
Last Updated: October 30, 2025
Introduction to X-Linked Sideroblastic Anemia, XLSA
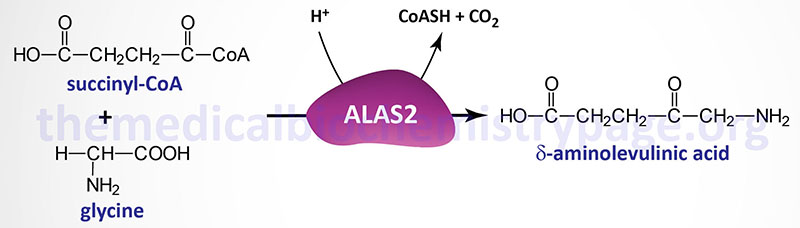
X-linked sideroblastic anemia (XLSA) is a recessive disorder that results from deficiencies in the erythroid-specific form of δ-aminolevulinic acid synthase (also called 5-aminolevulinic acid synthase) encoded by the ALAS2 gene. ALAS2 catalyzes the first reaction of heme biosynthesis. ALAS2 is related, but not identical, to an additional δ-aminolevulinic acid synthesizing enzyme encoded by the ALAS1 gene. Whereas, ALAS2 expression is restricted to fetal liver and adult bone marrow, ALAS1 is expressed in all cells.
Although not strictly considered one of the porphyrias, XLSA does involve a defect in heme metabolism so the disorder is grouped with the porphyria family of disorders. As a result of defective ALAS2 activity iron accumulates in the erythroid marrow. The excess iron deposits as non-ferritin iron in the mitochondria that surround the nuclei of erythroblast precursors (immature red blood cells). These deposits give the cells a distinctive histopathologic appearance referred to as ring sideroblasts.
XLSA has also been called congenital sideroblastic anemia, hereditary sideroblastic anemia, hereditary iron-loading anemia, X-linked hypochromic anemia, hereditary hypochromic anemia, and hereditary anemia.
Another X-linked sideroblastic anemia (abbreviated ASAT), that is associated with spinocerebellar ataxia, results from mutations in a gene unrelated to ALAS2. The gene, mutations in which cause ASAT, encodes the ATP-binding cassette (ABC) transporter family member ABCB7. The ABCB7 gene is located on chromosome Xq13.3.
Molecular Biology of X-Linked Sideroblastic Anemia
The δ-aminolevulinic acid synthase 2 gene (ALAS2) is located on the X chromosome at Xq11.21 spanning 22 kb and encompassing 12 exons that generate three alternatively spliced mRNAs, each of which encode a distinct protein isoform. ALAS2 isoform a is composed of 587 amino acids, ALAS2 isoform b is composed of 550 amino acids, and ALAS2 isoform c is composed of 574 amino acids.
The ALAS1 and ALAS2 proteins are only 59% homologous overall with no homology in the N-terminal regions of the proteins. After residue 197 in ALAS1 the two enzymes show 73% identity. At least 25 different mutations have been identified in the ALAS2 gene leading to XLSA.
Clinical Features of XLSA
The clinical manifestations of XLSA are the result of the anemia caused by the reduction in heme biosynthesis. The reduction in heme synthesis leads to the stimulation of erythropoiesis. As a result of the stimulated erythropoiesis there is an increase in iron absorption in an attempt to compensate. Thus, in addition to anemia, there is cellular toxicity due to the increased iron levels in the body. This iron overload is fatal if not properly treated.
Symptoms of XLSA usually appear in the second or third decade of life. Because this disorder is X-linked, affected females will present later in life (usually around 10 years later) than males. This phenomenon is partly due to the protection, in females, from iron overload due to blood loss during menstruation. Clinical manifestations of XLSA include hepatomegaly and/or splenomegaly, hepatocellular carcinoma, diabetes, body hair loss, nausea and abdominal pain, cirrhosis, growth delay, and arthropathy (disease of the joints). Weakness, fatigue, and palpitations are due to cardiac involvement. Iron toxicity leads to defective hormone production which can lead to growth delay.
Treatment of XLSA
Because ALAS is a pyridoxal phosphate (PLP)-requiring enzyme the defect in ALAS2 resulting in XLSA can be treated by administration of pyridoxine (vitamin B6) and folic acid which optimizes for heme synthesis. In addition, phlebotomy and/or iron chelation therapy are used to reduce the iron overload.