (BHB) also exerts cell signaling effects by direct binding and activation of the G-protein coupled receptor (GPCR) originally identified as GPR109A. GPR109A is a member of a family of hydroxycarboxylic acid (HCA)-responsive GPCR and as such is now identified as HCA2 (encoded by the HCAR2 gene). Indeed, β-hydroxybutyrate is the only identified endogenous ligand for HCA2.
The HCAR2 gene is expressed in both white and brown adipose tissue (WAT and BAT) and in cells of the immune system. HCA2 is a Gi-coupled GPCR and its activation in adipose tissue results in reduced activation of hormone-sensitive lipase, HSL. The activation of HCA2 by β-hydroxybutyrate in adipose tissue, therefore, serves as a negative feed-back loop on adipose tissue lipolysis by reducing the release of free fatty acids from adipocytes.
β-Hydroxybutyrate also exerts effects via binding to another GPCR, the free fatty acid receptor 3 (FFAR3; originally identified as GPR41). Interference with FFAR3 signaling by β-hydroxybutyrate has been shown to be responsible for the suppression of sympathetic nervous system activity exerted by β-hydroxybutyrate.