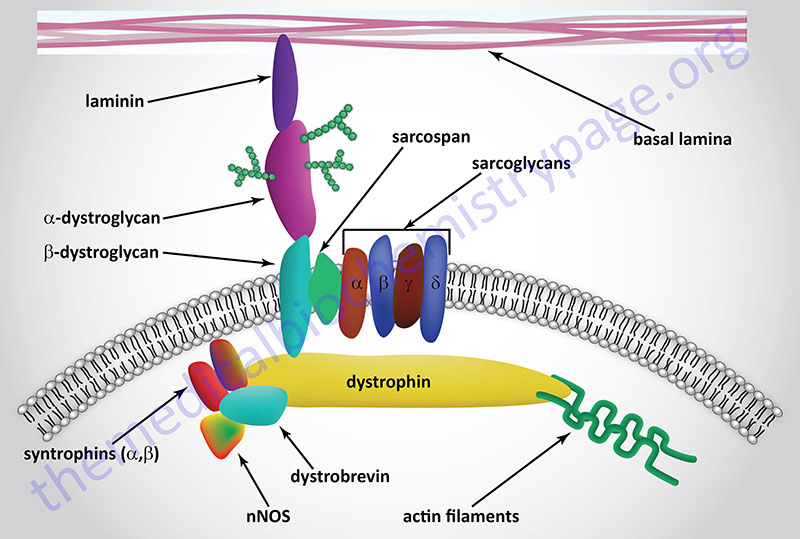
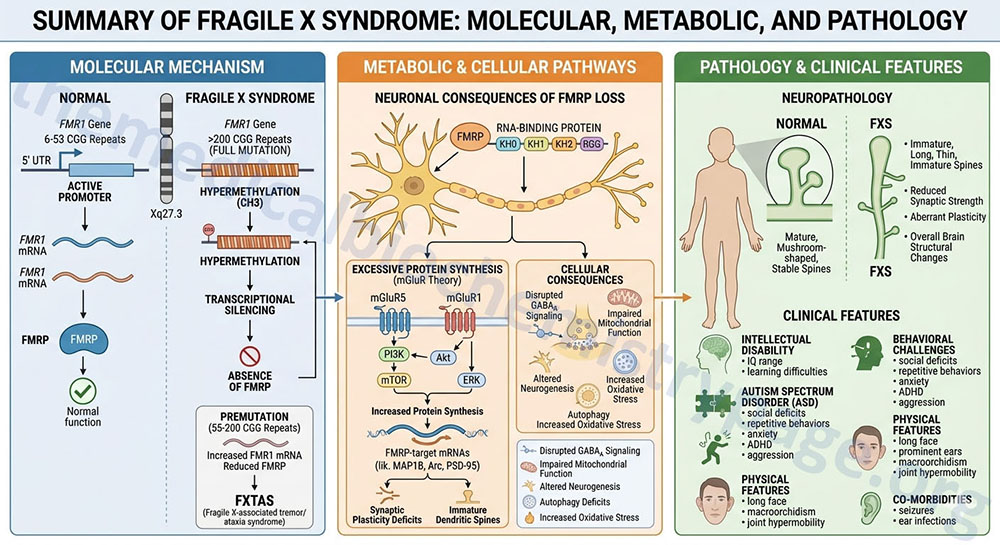
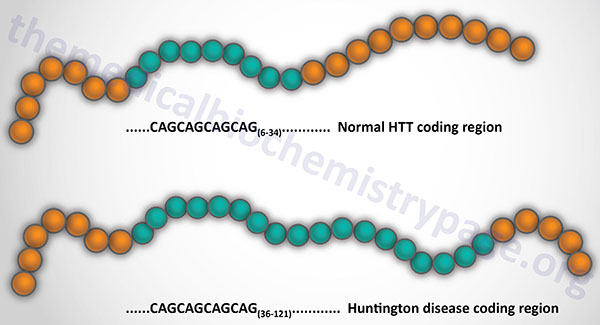
Last Updated: October 31, 2025 Introduction to Myotonic Dystrophy Type 1 myotonic dystrophy (originally abbreviated DM1 for the Latin dystrophia myotonica) is the most commonly occurring form of muscular dystrophy in adults. The disease manifests in nearly 1 in...