
 reactions depicted in detail")
Last Updated: July 5, 2026
Introduction to Pyruvate Metabolism and the TCA Cycle
The bulk of the ATP used by all cells (except mature red blood cells), to maintain homeostasis, is produced by the re-oxidation of the reduced electron carriers, NADH and FADH2, within the mitochondrial oxidative phosphorylation pathway. A large percentage of these two reduced electron carriers are generated in the TCA cycle by the oxidation of the acetyl-CoA derived from pyruvate via the actions of the pyruvate dehydrogenase complex (PDHc).
The fate of pyruvate depends on the cell energy charge. In liver, when the energy charge is high, pyruvate is directed toward gluconeogenesis. However, when the energy charge is low, pyruvate is preferentially oxidized to CO2 and H2O through the activities of the pyruvate dehydrogenase complex (PDHc) and the enzymes of the TCA cycle, with generation of approximately 15 moles of ATP per mole of pyruvate. The enzymatic activities of the TCA cycle (and of oxidative phosphorylation) are located within the inner mitochondrial matrix and inner mitochondrial membrane.
The synthesis of pyruvate occurs nearly exclusively in the cytosol and its oxidation occurs exclusively in the mitochondria, therefore transport into the mitochondria is critical for overall pyruvate homeostasis. The transport of pyruvate into the mitochondria involves two distinct sets of transport complexes. The outer mitochondrial membrane transports pyruvate into the intermembrane compartment via the action of a voltage-gated porin complex. Transport of pyruvate across the inner mitochondrial membrane requires the mitochondrial pyruvate carrier (MPC) complex. The MPC is a heterotetrameric complex composed of subunits encoded by the MPC1 and MPC2 genes. The MPC transporter proteins are members of the large superfamily of solute carrier (SLC) proteins and as such MPC1 is also identified as SLC54A1 and MPC2 is also identified as SLC54A2.
The MPC1 gene is located on chromosome 6q27 and is composed of 7 exons that generate seven alternatively spliced mRNAs encoding MPC1 isoform 1 (109 amino acids), MPC1 isoform 2 (66 amino acids), and MPC1 isoform 3 (70 amino acids).
The MPC2 gene is located on chromosome 1q24.2 and is composed of 7 exons that generate two alternatively spliced mRNAs that both encode the same 127 amino acid protein.
Role of Mitochondrial Pyruvate Transport in Disease
In animal studies, where the transport of pyruvate into the mitochondria via the MPC has been disrupted by gene deletion or by pharmacological inhibition, a beneficial effect is observed at the level of insulin resistance, blood glucose homeostasis, diabetes, and the development of metabolic dysfunction-associated steatohepatitis, MASH (commonly referred to as non-alcoholic steatohepatitis: NASH). These results imply that the function of the MPC contributes to metabolic dysfunction and that targeted pharmacological interference with the MPC in humans may exert beneficial health effects.
In addition to disturbances in mitochondrial pyruvate metabolism in diabetes, which contributes to excess hepatic gluconeogenesis, this disorder, as well as insulin resistance, is associated with disruptions in lipid and amino acid metabolic homeostasis.
In individuals with insulin resistance, as well as those with type 2 diabetes, there is an associated elevation in the circulating levels of the branched-chain amino acids (BCAA) leucine, isoleucine, and valine. Indeed, high plasma levels of BCAA has been shown to be predictive for the future development of diabetes. In addition, in animal models it has been shown that dietary restriction of the BCAA is associated with improve insulin sensitivity.
Pharmacologic inhibition of the MPC is associated with reduction in plasma levels of the BCAA. As described in detail in the Amino Acid Catabolism page, the metabolism of the BCAA begins with a transamination reaction that yields the corresponding branched-chain ketoacids (BCKA). The three BCKA are then oxidized by branched-chain keto acid dehydrogenase, BCKD.
The activity of the BCKD is controlled by phosphorylation, which inhibits the complex, and dephosphorylation, which activates the complex. The effects of MPC inhibition have been shown to be associated with increased oxidation of the BCKA via activation of the dephosphorylation of the BCKD by the phosphatase, PPM1K.
In addition to effects directly on the activity of BCKD, the inhibition of MPC leads to activation of AMPK with the result being alterations in various metabolic processes that, in turn, reduce the level of phosphorylation of BCKD with the consequences being increased metabolism of BCAA.
The Pyruvate Dehydrogenase Complex (PDHc)
When transported into the inner mitochondrial matrix, pyruvate encounters two principal metabolizing enzymes: pyruvate carboxylase, PC (a gluconeogenic enzyme) and pyruvate dehydrogenase (PDH), the first enzyme of the PDH complex (PDHc). With a high cell-energy charge within hepatocytes, co-enzyme A (CoA) exists principally as acetyl-CoA which potently activates pyruvate carboxylase, directing pyruvate toward gluconeogenesis. When the energy charge is low, CoASH is not acetylated, therefore, pyruvate carboxylase is inactive, and pyruvate is preferentially oxidized to acetyl-CoA via the PDHc. The PDHc reactions contribute to the generation of ATP through the concomitant reduction of NAD+ to NADH followed by the re-oxidation of NADH via complex I of the oxidative phosphorylation process.
Within the context of the TCA cycle the acetyl-CoA generated by the PDHc is completely oxidized to CO2 and H2O. During this oxidative cycle the reduced electron carriers (NADH and FADH2) that are generated can then be used to drive ATP synthesis via oxidative phosphorylation.
The pathway of PDHc-mediated oxidation of pyruvate to acetyl-CoA is diagrammed below:
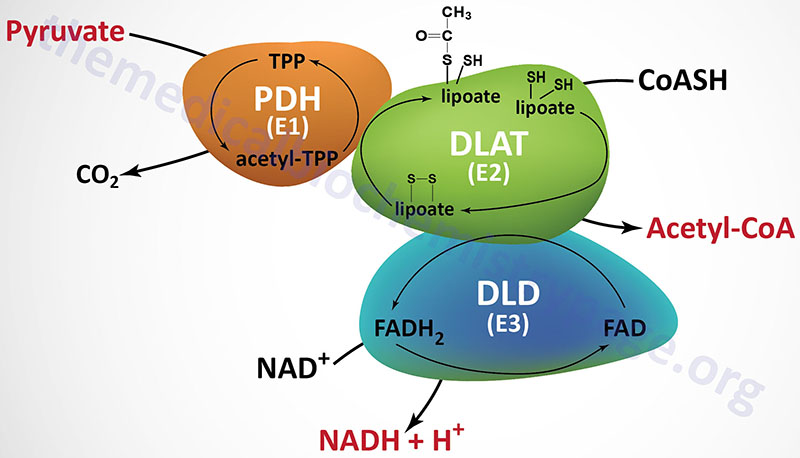
The PDHc is comprised of multiple copies of three distinct enzyme activities: pyruvate dehydrogenase (PDH, identified as the E1 component), dihydrolipoamide S-acetyltransferase, (DLAT, identified as the E2 component), and dihydrolipoamide dehydrogenase, (DLD, identified as the E3 component). The oxidation of pyruvate via the PDHc is highly exergonic with a free energy change on the order of –33 to –37.5 kJ/mol (–8 to –9 kcal/mol).
The PDH activity (E1) is a heterotetrameric complex composed of two α (alpha) and two β (beta) subunits. The α subunits are primarily encoded by the PDHA1 gene. The PDHA1 gene is located on chromosome Xp22.12 and is composed of 12 exons that generate four alternatively spliced mRNAs, each of which encodes a distinct protein isoform. The predominant PDHA1 derived mRNA (isoform 1) encodes a precursor protein of 390 amino acids. Another PDH α-subunit gene (PDHA2) encodes a testis-specific isoform. The β subunits are encoded by the PDHB gene. The PDHB gene is located on chromosome 3p14.3 and is composed of 9 exons that generate three alternatively spliced mRNAs, each of which encodes a distinct protein isoform.
The DLAT gene is located on chromosome 11q23.1 and it is composed of 14 exons that generate 13 alternatively spliced mRNAs. Each of these 13 mRNAs encode distinct protein isoforms of the DLAT enzyme.
The DLD gene is located on chromosome 7q31.1 and it is composed of 14 exons that generate four alternatively spliced mRNAs with the largest mRNA encoding a protein of 509 amino acids. The DLD encoded proteins function within the PDHc as homodimeric subunits. The DLD encoded proteins are also functional components of the 2-oxoglutarate dehydrogenase complex (more commonly known as α-ketoglutarate dehydrogenase) of the TCA cycle described below, and of the branched-chain keto acid dehydrogenase complex (BCKD, involved in branched-chain amino acid metabolism).
In addition to the catalytic components of the PDHc there is a non-catalytic component termed component X (PDHX) of the PDHc. PDHX is also called E3 binding protein (E3BP). The PDHc component X protein tethers the E3 subunits of the PDHc to the E2 subunits. The PDHX gene is located on chromosome 11p13 and is composed of 13 exons that generate three alternatively spliced mRNAs, each of which encode distinct protein isoforms. The protein encoded by the isoform 1 mRNA is the largest of the three proteins at 501 amino acids. The protein encoded by the isoform 2 mRNA is translated from an upstream in-frame AUG codon. The protein encoded by the isoform 3 mRNA lacks 227 amino acids that are present in the central region of the protein encoded by the isoform 1 mRNA.
In addition to the complexity of the PDHc at the level of the total number of protein subunits (30 subunits of E1, 60 subunits of E2, and 12 subunits of E3), the full activity of the PDHc requires five different vitamin-derived coenzymes: CoASH, NAD+, FAD, lipoic acid, and thiamine pyrophosphate (TPP). A mnemonic that helps one remember these five required co-factors/coenzymes is Tender (TPP) Loving (lipoic acid) Care (CoA) For (FAD) Nancy (NAD+). Three of the coenzymes of the PDHc are tightly bound to enzymes of the complex (TPP, lipoic acid, and FAD) and two are employed as carriers of the products of PDHc activity (CoASH and NAD+). Two additional dehydrogenase complexes require these same five coenzymes: the 2-oxoglutarate dehydrogenase complex (OGDH) and the BCKD complex.
The first enzyme of the PDHc is the pyruvate dehydrogenase activity (E1) which oxidatively decarboxylates pyruvate. During the course of the reaction the acetyl group derived from decarboxylation of pyruvate is bound to TPP. The next reaction of the complex (E2) is the transfer of the two carbon acetyl group from acetyl-TPP to lipoic acid, the covalently bound coenzyme of DLAT. The transfer of the acetyl group from acetyl-lipoamide to coenzyme A (CoASH) results in the formation of two free sulfhydryl (–SH) groups in lipoate. The -SH groups require re-oxidation to the disulfide (–S-S–) form to regenerate lipoate as a competent acetyl acceptor. The enzyme DLD (E3), with FAD as a cofactor, catalyzes that oxidation reaction generating FADH2. The final activity of the PDHc is the DLD-mediated transfer of reducing equivalents from the FADH2 to NAD+ generating NADH. The fate of the NADH is oxidation via mitochondrial electron transport, to produce approximately three equivalents of ATP:
The net result of the reactions of the PDHc are:
Pyruvate + CoA + NAD+ → CO2 + acetyl-CoA + NADH + H+
Regulation of the PDH Complex
The reactions of the PDHc serve to interconnect the metabolic pathways of glycolysis, gluconeogenesis and fatty acid oxidation to the TCA cycle. The activity of the PDHc is also important in regulating the flux from glucose to malonyl-CoA which is required for the de novo synthesis of fatty acids.
Acetyl-CoA, produced in the mitochondria via the action of the PDHc, will be transported out into the cytosol in the form of citrate when the energy charge of the cell rises. In the cytosol, the citrate is hydrolyzed by ATP-citrate lyase (gene symbol: ACLY) yielding oxaloacetate and acetyl-CoA.
In the cytosol the acetyl-CoA can be converted to malonyl-CoA via the action of acetyl-CoA carboxylase (ACC). This malonyl-CoA serves as the precursor for the synthesis of fatty acids. Accumulation of cytosolic malonyl-CoA, as will happen in energy rich conditions, partitions cytosolic fatty acids away from the oxidizing machinery of the mitochondria by inhibiting carnitine palmitoyltransferase 1 (CPT-1). This effect of malonyl-CoA, derived from acetyl-CoA, couples increased rates of glucose oxidation to inhibition of fatty acid oxidation.
The regulation of the PDHc is complex involving both allosteric activation and inhibition as well as post-translational modification. The principal post-translational modification affecting the activity of the PDHc is phosphorylation by a family of kinases called PDH kinases. The activity of these kinases is regulated by various allosteric effectors that can either increase or decrease the rate of PDHc phosphorylation. There are four PDH kinases (PDK1, PDK2, PDK3, and PDK4) and they are discussed in detail below the Figure.
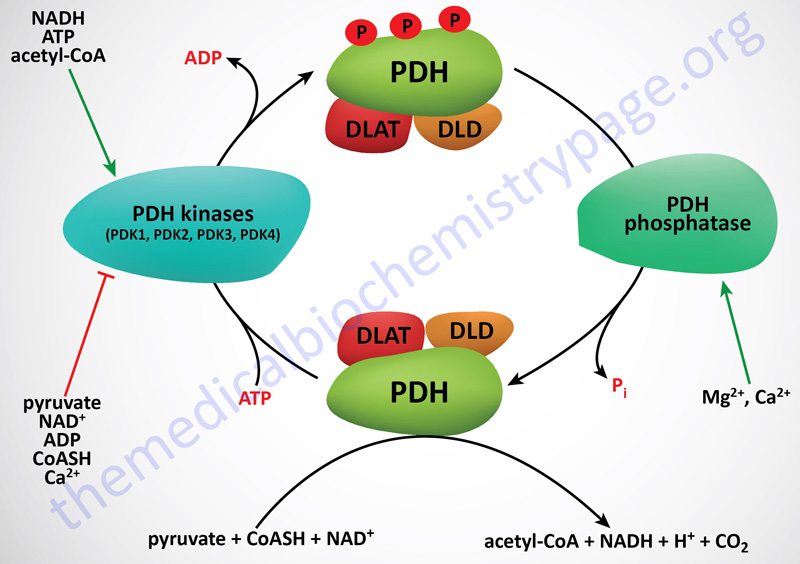
PDHc Kinases
The activity of PDH (E1) is rapidly regulated by phosphorylation and dephosphorylation events that are catalyzed by PDH kinases (PDK) and PDH phosphatases (PDP), respectively. Phosphorylation of PDH results in inhibition of activity, whereas, dephosphorylation increases it. The PDK have been shown to be tightly bound to the lipoyl domains of the DLAT (E2) subunits, anchoring the kinases to the PDHc. On average, there are two to three PDK bound to each PDHc.
Phosphorylation of the α-subunit of PDH (E1α; encoded by the PDHA1 gene) occurs at three different serine residues termed site 1 (Ser264), site 2 (Ser271), and site 3 (Ser203). Phosphorylation of any of these three serine residues results in the inhibition of the PDHc. However, data show that phosphorylation of site 1 occurs most rapidly and site 3 being the least rapidly phosphorylated. Given these differences in phosphorylation rate, site 1 is of primary importance for the acute regulation of the PDHc activity.
Four PDK isozymes have been identified in humans: PDK1, PDK2, PDK3, and PDK4, each of which is encoded by a distinct gene.
The PDK1 gene is located on chromosome 2q31.1 and is composed of 27 exons that generate two alternatively spliced mRNAs encoding two distinct protein isoforms. Isoform 1 is a 456 amino acid precursor protein and isoform 2 is a 436 amino acid precursor protein.
The PDK2 gene is located on chromosome 17q21.33 and is composed of 14 exons that generate four alternatively spliced mRNAs encoding three distinct protein isoforms.
The PDK3 gene is located on the X chromosome (Xp22.11) and is composed of 12 exons that generate two alternatively spliced mRNAs encoding two distinct protein isoforms. Isoform 1 is a 415 amino acid precursor protein and isoform 2 is a 406 amino acid precursor protein.
The PDK4 gene is located on chromosome 7q21.3 and is composed of 11 exons that encode a 411 amino acid protein.
Analysis of the patterns of expression of the PDK shows that PDK2 is the most widely expressed with the highest levels of expression seen in heart, liver, and kidney. PDK1 expression is highest in the heart and skeletal muscle. PDK3 expression predominates in lung and testis. PDK4 expression is high in heart, liver, pancreatic islets, and kidney and each of these tissues are capable of high rates of fatty acid oxidation and also express high levels of the transcription factors PPARα and PPARδ which are known to be critical regulators of the expression of genes encoding lipid metabolizing enzymes.
Utilizing mutant forms of PDH it has been possible to identify which sites are phosphorylated by which PDK. All four PDK have been shown to be able to phosphorylate sites 1 and 2. Site 3 is only phosphorylated by PDK1. PDK4 has a much higher preference for phosphorylation of site 2 when compared to the activities of PDK1, PDK2, and PDK3 at this site. Data from these types of experiments indicates that the activity of PDK2 is necessary for short-term inhibition of the PDHc via phosphorylation of site 1. On the other hand, PDK1 and PDK4 appear to be more important for allowing multisite phosphorylation which in turn prevents reactivation of the PDHc by PDP.
Allosteric Regulation of the PDHc and the PDK
Two products of the PDHc, NADH and acetyl-CoA (both of which are also products of fatty acid oxidation), are negative allosteric effectors on the non-phosphorylated and most active form of PDH. These effectors reduce the affinity of the enzyme for pyruvate, thus limiting the flow of carbon through the PDH complex. In addition, NADH and acetyl-CoA are powerful positive effectors on the activity of PDK2 and PDK4. Since NADH and acetyl-CoA accumulate when the cell energy charge is high, it is not surprising that high ATP levels also up-regulate PDK activity, reinforcing down-regulation of PDH activity in energy-rich cells. Note, however, that pyruvate is a potent negative effector on all PDKs, with the result that when pyruvate levels rise, active PDH will be favored even with high levels of NADH and acetyl-CoA.
Concentrations of pyruvate which maintain PDH in the active form are sufficiently high so that, in energy-rich cells, the allosterically down-regulated, high Km form of PDH is nonetheless capable of converting pyruvate to acetyl-CoA. With large amounts of pyruvate in cells having high energy charge and high NADH, pyruvate carbon will be directed to the two main storage forms of carbon, glycogen via gluconeogenesis and fat production via fatty acid synthesis, where acetyl-CoA is the principal carbon donor.
Regulation of the activity of the various PDK is manifest with isoform specificity. Pyruvate, generated from glycolysis, exerts differential inhibitory actions on the various PDK. PDK2 is the most sensitive to inhibition by pyruvate, whereas, PDK4 is relatively insensitive. As the ATP/ADP ratios falls (i.e. increases in ADP levels) ADP will exert an allosteric inhibition on the activity of the PDK. ADP exerts its allosteric inhibition of the PDK both independently and in synergy with pyruvate inhibition. In contrast to the acute inhibition of PDK by pyruvate, acute activation occurs via products of the PDHc as well as by fatty acid oxidation, namely acetyl-CoA and NADH. PDK2 is the most sensitive to increases in the level of acetyl-CoA in comparison to the other PDK isoforms. The ratio of NADH to NAD+ also exerts isoform-specific allosteric regulation of the PDK. PDK4 is highly responsive (activated) to an increased NADH/NAD+, whereas, PDK2 is much less sensitive to this activation.
PDHc Phosphatases
Two genetically and biochemically distinct PDP isoforms (PDP1 and PDP2) have been characterized. The PDP1 is a Mg2+-dependent and Ca2+-stimulated protein serine phosphatase of the protein phosphatase 2C (PP2C) superfamily. PDP1 is found bound to the inner mitochondrial membrane. Both the PDP1 and PDP2 phosphatases are heterodimeric enzymes composed of a catalytic subunit (PDPc) and a regulatory subunit (PDPr). There are two different catalytic subunit genes in humans identified as the PDP1 gene and the PDP2 gene. The PDP1 encoded protein (also known as PPM2C) is a component of the PDP1 phosphatase complex, whereas the PDP2 encoded protein (also known as PPM2C2) is a component of the PDP2 phosphatase complex.
The PDP1 gene is located on chromosome 8q22.1 and is composed of 6 exons that generate four alternatively spliced mRNAs encoding two distinct protein isoforms. Isoform 2 is a 562 amino acid protein. Isoform 3 is generated by translation initiation at a downstream AUG codon, relative to the AUG utilized in the translation of isoform 2. The isoform 3 protein is composed of 537 amino acids.
The PDP2 gene is located on chromosome 16q22.1 and is composed of 3 exons that encode a 529 amino acid protein.
The PDP regulatory subunit is encoded by the PDPR gene which is located on chromosome 16q22.1 and is composed of 23 exons that generate four alternatively spliced mRNAs that collectively encode three distinct precursor proteins.
The PDP1 phosphatase complex is widely distributed and is considered a major regulator of the activity of the PDH complex. The mechanism by which Ca2+ stimulates PDP catalytic (PDPc) activity is by increasing its interaction with the PDH complex while also decreasing the Km for Mg2+ of the catalytic subunits of the PDP1 complex. Calcium ion also increases the association of PDP1 catalytic subunits with the phosphorylated E1α subunit of the PDHc. In contrast, PDPr lowers the sensitivity of the catalytic subunits to Mg2+. Both PDP1 components are targeted by insulin, which enhances PDPc activity and decreases PDPr negative control, followed by enhanced PDPc sensitivity to Mg2+ and improved overall PDP1 efficiency. In addition, the level of PDP1 activity is affected by pathology with decreased levels of activity seen in diabetes.
The activity of the PDP2 phosphatase is the less well understood of the two PDP complexes but has been shown to consist of a catalytic subunit that is insensitive to Ca2+ and is 10-fold less sensitive to Mg2+ than the PDPc of PDP1. However, PDP2 is also considered a target of insulin action and the level of PDP2 activity is decreased in diabetes and during starvation.
Nuclear Localized Pyruvate Dehydrogenase Complex (nPDHc)
The primary enzyme in the nucleus that is responsible for the generation of acetyl-CoA is ATP-citrate lyase, ACLY. ACLY utilizes citrate as a substrate generating acetyl-CoA and oxaloacetate (OAA). However, components of the PDHc have been shown to translocate to the nucleus where they contribute to the generation of nuclear acetyl-CoA from pyruvate. The generation of acetyl-CoA in the nucleus is necessary for the acetylation of histone proteins, particularly histone H3. The acetylation of histone proteins represents a major epigenetic modification that regulates gene expression. The translocation of enzymes of the PDHc to the nucleus represents one of several mechanisms, that includes ACLY, that directly link metabolism to the regulation of gene expression.
Although all the proteins of the PDHc have been found to be localized to the nucleus, the major protein of the PDHc that has been shown to be transported into the nucleus is the α subunit of the E1 portion of the complex which is encoded by the PDHA1 gene. Within the nucleus, the PDHA1 encoded enzyme has been shown to be involved in the process of somatic cell reprogramming and the enhancement of the pluripotency of the reprogrammed cells.
While residing in the nucleus the PDHc is constitutively repressed by a scaffolding component of the mitotic deacetylase complex identified as ELMSAN1 (ELM2‐SANT domain‐containing scaffolding protein 1). ELM2 refers to Egl-27 and MTA1 homology 2 where the Egl-27 protein was characterized in Caenorhabditis elegans and stands for Egg-laying defective protein 27 and MTA1 stands for Metastasis-Associated Protein 1. The name of the SANT domain is derived from the four proteins in which the domain was originally identified. These four proteins (Swi3, Ada2, N-CoR, and TFIIIB) are involved in chromatin remodeling and transcriptional regulation.
In addition to the proteins of the PDHc, several enzymes of the TCA cycle are also localized to the nucleus under various conditions.
Clinical Significance of PDHc Mutations
There is no pathway in mammals for the conversion of acetyl-CoA to glucose and therefore, the activity of the PDHc is highly regulated by a variety of allosteric effectors and by covalent modification. The importance of the PDHc to the maintenance of homeostasis is evident from the fact that, although diseases associated with deficiencies of the PDHc have been observed, affected individuals often do not survive to maturity. Since the energy metabolism of highly aerobic tissues such as the brain, skeletal muscle, and the heart is dependent on normal conversion of pyruvate to acetyl-CoA, aerobic tissues are most sensitive to deficiencies in components of the PDHc. Most genetic diseases associated with PDHc deficiency are due to mutations in either of the two subunits (α encoded by the PDHA1 gene or β encoded by the PDHB gene) of the functional PDH heterotetramer of the PDHc. Mutations in the PDHA1 gene represent the largest percentage (80%) of PDH deficiencies.
Since the PDHA1 gene is on the X chromosome, mutations in this gene are inherited in an X-linked manner (recessive). Normally, females are very rarely affected by X-linked recessive disorders but this is not the case with PDHA1 mutations. The reason for this is that there is skewing of X chromosome inactivation such that females with one mutant PDHA1 gene appear to have the X chromosome harboring the wild-type PDHA1 gene inactivated at a higher rate than predicted based upon the normal random X-inactivation process. Mutations in the other genes of the PDHc, that result in PDH deficiency syndromes, are inherited in autosomal recessive patterns.
Mutations in genes encoding other components of the PDHc, including the PDHB gene and the DLAT gene, have been shown to be associated with PDH deficiency syndromes. In addition to components of the PDHc itself, mutations in the gene (PDP1) encoding one of the catalytic subunits of the major PDH phosphatase and mutations in the gene (PDHX) that encodes a E3 subunit binding protein, have been shown to be associated with PDH deficiency syndromes.
The main pathology associated with PDHc deficiencies is moderate to severe cerebral lactic acidosis that appears shortly after birth. Additional pathologies are neurological deficiencies that include intellectual disability, hypotonia, seizures, and difficulty walking.
The TCA Cycle
The TCA cycle is the major metabolic pathway through which large amounts of reduced electron carrier, in the form of NADH and FADH2, can be generated during the complete oxidation of acetyl-CoA to CO2 and H2O. Each mole of acetyl-CoA derives three moles of NADH and one mole of FADH2 which are re-oxidized in the electron transport chain (ETC) of oxidative phosphorylation generating ATP. The sources of the acetyl-CoA oxidized in the TCA cycle include the PDHc, fatty acid oxidation, and amino acid oxidation.
In addition to its central role in energy generation, the TCA cycle serves as a source of intermediates for the biosynthesis of numerous biomolecules. The carbons of malate can be diverted to the synthesis of glucose via the pathway of gluconeogenesis. The carbons of citrate are used to transport mitochondrial acetyl-CoA to the cytosol where they can be used for the synthesis of fatty acids, cholesterol, and bile acids. The carbons of succinyl-CoA are utilized in the synthesis of heme.
Unlike other metabolic pathways such as glycolysis, gluconeogenesis, and fatty acid synthesis for example, the TCA cycle is not directly controlled by hormonal influence. The major metabolic regulatory hormones, insulin, glucagon, and epinephrine, do not directly affect TCA cycle activity through changes in the levels of cAMP or by phosphorylation/dephosphorylation as is the case for almost all other metabolic pathways.
Anaplerosis
The carbons of numerous amino acids, as well as nucleotides, and certain fatty acids are utilized to “fill up” the TCA cycle in a process termed anaplerosis. These reactions are particularly important in periods of prolonged fasting allowing for the diversion of the carbons into the pathway of gluconeogenesis. With respect to fatty acid carbons it is only fatty acids with an odd number of carbon atoms that can contribute to the synthesis of glucose. The propionyl-CoA derived from the oxidation of these types a fats is converted to succinyl-CoA which then enters the TCA cycle. The catabolism of adenine nucleotides yields fumarate in a pathway referred to as the purine nucleotide cycle.
Cataplerosis
Given that metabolic intermediates from various pathways can be added to the TCA cycle via the process of anaplerosis, it is important that these intermediates be effectively oxidized and/or disposed of so as to prevent their accumulation in the mitochondrial matrix. The process of disposing of TCA cycle intermediates and diverting them to other metabolic pathways is referred to as cataplerosis. The primary cataplerotic reactions involve the enzymes glutamate dehydrogenase (GDH), aspartate transaminase (AST), and phosphoenolpyruvate carboxykinase (PEPCK).
TCA cycle 2-oxoglutarate (α-ketoglutarate) can be transaminated to glutamate which can itself be transaminated to glutamine. This provides a means to ensure effective nitrogen trafficking within the mitochondria. Oxaloacetate (OAA) can be transaminated to aspartate by aspartate transaminase which also provides a means for nitrogen trafficking. The aspartate can be used for purine nucleotide synthesis and, in the liver, the glutamine and aspartate can be used for nitrogen disposal via urea synthesis.
The role of PEPCK in cataplerosis in the liver and kidneys is of particular significance since it can generate PEP from oxaloacetate. The PEP can then serve as a substrate in the gluconeogenesis pathway. In skeletal muscle the PEP can be used for pyruvate synthesis which can then be oxidized by the PDHc and the resultant acetyl-CoA can be oxidized by the TCA cycle providing reduced electron carriers for ATP synthesis.
The cataplerotic reactions that involve conversion of PEP to pyruvate in the cytosol then pyruvate to oxaloacetate (OAA) then OAA to aspartate in the mitochondria then aspartate back to OAA in the cytosol and OAA back to PEP constitute what is referred to as pyruvate cycling. This cycle places the cytosolic form of PEPCK (encoded by the PCK1 gene) as the major cataplerotic enzyme.
Reactions of the TCA Cycle
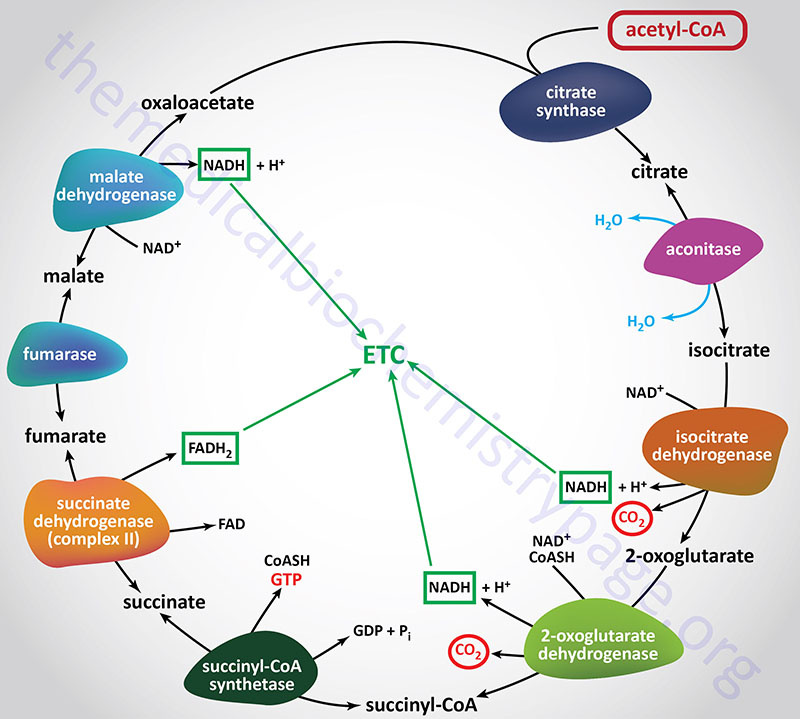
Citrate Synthase (Condensing enzyme):
Citrate synthase catalyzes the first reaction of the TCA cycle which involves the condensation of the methyl carbon of acetyl-CoA with the keto carbon (C-2) of oxaloacetate (OAA). Citrate synthase is encoded by the CS gene. The CS gene is located on chromosome 12q13.3 and is composed of 11 exons that encodes a precursor protein of 466 amino acids.
The citrate synthase reaction is often considered the rate-limiting step of the TCA cycle due to the large free energy change of the reaction. However, greater control over the overall rate of the cycle is exerted at the isocitrate dehydrogenase catalyzed reaction. The standard free energy of the citrate synthase reaction (–31.5 to –36.4 kJ/mol; –8.0 to –8.7 kcal/mol) drives it strongly in the forward direction. Since the formation of OAA from its precursor, malate, is thermodynamically unfavorable, the highly exergonic nature of the citrate synthase reaction is of central importance in keeping the entire cycle going in the forward direction, since it drives oxaloacetate formation by mass action principals.
When the cellular energy charge increases, the rate of flux through the TCA cycle will decline leading to a build-up of citrate. Excess citrate is used to transport acetyl-CoA carbons from the mitochondrion to the cytoplasm where they can be used for fatty acid synthesis and cholesterol biosynthesis. Citrate is transported across the inner mitochondrial membrane by the tricarboxylate transporter encoded by the SLC25A1 gene. The SLC25A1 encoded transporter is commonly identified as the mitochondrial citrate carrier, CiC.
Additionally, the increased levels of citrate in the cytoplasm activate the key regulatory enzyme of fatty acid biosynthesis, acetyl-CoA carboxylase (ACC) while at the same time citrate allosterically inhibits the rate-limiting enzyme of glycolysis, PFK-1.
Aconitase:
The isomerization of citrate to isocitrate by aconitase is stereospecific, with the migration of the –OH from the central carbon of citrate (formerly the keto carbon of OAA) being always to the adjacent carbon which is derived from the methylene group (–CH2–) of OAA. The stereospecific nature of the isomerization determines that the CO2 lost, as isocitrate is oxidized to succinyl-CoA, is derived from the oxaloacetate used in citrate synthesis. The standard free energy change of the aconitase reaction, in the normal direction of the TCA cycle, is on the order of +6.3 kJ/mol (+1.5 kcal/mol).
Humans produce two distinctly different aconitase enzymes, aconitase 1 and aconitase 2. One is a soluble form found within the cytosol and is involved in the iron-mediated regulation of protein synthesis, specifically the translation of several mRNAs including the ferritin and the transferrin receptor mRNAs (see the Protein Synthesis (Translation): Processes and Regulation page).
The soluble aconitase 1 is encoded by the ACO1 gene. The ACO1 gene is located on chromosome 9p21.1 and is composed of 23 exons that generate three alternatively spliced mRNAs, each of which encode the same protein of 889 amino acids.
The other aconitase enzyme (aconitase 2) is the mitochondrial enzyme involved in the TCA cycle. The mitochondrial aconitase 2 is encoded by the ACO2 gene. The ACO2 gene is located on chromosome 22q13.2 and is composed of 18 exons that encode a precursor protein of 780 amino acids.
Mitochondrial aconitase 2 is one of several mitochondrial enzymes known as non-heme-iron proteins. These proteins contain inorganic iron and sulfur, known as iron sulfur centers, in a coordination complex with sulfhydryls of cysteine residues in the protein. There are two prominent classes of non-heme-iron complexes, those containing two equivalents each of inorganic iron and sulfur, Fe2S2, and those containing four equivalents of each, Fe4S4. Aconitase 2 is a member of the Fe4S4 class. Its iron sulfur centers are often designated as Fe4S4Cys4, indicating that four cysteine sulfur atoms are involved in the complete structure of the complex. Within the iron sulfur compounds the iron is generally involved in the oxidation-reduction events.
Isocitrate Dehydrogenase:
Isocitrate is oxidatively decarboxylated to 2-oxoglutarate (α-ketoglutarate) by isocitrate dehydrogenase, IDH. There are three different isocitrate dehydrogenase (IDH) enzymes in humans identified as IDH1, IDH2, and IDH3 with only the IDH3 isoform being involved in the TCA cycle.
It is generally considered that control of carbon flow through the TCA cycle is regulated at IDH3 by the powerful negative allosteric effectors NADH and ATP and by the potent positive effectors; isocitrate, ADP and AMP. It is clear that cell energy charge is a key factor in regulating carbon flow through the TCA cycle.
The IDH1 and IDH2 enzymes are NADP+-dependent enzymes, whereas the IDH3 enzymes are NAD+-dependent. There are three IDH3 enzymes expressed in humans. The IDH3 isoforms are the primary mitochondrial enzymes responsible for oxidative decarboxylation of isocitrate within the context of the TCA cycle. The isocitrate dehydrogenase reaction is the rate-limiting step, as well as the first NADH-yielding reaction of the TCA cycle. The CO2 produced by the IDH3 reaction is from the original C-1 carbon of the oxaloacetate used in the citrate synthase reaction. The IDH3 reaction proceeds with a standard free energy change on the order of –3.3 kJ/mol (–0.8 kcal/mol).
Each of the functional IDH3 enzymes is a heterotetramer composed of three different subunits identified as the α, β, and γ subunits. The primary composition of IDH3 is α2β1γ1.
The α subunit is encoded by the IDH3A gene which is located on chromosome 15q25.1 and is composed of 12 exons that encode a precursor protein of 366 amino acids.
The β subunit is encoded by the IDH3B gene which located on chromosome 20p13 and is composed of 13 exons that generate four alternatively spliced mRNAs, each of which encodes a distinct precursor protein isoform.
The γ subunit is encoded by the IDH3G gene which is located on chromosome Xq28 and is composed of 12 exons that generate two alternatively spliced mRNAs encoding precursor proteins of 393 amino acids (isoform a) and 380 amino acids (isoform b).
Both IDH1 and IDH2 enzymes utilize NADP+ as their cofactor and generate NADPH via oxidative decarboxylation of isocitrate outside the context of the TCA cycle.
The IDH1 enzyme is a homodimeric enzyme encoded by the IDH1 gene. The IDH1 gene is located on chromosome 2q34 and is composed of 12 exons that generate three alternatively spliced mRNAs, all of which encode the same 414 amino acid protein. IDH1 is found primarily in the cytosol and the peroxisomes.
The IDH2 enzyme is also a homodimeric enzyme but is located in the mitochondria. IDH2 is encoded by the IDH2 gene which is located on chromosome 15q26.1 and is composed of 12 exons that generate three alternatively spliced mRNAs, each of which encode a distinct protein isoform.
The primary functions of IDH1 and IDH2 are to serve as producers of NADPH to enhance the cellular responses to oxidative stress and the generation of reactive oxygen species, ROS. The other principal sources of NADPH are from the glucose-6-phosphate dehydrogenase (G6PD) and 6-phosphogluconate dehydrogenase reactions in the pentose phosphate pathway and via the malic enzyme catalyzed reaction which is involved in the mobilization of mitochondrial acetyl-CoA to the cytosol.
The significance of IDH1 and IDH2 produced NADPH can be made clear by pointing out that cells with low level IDH1 and IDH2 expression are more sensitive to oxidative stress than cells with higher levels of expression of these two enzymes.
2-Oxoglutarate Dehydrogenase Complex (α-Ketoglutarate Dehydrogenase Complex):
2-Oxoglutarate (commonly called α-ketoglutarate) is oxidatively decarboxylated to succinyl-CoA by the 2-oxoglutarate dehydrogenase complex, OGDC. The OGDC is commonly referred to as the α-ketoglutarate dehydrogenase (α-KGDH) complex. This reaction generates the second TCA cycle equivalent of CO2 and NADH. As with the PDH complex described above, the reactions of the OGDH complex proceed with a large negative standard free energy change (–30.1 to –38.3 kJ/mol; –7.2 to –9.1 kcal/mol).
The OGDC is very similar to the PDH complex in the intricacy of its protein makeup (E1, E2, and E3 activities), cofactors (TPP, lipoic acid, CoA, FAD, and NAD+), and its mechanism of action. The three activities of OGDH complex are identified as oxoglutarate dehydrogenase (E1o), dihydrolipoamide S-succinyltransferase (DLST or E2o), and dihydrolipoamide dehydrogenase (DLD or E3).
Although the E1 activity of the OGDC is not subject to covalent modification as are the E1 components of the PDHc, allosteric regulation of the OGDC is indeed effected. The enzyme complex is also regulated by energy charge, the NAD+/NADH ratio, and effector activity of substrates and products.
The oxoglutarate dehydrogenase subunits of the OGDC are encoded by the OGDH gene. The OGDH gene is located on chromosome 7p13 and is composed of 29 exons that generate four alternatively spliced mRNAs that encode four distinct isoforms (isoform 1: 1023 amino acid precursor; isoform 2: 427 amino acid precursor; isoform 3: 1019 amino acid precursor; isoform 4: 1034 amino acid precursor).
The DLST subunits are encoded by the DLST gene. The DLST gene is located on chromosome 14q24.3 and is composed of 15 exons that generate two alternatively spliced mRNAs encoding isoform 1 (453 amino acid precursor) and isoform 2 (166 amino acid precursor).
The DLD subunits of the OGDC are the same as those found in the PDH complex.
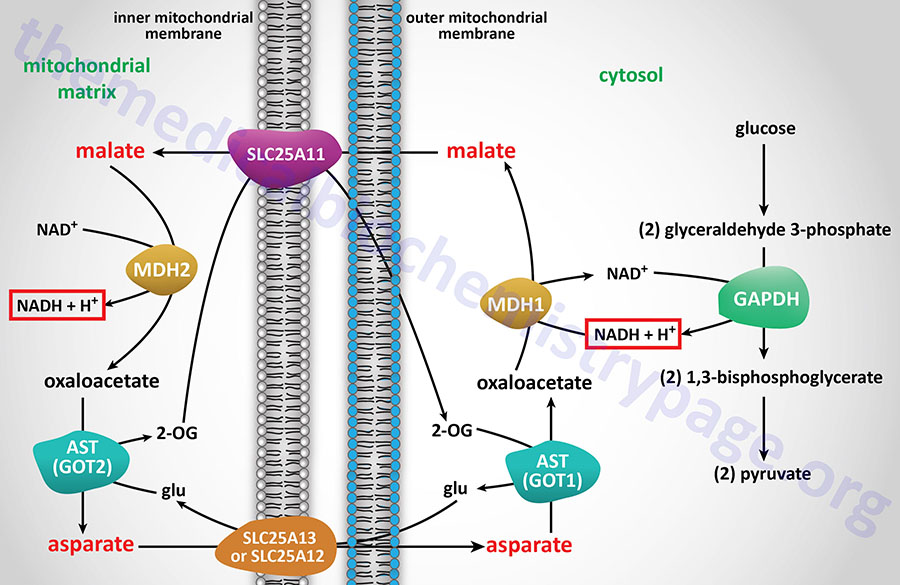
Succinyl-CoA and 2-oxoglutarate are also important metabolites outside the TCA cycle. In particular, 2-oxoglutarate represents a key anaplerotic metabolite linking the entry and exit of carbon atoms from the TCA cycle to pathways involved in amino acid metabolism. 2-Oxoglutarate is also important for driving the malate-aspartate shuttle. Succinyl-CoA, along with glycine, contributes all the carbon and nitrogen atoms required for the synthesis of heme and for non-hepatic tissue utilization of ketone bodies.
Succinyl-CoA Synthetase, SCS (succinate-CoA ligase, succinate thiokinase):
The conversion of succinyl-CoA to succinate, by succinyl-CoA synthetase (SCS), involves the use of the high-energy thioester of succinyl-CoA to drive synthesis of a high-energy nucleotide phosphate (GTP). This process is referred to as substrate-level phosphorylation. In this process, a high energy enzyme-phosphate intermediate is formed, with the phosphate subsequently being transferred to GDP. Mitochondrial GTP is used in a trans-phosphorylation reaction catalyzed by the mitochondrial enzyme, nucleoside diphosphate kinase, to phosphorylate ADP, producing ATP and regenerating GDP for the continued operation of succinyl-CoA synthetase. In addition to the use of GDP, SCS can also utilize ADP as a substrate which directly generates ATP during the course of the reaction. The use of GDP or ADP is determined by the presence of specific β-subunits.
The free energy change of the succinyl-CoA synthetase reaction is on the order of –3.4 kJ/mol (–0.8 kcal/mol).
Succinyl-CoA synthetase is a heterodimeric enzyme composed of an α (alpha) and a β (beta) subunit. The α-subunit is invariant in all forms of SCS and is encoded by the SUCLG1 (succinate-CoA ligase) gene. The SUCLG1 gene is located on chromosome 2p11.2 and is composed of 9 exons encoding a precursor protein of 346 amino acids.
The β-subunit of SCS determines the nucleotide specificity of the enzyme complex. There are two β-subunit genes encoding the A-beta and G-beta subunits. As indicated by the designations, the A-beta subunit determines specificity for ADP/ATP and the G-beta for GDP/GTP.
The A-beta subunit is encoded by the SUCLA2 gene. The SUCLA2 gene is located on chromosome 13q14.2 and is composed of 11 exons encoding a precursor protein of 463 amino acids.
The G-beta subunit is encoded by the SUCLG2 gene. The SUCLG2 gene located on chromosome 3p14.1 and is composed of 15 exons that generate two alternatively spliced mRNAs both of which encode distinct precursor protein isoforms of 440 amino acids (isoform 1) and 432 amino acids (isoform 2). The protein encoded by the SUCLG2 gene is also referred to as GTPSCS.
Recent studies have shown that the GTP-specific form of SCS can localize to the nucleus via a nuclear localization signal in the SUCLG1 encoded subunit. Within the nucleus SUCLG2 encoded subunit is acetylated which promotes the formation of a complex with the histone acetyltransferase, p300. This complex exhibits lactoyl-CoA synthetase and lactoyltransferase activities. The complex lactylates lysine 18 (K18) in histone H3 (H3K18la) which has been shown to lead to regulation of the expression of the GDF15 gene.
Succinate Dehydrogenase (SDH):
The multi-enzyme complex called succinate dehydrogenase (SDH) catalyzes the oxidation of succinate to fumarate with the sequential reduction of enzyme-bound FAD to FADH2 as well as the associated non-heme-iron. The SDH complex is the same as complex II of the electron transport chain (ETC) of oxidative phosphorylation. In mammalian cells, the electron acceptor is coenzyme Q (CoQ; written as CoQ10 to depict the major form in human cells), a mobile carrier of reducing equivalents that is restricted, by its lipophilic nature, to the lipid phase of the inner mitochondrial membrane and by its association with subunits of the SDH complex. The SDH complex is the only enzymatic activity of the TCA cycle that is embedded within the inner mitochondrial membrane, all of the other enzymes reside within the matrix of the mitochondria. The SDH reaction proceeds with a free energy change on the order of –3.1 kJ/mol (–0.7 kcal/mol).
The SDH complex is composed of four different proteins encoded by four different nuclear genes identified as SDHA, SDHB, SDHC, and SDHD. The SDH complex is the only component of the mitochondrial electron transport chain (ETC) that is composed solely of proteins encoded by nuclear genes. In addition to these four proteins, that constitute the functional SDH complex (oxidative phosphorylation complex II), at least two additional proteins are required for proper assembly of the complex. These assembly factors are called SDH complex assembly factor 1 (SDHAF1) and 2 (SDHAF2).
The A subunit of SDH is a flavoprotein encoded by the SDHA gene which is located on chromosome 5p15.33 and is composed of 17 exons the generate three alternatively spliced mRNAs encoding proteins of 664 amino acids (isoform 1), 616 amino acids (isoform 2), and 583 amino acids isoform 3). The SDHA encoded protein is the enzymatic activity that carries out the oxidation of succinate to fumarate with the simultaneous reduction of FAD to FADH2. The FAD is covalently linked to the active site of the SDHA subunits.
The B subunit of SDH is an iron-sulfur protein encoded by the SDHB gene which located on chromosome 1p36.13 and is composed of 8 exons that generate two alternatively spliced mRNAs encoding precursor proteins of 280 amino acids (isoform 1) and 262 amino acids (isoform 2). The SDHB encoded protein contains three distinct Fe-S (iron-sulfur) centers that facilitate the transfer of electrons from FADH2 to ubiquinone (coenzyme Q: CoQ). The Fe-S centers of SDHB consist of a 2Fe-2S center, which is near the FAD binding site, a 4Fe-4S center, and a 3Fe-4S center.
The C subunit of SDH is an integral membrane protein encoded by the SDHC gene which is located on chromosome 1q23.3 and is composed of 7 exons that generate 12 alternatively spliced mRNAs that collectively encode 11 distinct precursor protein isoforms.
The D subunit of SDH is also an integral membrane protein encoded by the SDHD gene located on chromosome 11q23.1 which is composed of 6 exons that generate four alternatively spliced mRNAs, each of which encode distinct precursor protein isoforms.
The inner mitochondrial membrane embedded SDHC and SDHD encoded proteins (referred to as the membrane domain of the SDH complex) anchor the SDHA and SDHB subunits to the inner mitochondrial membrane. The SDHC and SDHD subunits also facilitate interactions with CoQ and electron exchange from the Fe-S centers of the SDHB subunit. The membrane domain of the SDH complex contains a heme b moiety that is bound at the interface between the SDHC and SDHD subunits. The binding of CoQ to the SDHC and SDHD membrane domain occurs on the matrix side of the inner mitochondrial membrane.
Regulation of the SDH Complex
The catalytic activity of the SDH complex is controlled by post-translational modifications that include phosphorylation and acetylation. In addition, the catalytic activity of the SDHA subunit is allosterically inhibited by oxaloacetate. As succinate levels rise the inhibitory oxaloacetate is displaced, thereby activating the SDH complex.
Phosphorylation of the SDH complex has been shown to occur at two tyrosine residues via the action of FGR kinase, a SRC family member tyrosine kinase. However, these results were obtained via in vitro experiments and the identity of the kinase(s) that phosphorylate the SDH complex in vivo is still unknown. However, the significance of tyrosine phosphorylation, to the activity of the SDH complex, was demonstrated when it was shown that blocking the activity of the mitochondrial tyrosine phosphatase, PTPMT1, results in hyperphosphorylation and activation of the SDH complex.
Reversible lysine acetylation of the SDHA subunit has also been shown to be responsible for the attenuation of succinate oxidation to fumarate. When acetylated the activity of SDHA is reduced primarily due to reduction in substrate entry into the enzyme active site. Multiple Lys (K) residues in the SDHA subunit have been shown to be acetylated. However, similar to the situation for possible SDH kinases, no definitive SDH acetylase has been characterized. The principal enzyme responsible for the deacetylation of the SDHA subunit has been defined. One of the sirtuin family of deacetylases, SIRT3, has been shown to be the major mitochondrial deacetylase controlling the level of SDHA acetylation.
Fumarate Hydratase, FH (fumarase):
The fumarate hydratase (FH: commonly called fumarase) catalyzed reaction is specific for the trans form of fumarate. In the context of the TCA cycle the result of the FH reaction is that the hydration of fumarate proceeds stereospecifically with the production of L-malate. Due to the relatively low free energy change of the FH reaction, being around –0.8 kcal/mol (–3.8kJ/mol) in the direction of malate production, the reaction is freely reversible.
The fumarate hydratase gene (symbol: FH) is located on chromosome 1q43 and is composed of 10 exons that encode two distinct mRNAs as a result of alternative transcriptional initiation directed by a broad promoter region. It was originally thought that the two forms of human fumarate hydratase resulted from a single FH mRNA as a result of alternative translational start site utilization as is the case for the rat fumarate hydratase forms.
One form of the enzyme is localized to the cytosol while the TCA cycle enzyme is localized to the mitochondria. The mitochondrial version of fumarate hydratase (a 510 amino acid protein) is extended at the N-terminus, relative to the cytosolic form of the enzyme. The N-terminal extension contains a mitochondrial targeting sequence. The cytosolic fumarate hydratase is 43 amino acids shorter than the mitochondrial version and this shorter FH protein lacks the mitochondrial targeting sequence.
Fumarate Hydratase and DNA Repair
The cytosolic form of FH is involved in a process of cellular protection from DNA double strand breaks as a part of the DNA damage response pathway. In response to DNA damage the cytosolic FH migrates into the nucleus where its activity is required to inhibit histone demethylases. Histone demethylases belong to the large family of 2-oxoglutarate and Fe2+-dependent dioxygenases (also referred to as 2OG-oxygenases).
In response to DNA double strand breaks, such as occurs as a result of ionizing radiation, the nuclear localization of FH allows it to bind to the variant histone, H2A.Z, at regions of the breaks. The association of FH with histone H2A.Z is dependent on FH being phosphorylated on a threonine residue (T236) by the DNA-dependent protein kinase, DNA-PK. DNA-PK is encoded by the PRKDC gene. The association of FH with H2A.Z results in localized production of fumarate. This locally produced fumarate inhibits the activity of a lysine demethylase of the KDM2 family, specifically KDM2B. The inhibition of KDM2B by fumarate leads to accumulation of dimethylated histone H3 on lysine 36, designated H3K36me2. The dimethylated H3 promotes the accumulation of DNA-PK at regions of double strand breaks facilitating the process of non-homologous end joining (NHEJ) DNA repair.
Malate Dehydrogenase (MDH):
Mitochondrial malate dehydrogenase (MDH) catalyzes the “final” reaction of the TCA cycle. L-malate is the specific substrate for MDH. The forward reaction of the cycle, the oxidation of malate, yields oxaloacetate (OAA). In the forward direction the reaction has a standard free energy of about +7 kcal/mol, indicating the very unfavorable nature of the forward direction. As noted earlier, the citrate synthase reaction that condenses oxaloacetate with acetyl-CoA has a standard free energy of about –8 kcal/mol and is responsible for pulling the MDH reaction in the forward direction. The overall change in standard free energy change of the MDH reaction is about –1 kcal/mol (–4.2 kJ/mol) for the conversion of malate to oxaloacetate.
There are two malate dehydrogenases in humans encoded by two different genes. Both forms of the enzyme are required for the operation of the malate-aspartate shuttle. The cytosolic form of MDH is encoded by the MDH1 gene and the mitochondrial version is encoded by the MDH2 gene.
he MDH1 gene is located on chromosome 2p15 and is composed of 10 exons that generate four alternatively spliced mRNAs. Three of these MDH1-derived mRNAs encode distinct cytoplasmic enzymes while the fourth mRNA (designated MDH1x) encodes a peroxisomal enzyme. The MDH1x mRNA encodes two proteins through the use of alternative in-frame translation termination codons.
The MDH2 gene is located on chromosome 7q11.23 and is composed of 10 exons that generate three alternatively spliced mRNAs, each of which encode distinct protein isoforms.
Stoichiometry of the TCA Cycle
The overall stoichiometry of the TCA cycle is:
acetyl-CoA + 3NAD+ + FAD + GDP + Pi + 2H2O → 2CO2 + 3NADH + FADH2 + GTP + 2H+ + CoASH
Role of Lactate as Driver for TCA Cycle Activity
The lactate produced by anaerobic glycolysis does not simple serve as an intracellular carbon source for hepatic glucose production via gluconeogenesis. Lactate is transported out of cells transported out of the cells via transporters of the monocarboxylate transporter (MCT) family which are encoded by genes of the SLC16 family.
Extracellular lactate has been shown to be the major carbon source feeding the tricarboxylic acid (TCA) cycle within multiple different tissues, indeed in all tissues except the brain. Within cancer cells lactate is the primary substrate for TCA cycle activity as well. Lactate originating from anaerobic glycolysis, predominantly occurring in red blood cells and skeletal muscle, is transported via the blood to other tissues and transported into cells via SLC16 family member transporters. Upon uptake the lactate can be oxidized to pyruvate via the action of lactate dehydrogenase. The pyruvate is then oxidized by the pyruvate dehydrogenase complex (PDHc) and the resulting acetyl-CoA can enter the TCA cycle.
The metabolic benefit of lactate serving as the primary substrate for TCA cycle activity is that it allows decoupling of glycolysis from the TCA cycle allowing for independent tissue-specific regulation of both processes. This decoupling of glycolysis and TCA cycle activity allows glycolysis to be utilized to drive cell proliferation. This is accomplished by oxidation of glucose in the pentose phosphate pathway to yield the NADPH required for reductive biosynthetic processes. In addition, the decoupling allows more glucose be to utilized by the brain.
Regulation of the TCA Cycle
There is no direct hormonal regulation of TCA cycle activity. Substrate availability and allosteric regulation of enzyme activity controls the rate of flux through the cycle. Fuel enters the TCA cycle primarily as acetyl-CoA, albeit several intermediates in the cycle are replenished via the process referred to as anaplerosis. The generation of acetyl-CoA from carbohydrates is a major control point of the cycle. This is the reaction catalyzed by the PDH complex.
By way of review, the PDH complex is inhibited by acetyl-CoA and NADH and activated by non-acetylated CoA (CoASH) and NAD+. The pyruvate dehydrogenase activities of the PDH complex are regulated by their state of phosphorylation. This modification is carried out by specific kinases (PDK1–PDK4) and the phosphates are removed by a specific phosphatase (PDH phosphatase, PDP). The phosphorylation of PDH inhibits its activity and, therefore, leads to decreased oxidation of pyruvate. PDKs are activated by NADH and acetyl-CoA and inhibited by pyruvate, ADP, CoASH, Ca2+ and Mg2+. PDP, in contrast, is activated by Mg2+ and Ca2+.
Since three reactions of the TCA cycle as well as PDH utilize NAD+ as co-factor it is not difficult to understand why the cellular ratio of NAD+/NADH has a major impact on the flux of carbon through the TCA cycle. Substrate availability can also regulate TCA flux. This occurs at the citrate synthase reaction as a result of reduced availability of oxaloacetate. Product inhibition and energy state also controls the TCA flux.
The major control point in the TCA cycle occurs at the IDH3 catalyzed reaction. Both NADH and ATP allosterically inhibit the enzyme. The IDH3 catalyzed reaction is, therefore, considered the rate-limiting reaction of the cycle. Other points of control include citrate inhibition of citrate synthase and NADH and ATP allosteric inhibition of 2-oxoglutarate dehydrogenase (α-ketoglutarate dehydrogenase). Several key enzymes of the TCA cycle are also regulated allosterically by Ca2+, ATP, and ADP.
Cell Signaling by TCA Cycle Intermediates
Both succinate and 2-oxoglutarate (α-ketoglutarate) function as cellular signaling molecules through their ability to activate cell surface receptors of the GPCR family.
Succinate as Signaling Molecule
Mitochondrial succinate is transported to the cytosol via the concerted actions of the inner mitochondrial membrane dicarboxylate transporter encoded by the SLC25A10 gene and the outer mitochondrial membrane VDAC family transporters. Cytosolic succinate is transported out of the cell by the SLC16A1 encoded transporter.
Extracellular succinate is the ligand that activates the GPCR originally identified as GPR91 but is now referred to as the succinate receptor 1 and is encoded by the SUCNR1 gene. The succinate receptor is coupled to numerous different G-proteins including Gq, Gs, and Gi that allow it to activate a variety of divergent and convergent signal transduction pathways.
The SUCNR1-induced signaling pathways are involved in responses to local stress, immune responses, renin-induced hypertension, ischemia/reperfusion injury, platelet aggregation, and retinal angiogenesis. The SUCNR1-induced increase in blood pressure may contribute to diabetic nephropathy and cardiac hypertrophy.
Succinate Regulation of Gene Expression
Succinyl-CoA represents the substrate for lysine succinylation (Ksucc) of histone proteins as well as other non-histone proteins. Succinyl-CoA can be derived from several sources and pathways with the most prevalent being from the TCA cycle. Succinyl-CoA is also produced from propionyl-CoA which is an intermediate in the catabolism of the amino acids isoleucine, valine, methionine, and threonine, and from the catabolism of fatty acids with an odd number of carbon atoms, and from the peroxisomal oxidation of dicarboxylic acids. The predominant site of protein succinylation is within the mitochondria followed by the nucleus. However, there is ample evidence of cytoplasmic protein succinylation.
Succinyl-CoA is a sufficiently energetic compound that non-enzymatic succinylation can occur. Despite this, enzymatic succinylation has been described. The “writer” for enzymatic lysine succinylation has been shown to be the GCN5/PCAF family member protein GCN5 [also identified as KAT2A using the lysine (K) AcetlyTransferase nomenclature]. The nuclear OGDHc interacts with GCN5 allowing the succinyl-CoA that is generated to be directly accessible by the acetyltransferase.
De-succinylation of mitochondrial and nuclear succinylated proteins has been shown to be catalyzed by two members of the sirtuin family, SIRT5 and SIRT7. SIRT5 activity is the major mitochondrial de-succinylase but also functions within the nucleus. SIRT7 functions as a histone de-succinylase in the processes of DNA damage repair. SIRT7 is recruited to sites of DNA double-strand breaks (DSB) by polyADP-ribose polymerase 1 (PARP1) where it de-succinylates lysine 122 of histone H3 (H3K122). The de-succinylation of H3 promotes chromatin condensation and efficient DSB repair.
2-Oxoglutarate as Signaling Molecule
2-Oxoglutarate (α-ketoglutarate) is the ligand that activates the GPCR that was originally identified as GPR99/GPR80 but is now identified as the 2-oxoglutarate receptor 1 and is encoded by the OXGR1 gene. The OXGR1 encoded protein has also been shown to be the receptor for cysteinyl leukotrienes (peptidoleukotrienes), in particular leukotriene E4 (LTE4). Indeed, the affinity of OXGR1 for LTE4 is much higher than for 2-oxoglutarate.
Expression of the OXGR1 gene is highest in the kidneys and digestive system, but its expression in the heart and prostate gland is correlated with a potential for pathology. Activation of OXGR1 by binding 2-oxoglutarate leads to the regulation of a signal transduction pathway involving signal transducer and activator of transcription-3 (STAT-3). STAT-3 is an important regulator of numerous cellular processes, including the cell cycle, cell proliferation, apoptosis and cancer progression. Indeed, the persistent activation of STAT-3 is observed in numerous types of cancer, and a poor prognosis is associated with the level of phosphorylation STAT-3.
Within the heart, the activation of OXGR1 is associated with suppression of the role of STAT-3 in cardiac hypertrophy. This function of OXGR1 was demonstrated in mice in which the OXGR1 gene was knocked out which resulted in significant cardiac hypertrophy. Conversely, forced over-expression of OXGR1 protected cardiac myocytes from hypertrophy.
TCA Cycle Intermediates and Nuclear Function
Similar to the nuclear localization of the pyruvate dehydrogenase complex (PDHc), at least two enzymes and enzyme complexes of the TCA cycle are found in the nucleus. These include the 2-oxoglutarate dehydrogenase (α-ketoglutarate dehydrogenase) complex (OGDC), and fumarate hydratase (fumarase).
Other enzymes that are involved in the generation and/or utilization of TCA cycle intermediates within the nucleus include isocitrate dehydrogenase 3 (IDH3), isocitrate dehydrogenase 1 (IDH1), aconitase 2 (ACO2), the A (SDHA) and B (SDHB) subunits of the succinate dehydrogenase (SDH) complex, and several amino transferases such as glutamate-oxalate transaminase 1 (GOT1). GOT1 is the cytoplasmic version of the transaminase commonly called aspartate transaminase, AST.
With respect to the mitochondrially localized TCA cycle, glutamine and glutamate represent the main anaplerotic molecules feeding the cycle, predominantly through their conversion to 2-oxoglutarate (α-ketoglutarate) via the actions of glutaminase and glutamate dehydrogenase. Glutamine represents the major amino acid in the circulation as well as in several tissues. Nuclear glutamine has been shown to be the source for the generation of nuclear glutamate, succinate, fumarate, malate, citrate, and aspartate. Nuclear citrate is also a source for the synthesis of succinate. The TCA cycle metabolite, succinate, (as succinyl-CoA) is utilized for the succinylation of lysine (Ksucc) residues in histone proteins which represents a critical epigenetic modification.
TCA Cycle Enzyme Defects Associated with Cancer
At least three of the genes that encode TCA cycle enzymes (or homologues of TCA cycle enzymes), or protein subunits of enzyme complexes, have been shown to manifest mutations in certain types of familial cancer syndromes.
Mutations in two of the three isocitrate dehydrogenase (IDH) genes expressed in humans have been identified to harbor specific mutations and these mutations are correlated to the development of certain forms of cancer. The most common forms of cancer in which mutations in the IDH1 and IDH2 genes have been discovered are gliomas in the brain.
As indicated in the descriptions of the enzymes and enzyme complexes of the TCA cycle, the succinate dehydrogenase (SDH) complex is composed of four subunits encoded by four distinct genes, SDHA, SDHB, SDHC, and SDHD. Mutations in all four of these genes are associated with several familial cancer syndromes. In addition to the four functional components of the SDH complex, mutations in the SDH assembly factor gene, SDHAF2, are also found in certain forms of cancers. Although mutations in the SDH subunit assembly factor gene, SDHAF1, have not been associated with cancer syndromes, they are associated with mitochondrial deficiency syndromes. The SDH complex gene associated cancers include paragangliomas, pheochromocytomas, gastrointestinal stromal tumors (GIST), and an aggressive form of renal cancer identified as SDH-RCC (SDH-deficient renal cell carcinoma).
Cancer syndromes have also been associated with mutations in the fumarate hydratase (FH; also called fumarase) gene. The FH gene mutation associated cancers are cutaneous and uterine leiomyomas and an aggressive form of type 2 papillary renal cancer. The renal papillary carcinoma is characterized by a metabolic shift to aerobic glycolysis and glutamine-dependent reductive carboxylation.
Details of the TCA cycle gene mutations and their associations to cancer syndromes are covered in the Metabolic Alterations Associated with Cancer page and so will only be briefly reviewed in this page. It is important to note that succinate dehydrogenase is also complex II of the oxidative phosphorylation pathway and contributing to metabolic shifts seen in SDH-deficient cancers are deficiencies in mitochondrial energy generation via oxidative phosphorylation.
Isocitrate Dehydrogenase Mutations
As indicated earlier, both IDH1 and IDH2 enzymes utilize NADP+ as their cofactor and generate NADPH via oxidative decarboxylation of isocitrate outside the context of the TCA cycle. The primary function of IDH1 and IDH2 is to serve as producers of NADPH to enhance the cellular responses to oxidative stress and the generation of reactive oxygen species, ROS. The principal sources of NADPH are from the glucose-6-phosphate dehydrogenase (G6PD) reaction in the pentose phosphate pathway and via malic enzyme which is involved in the mobilization of mitochondrial acetyl-CoA to the cytosol. The significance of IDH1 and IDH2 produced NADPH can be made clear by pointing out that cells with low level IDH1 and IDH2 expression are more sensitive to oxidative stress than cells with higher levels of expression of these two enzymes.
Clinical significance is associated with the IDH1 and IDH2 genes as there has been a close association made between mutations in these two genes and the development of certain forms of cancer, specifically gliomas of the brain. All of the mutations in IDH1 that are associated with gliomagenesis are missense mutations that alter the normal Arg residue at position 132 to a histidine (R132H). The mutations in IDH2 are also single amino acid missense mutations at the comparable Arg residue at position 172 and are predominantly Lys substitutions (R172K). In some cases of IDH2 mutations another Arg residues is mutated to Glu (R140Q).
These mutations alter the activity of the encoded enzymes such that instead of converting isocitrate to 2-oxoglutarate (α-ketoglutarate), while also generating NADPH, the mutant IDH enzymes reduce 2-oxoglutarate (α-ketoglutarate) to 2-hydroxyglutarate (2-HG) while simultaneously oxidizing NADPH to NAPD+. Due to its effects as a competitive inhibitor of numerous 2-oxoglutarate and Fe2+-dependent dioxygenases, 2-HG is referred to as an oncometabolite.
Succinate Dehydrogenase Mutations
Mutations in the genes encoding the four proteins comprising the SDH complex result in a pathophysiological state defined as pseudo-hypoxia. This pathophysiologic state leads to a subsequent increase in angiogenesis, via activation of the HIF-1 hypoxia response pathway, resulting in the ability of tumors to acquire nutrients encouraging their aberrant growth. Activation of the HIF-1 pathway in SDH-deficient tumors is most likely the result of accumulating levels of succinate which is then transported into the cytosol via the action of the mitochondrial dicarboxylate transporter.
Accumulation of cytosolic succinate results the inhibition of the 2-oxoglutarate and Fe2+-dependent dioxygenases [prolyl hydroxylase domain (PDH) enzymes] responsible for the hydroxylation of the α-subunit (HIF-1α) of HIF-1. The hydroxylation of HIF-1α normally occurs continuously under normoxic conditions which stimulates ubiquitylation and proteasomal degradation of HIF-1α, thereby restricting the transcription factor function of HIF-1. The loss of HIF-1α hydroxylation results stabilization of the protein and consequent activation of the HIF-1 pathway in the absence of hypoxia. Due to its effects as a competitive inhibitor of numerous 2-oxoglutarate and Fe2+-dependent dioxygenases, succinate is referred to as an oncometabolite.
Fumarate Hydratase Mutations
Mutations in the fumarate hydratase (FH: also called fumarase) gene have been shown to be associated with hereditary leiomyomatosis and renal cell carcinoma (HLRCC). Leiomyoma is a form of smooth muscle tumor that can develop in any organ but is most commonly found in the uterus, esophagus, and small intestine. Uterine fibroids are a common form of leiomyoma of the uterine smooth muscle that are benign.
Similar to the alterations that occur in metabolic programs in SDH-deficient cancers, FH-deficient cells exhibit a substantial metabolic reorganization. As indicated in the Glycolysis and the Regulation of Blood Glucose page, as well as in the Metabolic Alterations Associated with Cancer page, and in the section above on SDH-deficient tumors, the common changes in metabolic profiles in cancer cells is, in part, the result of the activation of the hypoxia induced factor 1 (HIF-1) transcriptional program.
When fumarate accumulates in the mitochondria of FH-deficient cells the metabolite is transported to the cytosol by the dicarboxylate transporter. Within the cytosol the accumulating fumarate, similar to the effect of accumulating succinate, results in the inhibition of 2-oxoglutarate and Fe2+-dependent dioxygenases that are responsible for the hydroxylation of the α-subunit (HIF1α) of the HIF-1 complex. These effects provide a growth advantage to cells thereby promoting tumor cell growth and proliferation.
The Role of the TCA Cycle in Immune Modulation
The TCA cycle plays a central metabolic role in all cells, excluding red blood cells, and is of particular significance in cells of the immune system. Within activated macrophages there are distinct changes in TCA cycle activity that lead to the accumulation of TCA cycle intermediates, particularly fumarate and itaconate. These TCA cycle changes serve to regulate metabolic processes, immune system functions, cancer, and other diseases.
Fumarate Links Metabolism, Immune System Function, and Cancer
The major source of fumarate is the TCA cycle but in addition fumarate is generated in the context of the urea cycle, the biosynthesis of the purine nucleotides, and in the catabolism of the amino acids phenylalanine and tyrosine. Within the context of the urea cycle fumarate is generated by the actions of argininosuccinate lyase in the conversion of argininosuccinate to arginine. Within the context of purine nucleotide biosynthesis, fumarate is generated by the actions of adenylosuccinate lyase in the conversion of succinylaminoimidazole carboxamide ribotide (SAICAR) to 5-aminoimidazole-4-carboxamide ribotide (AICAR) and in the conversion of adenylosuccinate to AMP. Within the context of phenylalanine and tyrosine catabolism, fumarate is generated in the final reaction catalyzed by fumarylacetoacetate hydrolase which converts 4-fumarylacetoacetate to fumarate and acetoacetate.
As indicated in the Fumarate Hydratase Mutations section above, accumulation of fumarate in the cytosol leads to inhibition of 2-oxoglutarate and Fe2+-dependent dioxygenases and to the post-translational modification of numerous proteins. Fumarate is involved in these functions whether there are pre-existing mutations in the FH gene or not.
The ability of fumarate to non-enzymatically react with the sulfhydryl of cysteine residues in proteins, in a reaction referred to as “Michael addition”, generates a 2-(succino)cysteine (2SC) residue. This fumarate-mediated post-translational modification is referred to as succination.
The process of fumarate-mediated succination has been shown to occur on numerous proteins, resulting in modification of the activity of the modified proteins. The first protein shown to be modified by succination was glyceraldehyde-3-phosphate dehydrogenase (GAPDH). The succination of GAPDH leads to inhibition of its enzymatic activity. Although succination of GAPDH results in inhibition of its enzymatic activity, not all succination reactions result in inhibition.
Subsequent to the identification of GAPDH succination, numerous additional proteins, many being metabolic enzymes and anti-oxidant proteins, have been identified as undergoing succination. Other metabolic enzymes that are succinated include glucose-6-phosphate dehydrogenase and alcohol dehydrogenase. Anti-oxidant proteins that are succinated include KEAP1 (Kelch-like ECH-associated protein 1) and glutathione (GSH).
Numerous signaling proteins have been shown to be succinated including PTEN (phosphatase and tensin homolog). PTEN is a phosphoinositide phosphatase, specifically a phosphatidylinositol-3-phosphate phosphatase. Succination of PTEN inhibits its activity which results in increased signaling via the PI3K pathway.
Succinate Modulation of Immune Functions
As discussed in detail in the Metabolic Alterations Associated with Cancer page, and briefly above, increased levels of succinate in the cytosol result in the inhibition of the PHD enzymes that hydroxylate, and thereby destabilize, the HIF-1α subunit of the HIF-1 transcription factor complex. When HIF-1α is not hydroxylated, and is thus not targeted for degradation by ubiquitylation, it migrates to the nucleus leading to an increase in the transcription of HIF-1 target genes. In response to inflammatory processes, macrophages increase the rate of glycolysis and this change is, in part, due to the stabilization of HIF-1α as a consequence of succinate.
Succinate also enhances immune functions via the activation of the succinate receptor encoded by the SUCNR1 gene. Upon binding to its receptor, succinate initiates a signaling cascade that includes the activation of PLCβ. Activation of PLCβ results in the hydrolysis of membrane phosphatidylinositol-4,5-bisphosphate (PIP2; PtdIns-4,5-P2) generating inositol-1,4,5-trisphosphate (IP3; Ins-1,4,5-P3) diacylglycerol (DAG). The generation of IP3 results in release of endoplasmic reticulum (ER) stored Ca2+ and the Ca2+, along with DAG, leads to activation of PKC.
The activation of PKC results in phosphorylation of downstream kinases leading to activation of the MAPK signaling pathway. In the context of immune responses, the MAPK pathway results ultimately, in the enhanced transcription of pro-inflammatory genes. Activated PKC also leads to enhanced transcriptional activity of the critical immune modulatory transcription factor, NFκB.
Itaconate Synthesis and Immune System Modulation
One metabolite that accumulates in activated macrophages, as a consequence of alterations in the TCA cycle, is itaconate. Itaconate is a dicarboxylic acid that is produced from the TCA cycle intermediate, cis-aconitate, by the enzyme aconitate decarboxylase 1 which is encoded by the ACOD1 gene. The ACOD1 gene was originally identified as immunoresponsive gene 1 (IRG1). In addition to intracellular effects, itaconate is secreted from activated macrophages, particularly within the tumor microenvironment, where it exerts numerous additional effects. Itaconate is a critical metabolite that contributes to the negative feedback suppression of inflammatory cytokines.
The ACOD1 gene is located on chromosome 13q22.3 and is composed of 5 exons that encode a protein of 481 amino acids. Expression of the ACOD1 gene is near exclusive to macrophages and myeloid cells.
Itaconate produced by macrophages has antimicrobial activity which is crucial in the killing of phagocytosed bacteria. In vacuoles of macrophages where phagocytosed bacteria are killed, itaconate inhibits the bacterial enzyme, isocitrate lyase (ICL), a bacterial enzyme required for the glyoxylate shunt that functions during host infection by bacteria. The itaconate metabolite, itaconyl-CoA, inhibits bacterial methylmalonyl-CoA mutase (MCM) which in turn blocks propionyl-CoA–dependent bacterial growth. Itaconyl-CoA has also been shown to be a competitive inhibitor of mammalian methylmalonyl-CoA mutase and of the erythroid-specific form of δ-aminolevulinic (5-aminolevulinic) acid synthase encoded by the ALAS2 gene.
When macrophages are stimulated, for example by ligand interaction with the toll-like receptor 4 (TLR4), a signal transduction cascade is activated leading to enhanced expression of the ACOD1 gene. In addition to activation of ACOD1, the TLR4 signal transduction cascade results in enhanced production of interferon-alpha and -beta (INFα and INFβ). These interferons activate interferon receptors whose signal transduction cascades contribute to the activation of ACOD1 expression.
The increased levels of aconitate decarboxylase 1 result in diversion of cis-aconitate away from the TCA cycle via its metabolism to itaconate. Within the mitochondria itaconate inhibits the succinate dehydrogenase (SDH) complex which results in reduced flow of carbons through the TCA cycle as well as inhibition of oxidative phosphorylation since TCA cycle SDH is also complex II of the electron transport chain.
The inhibition of SDH reduces the generation of reactive oxygen species (ROS) at complex I. Decreased ROS production in the mitochondria allows for prolyl hydroxylase domain (PHD) enzymes to hydroxylate HIF1α. The prolyl hydroxylases that hydroxylate HIF α-subunits (HIF1α and HIF2α) are all 2-oxoglutarate (α-ketoglutarate) and Fe2+-dependent enzymes (2OG-oxygenases). Hydroxylation of HIF α-subunits renders the proteins susceptible to proteasomal degradation. The reduced levels of active HIF1α result in reduced transcription of genes encoding pro-inflammatory cytokines such as IL-1β. The net effect of itaconate at the level of SDH is anti-inflammatory.
Itaconate is transported to the cytosol where several enzymes of glucose metabolism are inhibited including aldolase A (encoded by the ALDOA gene), glyceraldehyde-3-phosphate dehydrogenase (encoded by the GAPDH gene), lactate dehydrogenase (encoded by the LDHA gene), enolase (encoded by the ENO1 gene), and the PKM2 isoform of pyruvate kinase (encoded by the PKM gene). The mechanism of itaconate-mediated inhibition of these enzymes is via alkylation of cysteine residues as well as by acylation of lysine residues. These forms of post-translational modification are collectively referred to as itaconylation (also referred to as itaconation).
Cytosolic itaconate has been shown to inhibit the NLRP3 inflammasome. NLRP3 refers to Nucleotide-binding oligomerization domain and Leucine-rich repeat-containing Receptor Protein 3. The NLRP3 inflammasome is a complex consists of a sensor (NLRP3 protein), an adaptor (apoptosis-associated speck-like protein, ASC), and an effector (caspase-1). Normally, one function of the NLRP3 inflammasome is to convert proinflammatory IL-1β (pro-IL-1β) to its active inflammatory state, IL-1β. The activity of the NLRP3 protein of the inflammasome is inhibited as a result of direct itaconation. Thus, inhibition of the NLRP3 inflammasome by itaconate represents an anti-inflammatory function of itaconate.
The NLRP3 inflammasome is also inhibited as a result of itaconate complexing with homocysteine forming homocysitaconate. Homocysteine is generated in the context of the methionine cycle which is the pathway by which methionine is utilized in the synthesis of S-adenosylmethionine (SAM or AdoMet) as well for diversion into cysteine biosynthesis.
During the process of protein synthesis, methionyl tRNA synthetase (MARS) catalyzes the conversion of homocysteine to homocysteine thiolactone. Homocysteine thiolactone is reactive towards lysine residues in a process termed N-homocysteinylation. The NLRP3 protein is one substrate for N-homocysteinylation, the consequences of which are enhanced inflammatory responses. The effect of homocysitaconate in this process is inhibition of the the MARS enzyme.
Thus, increased levels of itaconate repress inflammatory process mediated by the NLRP3 inflammasome by direct itaconation as well as through the inhibitory effects of homocystitaconate on the activity of the NLRP3 inflammasome.
Additional anti-inflammatory effects exerted by itaconate include activation of the basic region leucine zipper (bZIP) transcription factor identified as nuclear factor erythroid erythroid 2 (NEF2)-related factor 2 (NRF2). The human NRF2 protein is encoded by the NFE2L2 gene.
The transcriptional activity of NRF2 is regulated post-translationally by Kelch-like ECH-associated protein 1 (KEAP1) which mediates the degradation of NRF2. The activity of KEAP1 is inhibited by itaconylation. The itaconylation of KEAP1 allows NRF2 to activate genes whose encoded proteins serve antioxidant functions. The reduction in ROS production, as a result of NRF2 activity, contributes to the destabilization of HIF1α, furthering the anti-inflammatory activity of itaconate.
Two important targets of NRF2 are the HMOX1 (heme oxygenase 1) and NQO1 [NAD(P)H quinone oxidoreductase 1] genes. NRF2 also actively represses the transcription of pro-inflammatory genes in macrophages. Heme oxygenase 1 catalyzes the degradation of heme to free iron, biliverdin, and carbon monoxide (CO) thereby removing free heme which is pro-oxidant. The function of NQO1 is to catalyze the NADPH-dependent reduction of quinones to hydroquinones, thereby preventing the formation of highly reactive semiquinones and associated reactive oxygen species (ROS).
In addition to the effects of itaconate on macrophage functions, it is transported out of macrophages via the action of the ABCG2 encoded transporter. When exported from macrophages, itaconate limits the extent of anti-bacterial immunity.
Anti-Tumor Effects of Itaconate
When macrophage produced itaconate is transported to the tumor microenvironment, via the ABCG2 transporter, it is taken up by tumor cells. Within tumor cells, itaconate exerts anti-tumor effects predominantly as a result of the inhibition of the pentose phosphate pathway (PPP) enzyme glucose-6-phosphate dehydrogenase, G6PDH (encoded by the G6PD gene). Inhibition of G6PDH by itaconate occurs through direct binding of itaconate to the enzyme. As a consequence of the inhibition of G6PDH, tumor cells are less able to produce the NADPH necessary to handle the increased oxidative stress of the tumor microenvironment.
In addition, when produced by tumor cells, as opposed to macrophages or tumor infiltrating macrophages, itaconate enhances the presentation of tumor antigens which, in turn, enhances tumor cell recognition by T cells. In addition, the inhibition of G6PDH in tumor macrophages results in their conversion to anti-tumor macrophages.
Tumor Promoting Effects of Itaconate
Itaconate that is excreted from activated macrophages is transported into cells via the SLC13A3 encoded Na+-dicarboxylate cotransporter. In the context of tumor cells, the uptake of itaconate has been shown to result in the itaconylation of PD-L1 (programmed death ligand 1). The itaconylation of PD-L1 stabilizes the protein by preventing the protein from being ubiquitylated. Enhanced PD-L1 activity in tumor cells promotes immune evasion, thereby enhancing tumor cell proliferation.
Metabolism of Itaconate
Metabolism of itaconate generates acetyl-CoA and pyruvate. Succinyl coenzyme A synthetase converts itaconate to itaconyl-CoA. 3-Methylglutaconyl-CoA hydratase (encoded by the AUH gene) then converts itaconyl-CoA to citramalyl-CoA. Finally, citramalyl-CoA lyase (encoded by the CLYBL gene, also known as citrate lyase subunit beta-like ) converts citramlyl-CoA to acetyl-CoA and pyruvate.