
Last Updated: July 13, 2026
Enzymes of Type 1 Glycogen Storage Disease
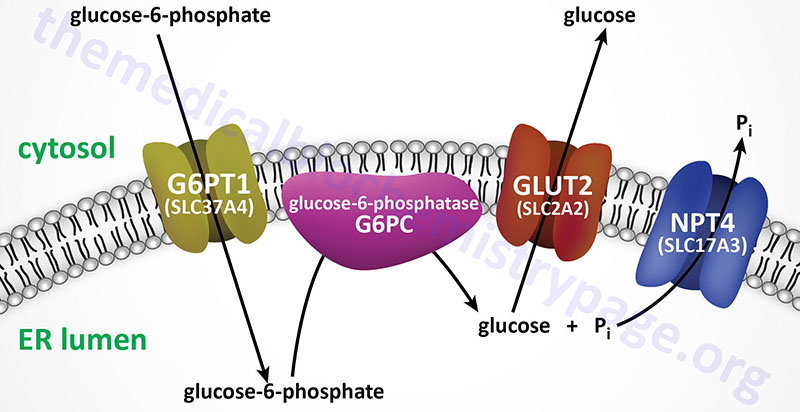
The mechanism by which free glucose is released from glucose-6-phosphate involves several different steps. Glucose-6-phosphate must first be transported into the lumen of the endoplasmic reticulum (ER) from the cytosol where it is generated either through phosphorylation of free glucose, from gluconeogenesis, or from glycogenolysis. Inside the ER the phosphate is removed through the action of ER membrane-localized glucose 6-phosphatase. The free glucose must then be transported back to the cytosol as well as the released inorganic phosphate, Pi. Defects in the process of glucose release from glucose-6-phosphate result in elevations in cytosolic glucose-6-phosphate which then leads to increases in incorporation into glycogen and subsequent excessive storage.
von Gierke Disease
Type 1 glycogen storage disease (GSD1) is an autosomal recessive disorder that was first described in 1929 by E. von Gierke as a “hepato-nephromegalia glycogenica”. For this reason the disease is still more commonly referred to von Gierke disease. In 1952, G. Cori and C. Cori identified that the absence of glucose 6-phosphatase activity was the cause of von Gierke disease. This discovery was the first ever identification of an enzyme defect in a metabolic disorder.
Subsequent to the identification of the pathway of glucose release from glucose-6-phosphate, additional patients with similar clinical manifestations to von Gierke disease were identified. However, these patients were not deficient in glucose-6-phosphatase activity. These latter patients were identified as having deficiency in the endoplasmic reticulum (ER) membrane-localized glucose-6-phosphate transporter (identified as G6PT1; encoded by the SLC37A4 gene) and so the disease was designated as type 1b GSD. The designation, GSD type 1a was then used to designate the form of the disease caused by defects in the ER localized glucose-6-phosphatase.
A novel form of type 1 GSD was identified in 1983 and since it was thought to be the result of defects in the microsomal inorganic phosphate transporter it was designated as type 1c GSD. However, later kinetic studies demonstrated that the defect was actually in the G6PT1 encoded protein. Current nomenclature classifies GSD type 1 patients as either type 1a or type non-1a. However, there is still common usage of the two designations GSD type 1a and GSD type 1b.
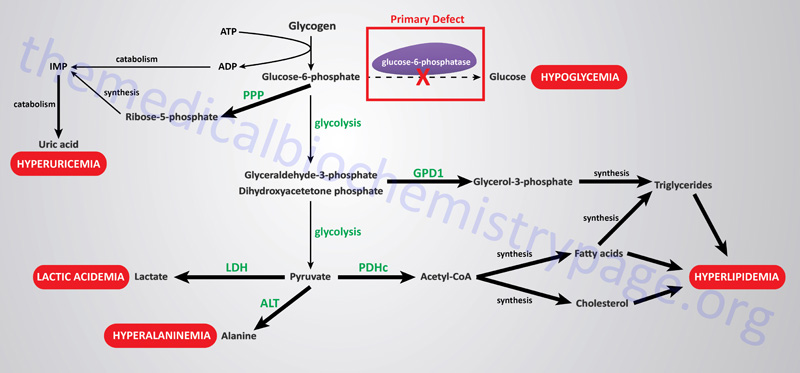
The metabolic consequences of the hepatic glucose-6-phosphate deficiency of von Gierke disease extend well beyond just the obvious hypoglycemia that results from the deficiency in liver being able to deliver free glucose to the blood. The inability to release the phosphate from glucose-6-phosphate results in diversion into glycolysis and production of pyruvate as well as increased diversion into the pentose phosphate pathway. The production of excess pyruvate, at levels above of the capacity of the TCA cycle to completely oxidize it, results in its reduction to lactate resulting in lactic acidemia. In addition, some of the pyruvate is transaminated to alanine leading to hyperalaninemia.
Some of the pyruvate will be oxidized to acetyl-CoA which can’t be fully oxidized in the TCA cycle and so the acetyl-CoA will end up in the cytosol where it will serve as a substrate for triglyceride and cholesterol synthesis resulting in hyperlipidemia.
The oxidation of glucose-6-phophate via the pentose phosphate pathway leads to increased production of ribose-5-phosphate which then activates the de novo synthesis of the purine nucleotides. In excess of the need, these purine nucleotides will ultimately be catabolized to uric acid resulting in hyperuricemia and consequent symptoms of gout. The interrelationships of these metabolic pathways is diagrammed in the Figure below.
Genetics of Type I Glycogen Storage Diseases
Humans express three glucose 6-phosphatase catalytic subunit genes identified as G6PC1 (also identified as G6PC), G6PC2, and G6PC3. Pathogenic variants in the G6PC1 gene are the cause of type 1a GSD (classic von Gierke disease).
The G6PC1 gene is located on chromosome 17q21.31 spanning 12.5 kb and is composed of 5 exons that generate two alternatively spliced mRNAs encoding proteins of 357 amino acids (isoform 1) and 176 amino acids (isoform 2). Pathogenic variants in the G6PC1 gene account for approximately 80% of all type 1 GSD cases.
The glucose-6-phosphate transporter 1 protein is a member of the solute carrier family of membrane transporters. The gene that encodes this transporter is the solute carrier family 37, member 4 (SLC37A4) gene. Pathogenic variants in the SLC37A4 gene result in what was originally designated as type 1b GSD (GSD1b). Current nomenclature designates the forms of GSD1 that result from SLC37A4 variants, and all other causes not related to pathogenic variants in the G6PC1 gene as type non-1a GSD1. However, usage of the designations, GSD type 1a and GSD type 1b, remain highly common in the literature.
The SLC37A4 gene is located on chromosome 11q23.3 and is composed of 12 exons that generate five alternatively spliced mRNAs that collectively encode three distinct isoforms of the protein. The mRNA found in the brain contains exon 7 whereas the one in the liver does not contain this exon. Pathogenic variants in the SLC37A4 gene account for approximately 20% of all type non-1a GSD cases.
Analysis of pathogenic variants resulting in type 1 GSD have been most extensively studied in von Gierke (type 1a) disease. The 130X pathogenic variant is the result of a 2-base pair (bp) insertion in exon 3 of the G6PC1 gene resulting in a translation stop codon. This pathogenic variant is identified as FS130TER. This pathogenic variant has, thus far, only been found in Hispanic patients. All Ashkenazi Jews with type 1a GSD have been shown to harbor a pathogenic variant in exon 2 resulting in the alteration of arginine 83 to a cysteine (R83C). One compound heterozygote patient has been identified harboring two variants in G6PC1 where one was the R83C variant and the second was arginine 295 altered to a cysteine (R295C). Twelve additional pathogenic variants have been mapped to the G6PC1 gene: R83H, I341D, G188R, A124T, IVS1DS (variant in the splice donor site of intron 1), W77R, D38V, IVS4AS (variant in intron 4 resulting in an alternative splice site), Q347X (variant resulting in a termination codon), V166G, G184E, E110K.
Clinical Findings of Type 1 Glycogen Storage Diseases
Patients with type 1 glycogen storage disease can present during the neonatal period with lactic acidosis and hypoglycemia. More commonly though, infants of 3–4 months of age will manifest with hepatomegaly and hypoglycemic seizures. The hallmark features of this disease are hypoglycemia, lactic acidosis, hyperuricemia, and hyperlipidemia. The severity of the hypoglycemia and lactic acidosis can be such that in the past affected individuals died in infancy. Infants often have a doll-like facial appearance due to excess adipose tissue in the cheeks. In addition, patients have thin extremities, a short stature, and protuberant abdomens (due to the severe hepatomegaly).
Long-term complications are usually only seen now in adults whose disease was poorly treated early on. The main problem is associated with liver function but multiple organ systems are also involved, in particular the intestines and kidneys. Growth will continue to be impaired and puberty is often delayed. Affected female patients will have polycystic ovaries but none of the other symptoms of polycystic ovarian syndrome (PCOS) such as hirsutism.
Similar to the re-identification of NAFLD as MASLD due to the realization of the broad metabolic dysfunction of the disorder, PCOS has also undergone a re-imagining of the best way to describe the disorder. It has long been understood that PCOS, as a disorder, is much more than just pathological ovarian cysts and in fact encompasses diverse endocrine and metabolic features and that these features contribute to delayed definitive diagnosis and a resulting fragmentation in delivery of care. As a result it has been suggested that a more useful and informative descriptor for this disorder should be polyendocrine metabolic ovarian syndrome (PMOS).
Treatment of Von Gierke Disease
The common treatment for type 1 glycogen storage disease is to maintain normal blood glucose concentration. With normoglycemia will come reduced metabolic disruption and a reduced morbidity associated with the disease. To attain normoglycemia patients are usually treated in infancy with nocturnal nasogastric infusion of glucose. Total parenteral nutrition or the oral feeding of uncooked cornstarch can also achieve the desired results.
In the past the prognosis for type 1 glycogen storage disease patients was poor. However, with proper nutritional intervention growth will improve and the lactic acidosis, hypercholesterolemia, and lipidosis will decrease.