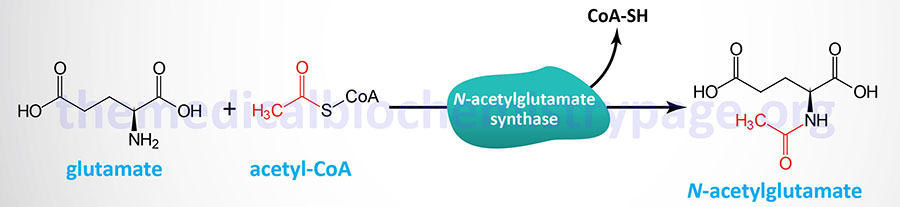
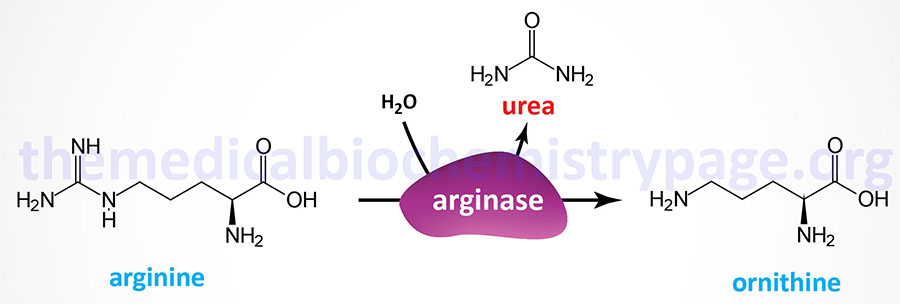
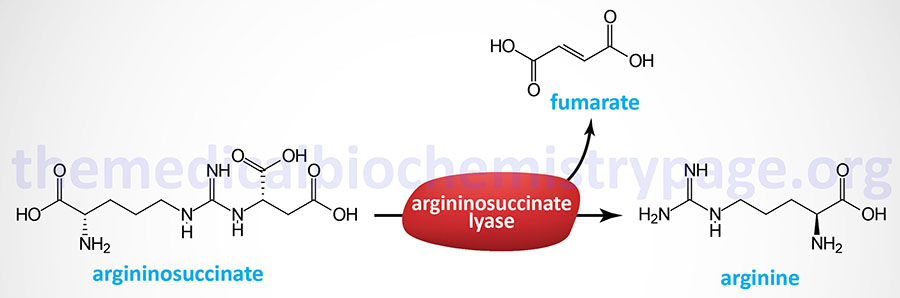
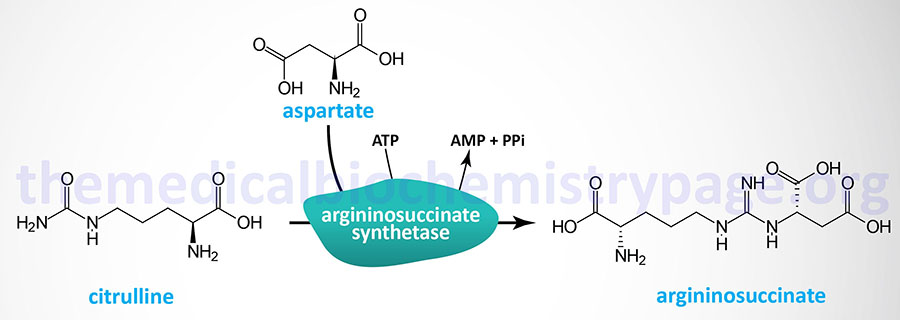
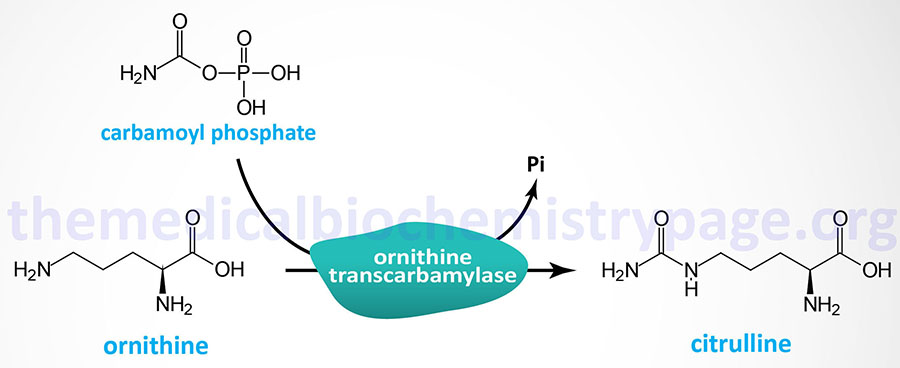
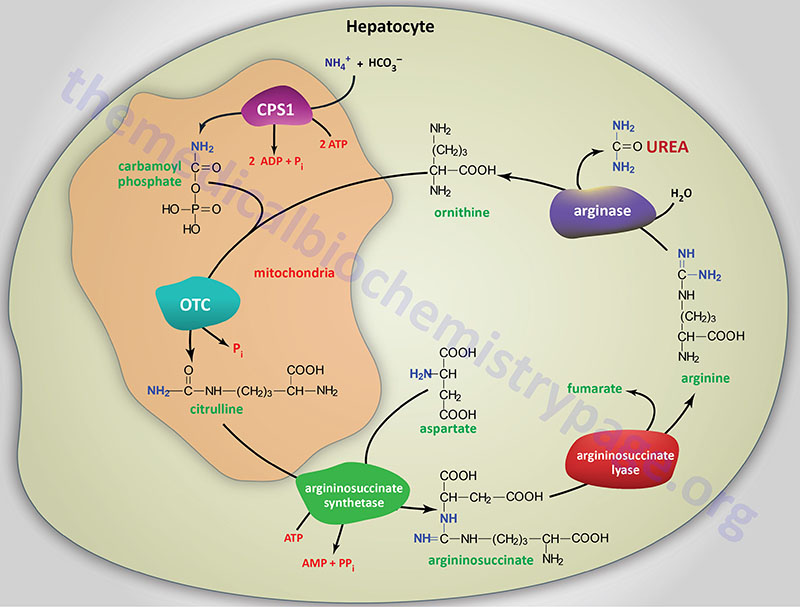
Last Updated: March 25, 2026 Introduction to NAGS Deficiency N-acetylglutamate synthase deficiency (NAGSD) is most commonly the result of the inheritance of mutations in the NAGS gene which encodes N-acetylglutamate synthase. NAGSD manifests in two forms identified as...