
Last Updated: June 15, 2026
Introduction to Sphingosine and the Ceramides
The sphingolipids, like the phospholipids, are composed of a polar head group and two nonpolar tails. The core of sphingolipids is the long-chain amino alcohol, sphingosine. The sphingolipids include the sphingomyelins and glycosphingolipids (the cerebrosides, sulfatides, globosides, and gangliosides). Sphingomyelins are the only sphingolipid class that are also phospholipids. Sphingolipids are components of all membranes but are particularly abundant in the myelin sheath.
In addition to their roles in membrane organization and structure, numerous sphingolipids function as bioactive lipids exerting effects following binding to cell surface receptors. Ceramides are also critical regulators of mitochondrial homeostasis and contribute to the induction of apoptosis. Ceramides self-assemble in the outer mitochondrial membrane (OMM) forming channels. The formation of these ceramide channels enhances what is termed mitochondrial outer membrane permeabilization, MOMP. Associated with enhanced MOMP is the release of cytochrome c to the cytosol which is the trigger for mitochondria-induced apoptosis.
Synthesis of Sphingosine and Ceramides
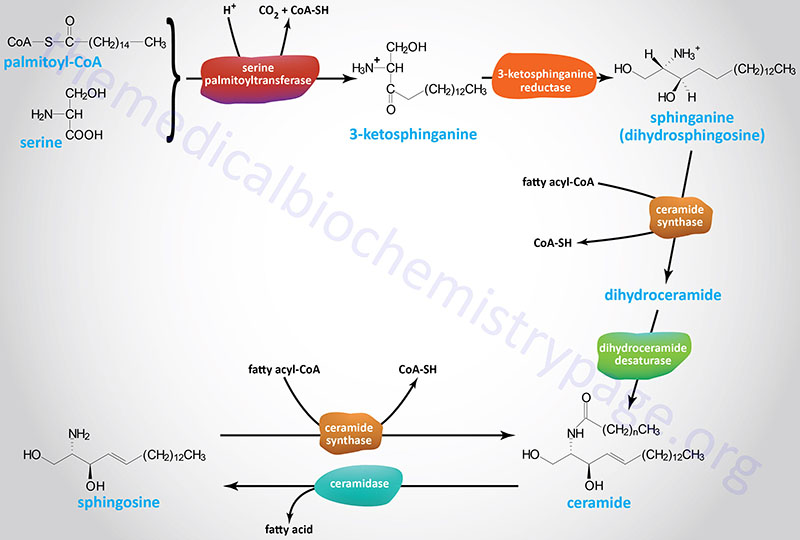
The initiation of the synthesis of the sphingoid bases (sphingosine, dihydrosphingosine, and the various ceramides) takes place via the condensation of palmitoyl-CoA and serine as shown in the Figure below. This initial reaction occurs on the cytoplasmic face of the endoplasmic reticulum (ER) and is catalyzed by the pyridoxal phosphate-dependent enzyme, serine palmitoyltransferase (SPT). The product of this reaction is 3-ketosphinganine (3-ketodihydrosphingosine). SPT is the rate-limiting enzyme of the sphingolipid biosynthesis pathway.
Serine Palmitoyltransferase Complex
Active SPT is a heterodimeric enzyme composed of two main catalytic subunits. The two main catalytic subunits of SPT are SPTLC1 and SPTLC2, or the SPTLC3 isoform which is also called SPTLC2L. The SPTLC1 subunit is present in all catalytically active SPT enzymes. Depending on the tissue in which the SPT complex is expressed there will be either the SPTLC2 or the SPTLC3 subunit in the complex. The LC in the enzyme designations refers to long-chain base subunit of SPT.
The SPTLC1 gene is located on chromosome 9q22.31 and is composed of 22 exons that generate five alternatively spliced mRNAs, each of which encode a distinct protein isoform .
The SPTLC2 gene is located on chromosome 14q24.3 and is composed of 13 exons that encode a 562 amino acid protein.
The SPTLC3 gene is located on chromosome 20p12.1 and is composed of 14 exons that generate two alternatively spliced mRNAs, both of which encode the same 552 amino acid protein.
An additional protein associates with the catalytic subunits to greatly enhance the activity of the enzyme complex as well as to confer acyl-CoA preference to the complex. There are two distinct activity enhancing subunits of SPT referred to as SPT small subunit A (SPTSSA) and SPT small subunit B (SPTSSB).
The SPTSSA gene is located on chromosome 14q13.1 and is composed of 2 exons that encode a protein of 71 amino acids.
The SPTSSB gene is located on chromosome 3q26.1 and is composed of 6 exons that generate two alternatively spliced mRNAs, both of which encode the same protein of 76 amino acids.
3-Ketosphinganine Reductase
Following formation of 3-ketosphinganine this compound is reduced to sphinganine (dihydrosphingosine) via the action of 3-ketosphinganine reductase (3KR; also known as 3-ketodihydrosphingosine reductase).
The 3-ketosphinganine reductase (3KR) enzyme is encoded by the KDSR gene. The KDSR gene is located on chromosome 18q21.33 and is composed of 10 exons that encode a precursor protein of 332 amino acids.
Ceramide Synthases
Following the formation of sphinganine, it is fatty N-acylated generating dihydroceramide. The N-acylation of sphinganine (also called dihydrosphingosine) occurs through the activities of six different ceramide synthases (CerS) in humans. These CerS enzymes introduce fatty acids of varying lengths [designated by the –(CH2)n– in the structure] and degrees of unsaturation. The products of the CerS enzymes are dihydroceramides. In other organisms the ceramide synthases are referred to as sphinganine N-acyl transferases. The actions of CerS and the role of ceramide in biological responses is covered in the section below on Ceramide Metabolism.
Dihydroceramide Desaturase
Dihydroceramide is then unsaturated in the original palmitic acid portion of the molecule by the enzyme dihydroceramide desaturase 1 (DES1). The official designation for DES1 is delta(4)-desaturase, sphingolipid 1 which is encoded by the DEGS1 gene. The DEGS1 gene belongs to the family of fatty acid desaturase encoding genes.
The DEGS1 gene is located on chromosome 1q42.11 and is composed of 5 exons that generate three alternatively spliced mRNAs encoding proteins of 323 amino acids (isoform 1), 288 amino acids (isoform 2), and 287 amino acids (isoform 3).
Humans express eight genes that encode enzymes of the fatty acid desaturase family that includes the DEGS1 (also identified as FADS7) and DEGS2 (also identified as FADS8) genes. The other genes in the family include FADS1, FADS2, FADS3, FADS6, SCD (also identified as FADS5), and SCD5 (also identified as FADS4) genes. The FADS1, FADS2, and SCD genes encode the desaturases, Δ5-eicosatrienoyl-CoA desaturase (D5D), the Δ6-oleoyl(linolenoyl)-CoA desaturase (D6D), and the Δ9-stearoyl-CoA desaturase (SCD), respectively.
The DEGS2 (delta(4)-desaturase, sphingolipid 2) encoded enzyme is a bifunctional enzyme that possesses both dihydroceramide desaturase and dihydroceramide hydroxylase (C4-hydroxylase) activities. The dihydroceramide hydroxylase activity converts dihydroceramide to phytoceramide. The DEGS2 encoded enzyme has important functions in epithelial and intestinal homeostasis. In the skin, phytoceramide plays an important role in barrier functions. Mutations in the DEGS2 gene are associated with susceptibility to colitis.
The FADS3 encoded enzyme functions as a ceramide desaturase in the synthesis of the ceramide derivatives of the sphingadiene (SD) class. Sphingadienes are long chain sphingoid bases that contain an additional carbon-carbon double bond in the long-chain acyl moiety when compared to sphingosine. Sphingosine is the most common mammalian sphingoid base. FADS3 has been shown to be required for the synthesis of 4,14-sphingadiene (specifically: 4E,14Z-sphingadiene). The sphingadienes constitute a class of lipid that has been shown to exert cytotoxic effects on certain types of cancers through their ability to inhibit the activity of the AKT/PKB signaling pathway.
The FADS6 and SCD5 (also identified as FADS4) genes encode enzymes that function similar to the FADS2 and SCD encoded enzymes, respectively.
Ceramidases
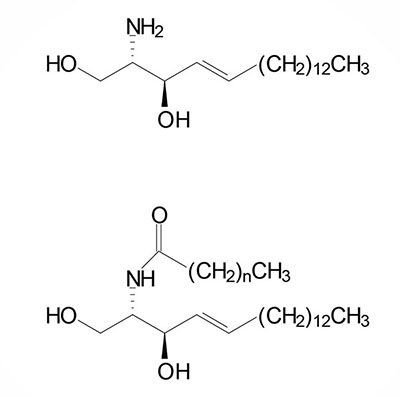
Sphingosine is derived from a ceramide via the action of ceramidases. The chemical designation for human sphingosine is d18:1 where the “d” refers to D-erythro reflecting the stereospecificity of the molecule and 18:1 designates oleic acid, an 18 carbon fatty acid with one site of unsaturation. The two carbons added to the initial palmitic acid generating the 18 carbon fatty acid of 3-ketosphinganine are derived from the amino acid serine. The destaturation of dihydroceramide to a ceramide generates the single site of unsaturation designated by the 18:1 nomenclature.
Ceramide Metabolism
Following the synthesis of a ceramide the molecule is transferred from the ER to the Golgi apparatus. It is within the membranes of the Golgi that ceramides serve as the precursors for the synthesis of the more complex sphingolipids that includes the sphingomyelins and the glycosphingolipids.
The transfer of ceramides from the ER to the Golgi is carried out by both vesicular and non-vesicular pathways. The ceramides that will be used for the synthesis of the glycosphingolipids are transferred to the Golgi while associated with the vesicle-associated lipid binding protein identified as four-phosphate adaptor protein, FAPP2. The FAPP2 protein is a pleckstrin homology domain-containing protein and as such is encoded by the PLEKHA8 (pleckstrin homology domain containing A8) gene.
Ceramides that will be utilized for sphingomyelin synthesis are transferred via a non-vesicular process that involves the protein identified as ceramide transport protein, CERT. The CERT protein is encoded by the CERT1 (ceramide transporter 1) gene.
Sphingosine can be released from ceramides via the action of ceramidases. Sphingosine can be re-converted to a ceramide by condensation with a fatty-acyl-CoA catalyzed by the various ceramide synthase (CerS) enzymes. There are at least five ceramidase genes in humans defined by their pH range of activity: acid, alkaline, and neutral.
Acid ceramidase (N-acylsphingosine amidohydrolase) is encoded by the ASAH1 gene. The ASAH1 gene is located on chromosome 8p22 and is composed of 17 exons that generate four alternatively spliced mRNAs, each of which encode a distinct protein isoform.
When studied in mice it has been shown that the ASAH1 gene is critical for early embryo survival and for removing ceramide from the embryo to prevent the default apoptosis pathway. Defects in the human ASAH1 gene result in the lysosomal storage disease (lysosomal storage disorder) identified as Farber lipogranulomatosis.
Neutral ceramidase (non-lysosomal ceramidase) is encoded by the ASAH2 gene and the enzyme is expressed in the apical membranes of the proximal and distal tubules of the kidney, endosome-like organelles in hepatocytes, and in the epithelial cells of the gut. The ASAH2 gene is located on chromosome 10q11.23 and is composed of 21 exons that generate two alternatively spliced mRNAs, both of which encode distinct protein isoforms. By studying the effects of ASAH2 knock-out mice it has been determined that neutral ceramidase is involved in the catabolism of dietary sphingolipids and the regulation of bioactive sphingolipid metabolites in the intestinal tract.
Humans express three alkaline ceramidases encoded by the ACER1 (alkaline ceramidase 1), ACER2, and ACER3 genes.
In 2011 it was reported that both adiponectin receptors, ADIPOR1 and ADIPOR2, posses ceramidase activity. Upon binding adiponectin the induced conformational change in the receptor enhances its ceramidase activity. This enzymatic activity of adiponectin receptors is likely to contribute to the cardioprotective and anti-diabetic effects of adiponectin.
Ceramide Synthases
The overall level of ceramides in a cell is a balance between the need for sphingosine and sphingosine derivatives, such as sphingosine-1-phosphate, ceramide-1-phosphate, and the sphingomyelins. With respect to the sphingomyelins, they serve a dual purpose of being important membrane phospholipids and as a reservoir for ceramides.
The conversion of both dihydrosphingosine (sphinganine) and sphingosine to ceramide is catalyzed by the ceramide synthases (CerS) that attach fatty acids of various lengths to the nitrogen of sphinganine, a process of N-acylation. As indicated above there are six CerS in humans identified as CerS1 through CerS6 encoded for by six different genes.
CerS genes were originally referred to as Lass genes (for Longevity Assurance) based on their homology to the yeast gene, longevity assurance gene-1 (LAG1). LAG1 was so-called because deletion of the gene in yeast prolonged their life-span. An additional related gene in yeast is referred to as LAC1 and when both genes are deleted yeast exhibit poor growth or die. A human gene, originally identified as UOG-1 (upstream of growth and differentiation factor-1), was shown to complement a LAG1 deletion in yeast and when overexpressed in mammalian cells resulted in increased ceramide synthesis. The ceramides synthesized by the enzyme contained exclusively stearic acid (C18:0 saturated fatty acid). Subsequent studies demonstrated that other human LAG homologs, originally identified as translocating chain-associating membrane (TRAM) protein homologs (TRH), synthesized ceramides with varying fatty acyl chain length. Each of these genes are now identified as CerS genes.
Each CerS exhibits fatty acyl chain length specificity as well as differential tissue distribution. CerS1 is structurally and functionally distinct from the other five CerS, all of which contain a homeobox-like domain.
CerS1 is specific for stearic acid (C18:0) and is expressed in brain, skeletal muscle, and testis. The CERS1 gene is located on chromosome 19p13.11 and is composed of 10 exons that generate ten different mRNAs due to alternative promoter usage and alternative splicing. These ten mRNAs collectively encode five distinct protein isoforms. One of the CERS1 mRNAs is a bicistronic transcript that encodes the CerS1 isoform 1 protein (350 amino acids) as well as the protein identified as growth and differentiation factor 1, GDF1. Mutations in the CERS1 gene are associated with a form of epilepsy termed epilepsy, progressive myoclonic 8 (EPM8).
CerS2 is specific for C20:0–C26:0 fatty acids and is expressed in the liver and kidney. The CERS2 gene is located on chromosome 1q21.3 and is composed of 13 exons that generate two alternatively spliced mRNAs, both of which encode the same 380 amino acid protein.
CerS3 is specific for C22:0–C26:0 fatty acids and is expressed in the skin and testis. The predominant ceramides produced via the actions of the CerS3 enzyme contain a C26:0 fatty acid. The CERS3 gene is located on chromosome 15q26.3 and is composed of 15 exons that generate five alternatively spliced mRNAs that collectively encode two distinct protein isoforms of 394 amino acids (isoform 1) and 383 amino acids (isoform 2).
CerS4 is specific for C18:0 and C20:0 fatty acids and is ubiquitously expressed but with highest levels in liver, heart, skin, and leukocytes. The CERS4 gene is located on chromosome 19p13.2 and is composed of 17 exons that encode a 394 amino acid protein. CERS4 was originally identified as TRH1 [translocating chain-associating membrane (TRAM) protein homolog 1].
CerS5 is specific for palmitic acid (C16:0) and is ubiquitously expressed at low levels. The CERS5 gene is located on chromosome 12q13.12 and is composed of 15 exons that generate seven alternatively spliced mRNAs, each of which encode a distinct protein isoform. CERS5 was originally identified as TRH4 [translocating chain-associating membrane (TRAM) protein homolog 4]. Expression levels of CERS5, particularly within the intestines, correlates with accelerated tumor growth in the context of colorectal carcinoma.
CerS6 is specific for myristic (C14:0) and palmitic acid (C16:0) and is expressed at low levels in all tissues. The CERS6 gene is located on chromosome 2q24.3 and is composed of 13 exons that generate two alternatively spliced mRNAs, each of which encode distinct protein isoforms.
The biological significance of ceramide synthesis and the activity of the CerS is demonstrated by studies in several different types of human cancers. In this regard, CerS1 appears to be most significant. Head and neck squamous cell carcinomas (HNSCC) exhibit a downregulation of C18-ceramide levels when compared to adjacent normal tissue. In addition, a balance between the levels of C16- and C18-ceramides is associated with the state of clinical progression of HNSCC.
In the chemotherapy of certain cancers, CerS1 activity may also play a role. Enhanced expression of CerS1 has been shown to sensitize cells to a variety of chemotherapeutic drugs such as cisplatin, vincristine, and doxorubicin. In HNSCC CerS1 is involved in regulating apoptotic cell death via doxorubicin-induced caspase activation. When myeloid leukemia cells are treated with Gleevec® (imatinib) there is an increased production of C18-ceramide which is involved in the induction of apoptosis.
The proposed mechanism for ceramide involvement in apoptotic processes involves the activation of the aspartate protease cathepsin D. Cathepsin D is associated with membranes and when activated by ceramides is released to the cytosol where it triggers the mitochondrial apoptosis pathway. Further evidence for the role of ceramides in negative growth responses is seen in cell cultures to which ceramide analogues are added. These types of assays demonstrate that ceramides induce oxidative stress, growth arrest, and apoptosis and/or necrosis.
Studies have shown that high levels of CERS3 expression are associated with enhanced tumor cell proliferation, particularly colorectal cancers. Gut microbiota have been shown to be involved in the suppression of colorectal cancer progression via the production of riboflavin. Specifically it is Bacteroides cellulosilyticus of the Bacteroidetes phylum that has been shown to produce the riboflavin. The effect of the bacterial riboflavin is the inhibition of the ceramide designated d18:1/26:0.
When derived from the sphingomyelins, ceramides are the products of the action of acid sphingomyelinase (ASMase). The importance of sphingomyelin as a source of ceramide can be evidenced by the fact that the activation of the ASMase pathway is a shared response to the effects of cytokines, stress, radiation, chemotherapeutic drugs, and pathogenic and cytotoxic agents. Several receptor- and non-receptor-mediated pathways, activated in response to stress, such as those involving death ligands like TNFα and TNF-related apoptosis inducing ligands (TRAIL), are coupled to the activation of ASMase activity. The induction of ASMase, in response to apoptotic triggers, results in increased production of ceramides which then can initiate aspects of the pathways of apoptosis.
Mitochondrial Ceramide Synthesis
The significance of mitochondrial ceramides is that evidence has demonstrated that ceramides in the inner mitochondrial membrane (IMM) containing C16 fatty acids influence the processes of electron transport and oxidative phosphorylation. Mitochondrial ceramides can be synthesized from sphinganine (dihydrosphingosine) and from sphingomyelins or they can be transported into the mitochondria from the endoplasmic reticulum (ER) by an as yet unidentified transporter.
Within the mitochondria sphinganine and acyl-CoAs are converted to various dihydroceramides through the action of ceramide synthases (CerS1, CerS2, CerS4, or CerS6). As for the conversion of dihydroceramides to ceramides in the ER, mitochondrial dihydroceramides are converted to ceramides via the action of the DEGS1 encoded dihydroceramide desaturase.
Sphingomyelins are also transported into the mitochondria by an as yet unidentified transporter where they are substrates for a neutral sphingomyelinase converting them to ceramides. The mitochondria-localized neutral sphingomyelinase is likely encoded by the SMPD5 gene which has been designated a pseudogene in humans but, as discussed in the Sphingomyelinases section below, does contain an open reading frame that possesses a mitochondrial localization sequence.
The most significant mitochondrial ceramides are C16-ceramides because they have been found to be associated with decreased electron transport chain (ETC) activity, increased mitochondrial fission, and increased mitochondrial outer membrane permeabilization (MOMP), the latter contributing to release of cytochrome c and activation of mitochondrial apoptosis.
2-Hydroxy Fatty Acid Containing Sphingolipids
Sphingolipids that contain a 2-hydroxylated fatty acid (hFA or 2hFA) represent a special class of sphingolipids that are particularly important for neuronal functions in the brain. The presence of hFA in ceramides was originally identified in galactosylceramides isolated from brain. Galactosylceramides are important constituents of the myelin sheath. Subsequent to the initial identification of hFA in galactosylceramides, this type of sphingolipid was identified in various other tissues.
The 2-hydroxylation of fatty acids, required for their incorporation into the sphingosine base and the N-acyl chain of ceramides requires the action of the fatty acid 2-hydroxylase enzyme encoded by the FA2H gene.
The FA2H gene is located on chromosome 16q23.1 and is composed of 8 exons that encode a 372 amino acid protein. The FA2H encoded enzyme is an NADPH-dependent monooxygenase that is localized to the membranes of the endoplasmic reticulum (ER) with the catalytic domain facing into the cytosol. The highest levels of expression of the FA2H gene are in the brain and the stomach.
Following the 2-hydroxylation reaction, the hFA are activated by the same family of fatty acyl-CoA synthetases involved in ceramide synthesis as outlined in the Figure above. The CoA activated hFA is then used as a substrate for the ceramide synthases (CerS) for the N-acylation of sphinganine (dihydrosphingosine) generating a dihydroceramide. The hFA containing dihydroceramides are then substrates of dihydroceramide desaturase which generates the hFA containing ceramides.

The hFA containing ceramides are the substrates for all of the various hFA containing sphingolipids. The actions of sphingomyelin synthases generate the various hFA containing sphingomyelins. The hFA containing ceramides are also substrates for the synthesis of hFA containing galactosylceramides, hFA containing sulfatides, as well as hFA containing glycosphingolipids.
The hFA found in sphingolipids include, but is not limited to, palmitic acid, (C16:0), stearic acid (C18:0), docosanoic acid (C22:0; common name behenic acid), tetracosanoic acid (C24:0; common name lignoceric acid), and nervonic acid [C22:1; an omega-9 monounsaturated fatty acid (MUFA)]. The most common hFA found in hFA-galactosylceramides is 2-hydroxytetracosanoic acid. The most common hFA in hFA-GM3 gangliosides is 2-hydroxydocosanoic acid.
Likely the most clinically significant hFA containing sphingolipids are the hFA containing galactosylceramides. This is demonstrated by the fact that mutations in the FA2H gene are associated with spastic paraparesis and leukodystrophy. The form of spastic paraparesis caused by FA2H mutations is identified as spastic paraplegia 35 (SPG35).
As described in the Cerebrosides section below, the attachment of galactose to ceramides, including hFA-ceramides, is catalyzed by the UGT8 encoded glycosyltransferase. Sulfation of a hFA-galactocerebroside by cerebroside sulfotransferase encoded by the GAL3ST1 (galactose-3-O-sulfotransferase 1) gene will generate a hFA-sulfatide.
The hFA containing sphingolipids are most likely involved in the structure and function of membrane microdomains whose functions are in the trafficking of membrane constituents such as proteins and other lipids. Within the brain, hFA-galactosylceramides and hFA-sulfatides are the predominant hFA containing sphingolipids and these are components of the myelin sheaths. In addition to the brain the skin is also rich in hFA sphingolipids where they are likely involved in the development and maintenance of the epidermal permeability barrier.
ω-Hydroxy Fatty Acid Containing Sphingolipids
A specialized type of sphingolipid molecule are found in the epidermis where they participate in the formation of the barrier function of the skin. These sphingolipids contain ω-hydroxy fatty acids generated from a unique class of fatty acids that contain more than 28 carbon atoms. This class of class of fatty acid is referred to as the ultra-long-chain fatty acids (ULCFA).
Human fatty acid ω-hydroxylases are all members of the cytochrome P450 (CYP) family of enzymes. These enzymes are abundant in the liver and kidneys. Specifically, the human ω-hydroxylase enzymes are members of the CYP4A and CYP4F families that preferentially ω-hydroxylate the terminal methyl group of C10–C26 length fatty acids. The CYP4F22 encoded enzyme is one that is known to ω-hydroxylate ULCFA.
CYP4A11 is the human homolog of the rat liver CYP4A1 gene whose encoded enzyme was the first ω-hydroxylase characterized. CYP4A11 utilizes NADPH and O2 to introduce an alcohol to the ω-CH3– of several fatty acids including lauric (12:0), myristic (14:0), palmitic (16:0), oleic (18:1), and arachidonic acid (20:4).
Several ω-hydroxy fatty acids are substrates for the synthesis of a specialized class of sphingolipids identified as the ω-hydroxyceramides and the ω-O-acylceramides. The ω-O-acylceramides are the specialized sphingolipids that are particularly important to the skin barrier formation.
The synthesis of the ω-O-acylceramides involves several steps that first begins with a very long-chain fatty acyl-CoA. The initial steps all take place in microsomal membranes such as the endoplasmic reticulum (ER) membrane. A VLCFA-CoA is first elongated by an enzyme of the 3-keto acyl-CoA synthase family. 3-keto acyl-CoA synthase encoding genes. Humans express seven distinct 3-keto acyl-CoA synthase encoding genes. These seven genes are designated ELOVL1–ELOVL7 which stands for ELOngation of Very Long fatty acids.
ELOVL4 elongates a VLCFA-CoA to a ULCFA-CoA which then has the CoA is removed by a thioesterase. The ULCFA is a substrate for the CYP4F22 encoded ω-hydroxylase which introduces the ω-hydroxyl group. The ω-hydroxy ULCFA is then re-esterified to CoA by the SLC27A4 encoded acyl-CoA synthetase. The SLC27A4 encoded enzyme is more commonly referred to as FATP4 (fatty acid transport protein 4). Sphinganine is then added to the ω-hydroxy ULCFA-CoA via the CerS3 ceramide synthase. This molecule is then a substrate for dihydroceramide reductase which yields a ω-hydroxy ULCFA containing ceramide. Linoleic acid is then esterified to the ω-hydroxyl by the PNPLA1 encoded enzyme of the PLA2 family of enzymes generating an ω-O-acylceramide.
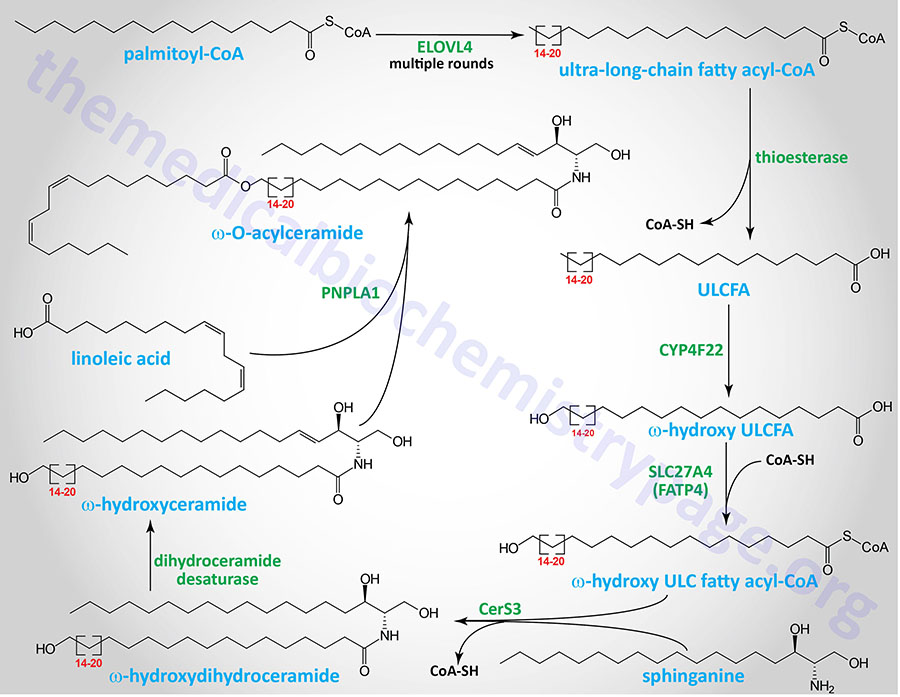
The ω-O-acylceramide is then transferred to the Golgi complex where carbohydrates are added. The UGCG encoded glucosylceramide synthase adds a molecule of glucose from UDP-glucose, similar to the process of glucosylceramide (glucocerebroside) synthesis described below. The product of this UGCG catalyzed reaction is termed an ω-O-acyl-glucocerebroside.
The ω-O-acyl-glucocerebrosides are then transferred to secretory vesicles via the action of the ABC family transporter, ABCA12. When transferred to the extracellular space of the stratum corneum (the outermost layer of the skin), the ω-O-acyl-glucocerebrosides are substrates for a series of reactions that generate an epoxy-enone in the linoleic acid portion of the molecule followed by deglucosylation. These reaction are carried out by the ALOX12B encoded arachidonate 12-lipoxygenase, 12R type (commonly designated 12R-LOX) which generates a 9-hydroperoxide in the linoleic acid. The ALOXE3 encoded arachidonate lipoxygenase 3 (commonly designated E-LOX) then converts the 9-hydroperoxide to an epoxy-alcohol. The epoxy-alcohol is converted to an epoxy-enone by the SDR9C7 encoded dehydrogenase. SDR9C7 is a member of the short-chain dehydrogenase/reductase (SDR) family of dehydrogenases. Either before the epoxy-enone formation or after, the glucose is removed by the action of the GBA1 encoded acid β-glucosidase (commonly called glucocerebrosidase). The linkage of the resultant ω-O-acylceramide to corneocyte surface proteins occurs at some point in the described synthesis pathway.
The clinical significance of the synthesis of ω-O-acylceramides can be evidenced by the disorders that result from mutations in several of the genes in this synthesis process. Mutations in the SDR9C7 gene result in a disorder termed autosomal recessive congenital ichthyosis 13, ARCI13. The ARCI disorders are associated with dry, scaly skin due to a defective skin barrier. Mutations in the ALOXE3 gene are the causes of nonbullous congenital ichthyosiform erythroderma (NBCIE) which is a form of ARCI (specifically ARCI3). Mutations in the ALOX12B gene also cause a form of NBCIE (also identified as ARCI2) with the mildest form being referred to as self-healing collodion baby (SICI). Complete loss of function of the ABCA12 encoded transporter is the cause of a form of ARCI (ARCI4) referred to as Harlequin ichthyosis.
Mutations in several other genes whose encoded enzymes are involved in the synthesis of complex ceramides are also associated ARCI syndromes. Mutations in the CERS3 gene are associated with ARCI9. Mutations in the PNPLA1 gene are associated with ARCI10.
Ceramides in Cardiovascular Disease Risk
Numerous lines of evidence have demonstrated a linkage between the levels of various ceramides in the blood and the potential for the development of atherosclerosis and associated cardiovascular disease. In particular, elevated levels of C16:0, C18:0, and C24:1 ceramides have been linked to an increased risk for cardiovascular disease with the level of C18:0 being the most significantly associated.
Role of Sphingomyelinases in Cardiovascular Disease
The activities of both the SMPD1 (acid sphingomyelinase) and SMPD3 (neutral sphingomyelinase) encoded genes have been shown to be associated with the development of atherosclerotic lesions. This was demonstrated in mice where knock-out of either the SMPD1 or SMPD3 genes resulted in a reduction of atherosclerotic plaque formation in atherosclerosis-prone mice. In addition, the tricyclic antidepressant, amitriptyline, which inhibits the activity of the SMPD1 encoded enzyme, is associated with reduction of atherosclerotic plaque formation in these same atherosclerosis-prone mice.
Ceramides and Insulin Resistance
Numerous lines of evidence have shown that various inducers of cellular stress such as inflammatory activation, excess saturated fatty acid intake, and chemotherapeutics, result in increased rates of ceramide synthesis. In addition, there is ample evidence demonstrating that the accumulation of cellular ceramides is associated with the pathogenesis of diseases such as obesity, diabetes, atherosclerosis, and cardiomyopathy.
For example, studies in mice have correlated endogenous ceramides and glucosylceramides with the antagonism of insulin-stimulated glucose uptake and synthesis. In animal models of obesity, evidence shows that genetic or pharmacological inhibition of ceramide or glucosylceramide biosynthesis leads to increased peripheral insulin sensitivity while at the same time reducing the severity of pathologies associated with insulin resistance including diabetes, atherosclerosis, hepatic steatosis, and/or cardiomyopathy. With respect to overall lipid homeostasis and the role of adipose tissue in disease pathology, studies have revealed roles for the adipokines leptin, adiponectin, and TNFα in the modulation of cellular ceramide levels.
An enhanced systemic inflammatory status as well as cellular stress have both been associated with insulin resistance. With respect to biological lipids, excess lipid intake, especially saturated fatty acids, leads to mitochondrial and endoplasmic reticulum (ER) stress. Increased fat oxidation in mitochondria leads to the production of reactive oxygen species (ROS) which are known to result in insulin resistance. Both mitochondrial and ER stress can result in apoptosis.
Excess fatty acid intake also interferes with normal insulin receptor-mediated signal transduction resulting in insulin resistance. Excess saturated fatty acid intake, particularly palmitic acid, results in increased ceramide synthesis which has been shown to be both a cause and effector of pancreatic β-cell stress resulting in impaired insulin secretion. For more information on the role of fats and mitochondrial stress in insulin resistance visit the Insulin Function, Insulin Resistance, and Food Intake Control of Secretion page. Obesity, which results in insulin resistance and development of type 2 diabetes, has long been associated with low-grade systemic inflammation. The correlation between obesity, ceramide synthesis, and insulin resistance is discussed below.
The ability of ceramides to interfere with insulin receptor signaling is the result of blocking the receptors ability to activate the downstream effector kinase, PKB/AKT. PKB (protein kinase B)/AKT (AK strain transforming) was originally identified as the tumor inducing gene in the AKT8 retrovirus found in the AKR strain of mice. Humans express three genes in the AKT family identified as AKT1 (PKBα), AKT2 (PKBβ), and AKT3 (PKBγ).
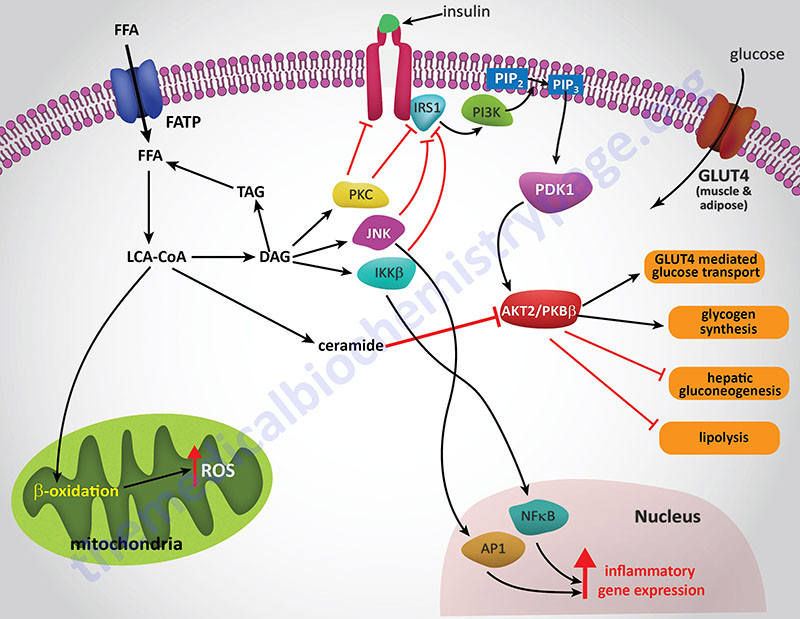
Experiments in cell culture, involving both adipocytes and skeletal muscle cells, have shown that ceramides inhibit insulin-stimulated glucose uptake by blocking translocation of GLUT4 to the plasma membrane as well as by interfering with glycogen synthesis. The blockade of PKB/AKT activation is central to the effects of ceramides and can be demonstrated by constitutive overexpression of the kinase which negates the effects of ceramides. This PKB/AKT-blocking action of ceramides has been shown in all cell types tested.
Several lines of evidence have solidified the model of ceramides leading to insulin resistance as a consequence of blockade of PKB/AKT activation. Administration of ceramide to cells in culture blocks the translocation of PKB/AKT to the plasma membrane. This inhibition of translocation is the result of the phosphorylation of a regulatory site in the plekstrin homology (PH domain) of PKB/AKT. The phosphorylation leads to reduced affinity of the kinase for phosphoinositides. The kinase responsible for the ceramide-induced phosphorylation of PKB/AKT is likely to be the atypical PKC isoform PKCζ since this kinase is activated by ceramides in vitro.
Additional evidence pointing to a link between ceramides and activation of PKCζ is that mutation of a target serine (S34) in the kinase, to alanine, confers resistance to ceramide action. Also, ceramide addition has been shown to stabilize interactions between PKB/AKT and PKCζ via their recruitment to plasma membrane rafts or caveolae.
Another mechanism by which ceramides impact the activity of PKB/AKT is by activating protein phosphatase 2A (PP2A) to dephosphorylate the kinase. Experiments that were designed to specifically inhibit PP2A were shown to prevent the effects of ceramide on PKB/AKT in a number of different cell types. In some cell types, both mechanisms are functional, while in other cell culture systems either PKCζ or PP2A is the central mediator of ceramide effects.
Palmitic acid (C16:0) is the most abundant saturated fatty aid in the circulation. The role of saturated fatty acids in increased levels of ceramides has been demonstrated by adding palmitate to cultured muscle cells. In this system the addition of palmitate results in increased ceramide accumulation while simultaneously inhibiting PKB/AKT. Ceramide synthesis was indeed required for the effect of palmitate addition on the activity of PKB/AKT since pharmacological inhibition of ceramide synthesis or siRNA-mediated knockdown of several enzymes required for ceramide biosynthesis (serine palmitoyltransferase, ceramide synthases, or dihydroceramide desaturase) completely blocks the effects of palmitate on insulin signaling.
An alternative means to examine the effects of ceramides on insulin sensitivity is to block the pathways of ceramide metabolism. Treatment of cells with acid ceramidase inhibitors results in increased endogenous ceramide levels while simultaneously blocking insulin-mediated activation of PKB/AKT. Under conditions of ceramidase inhibition there is an exaggerated effect of palmitic acid addition on insulin resistance. Conversely, if one overexpresses acid ceramidase, the inhibition of insulin signaling induced by palmitate addition is completely blocked.
The cellular effects of glucosylceramide (glucocerebroside), although similar to ceramides themselves, does exhibit cell-type specificity. Glucosylceramide is the precursor for synthesis of the complex glycosphingolipid family of gangliosides, for example the GM3 gangliosides. Adipocytes are highly sensitive to the inhibitory effects of glycosylated sphingolipids on insulin actions, whereas muscle cells are unaffected. Addition of GM3 ganglioside to adipocytes inhibits insulin activation of IRS-1.
In addition, TNFα treatment induces GM3 accumulation in membrane lipid rafts allowing for association with the insulin receptor through caveolin-1 present in the rafts. The antagonistic effects of TNFα can be prevented by first depleting cells of glycosylated ceramides.
Obesity is associated with adipose tissue enrichment in the complex gangliosides, GM2, GM1, and GD1a. The significance of the accumulation of these gangliosides has been demonstrated in mice lacking GM3 synthase which generates the major ganglioside precursor. GM3 synthase is encoded by the ST3GAL5 (ST3 beta-galactoside alpha-2,3-sialyltransferase 5) gene. These mice are protected from insulin resistance and glucose intolerance when fed a high-fat diet.
Although knock-out of the ST3GAL5 gene in mice protects these mice from insulin resistance under conditions of a high-fat diet, the loss of this gene in humans results in a severe infantile disorder. Mutations in the human ST3GAL5 gene are associated with infantile onset of severe irritability, feeding difficulties, hypotonia, failure to grow, microcephaly, sensorineural hearing impairment, and poor visual function. The disorder associated with ST3GAL5 mutations is also known as Amish infantile epilepsy syndrome and as ST3GAL5-CDG (congenital disorder of glycosylation).
Ceramides can also be glucosylated by the enzyme, UDP-glucose:N-acylsphingosine D-glucosyltransferase which is encoded by the UGCG gene. This enzyme is commonly called glucosylceramide synthase, GCS. Attachment of glucose to ceramide forms glucosylceramides (glucocerebrosides; also termed glucosylcerebrosides) which then serve as the building blocks of complex glycosphingolipids.
Treatment of genetically obese or diet-induced obese mice with highly specific glucosylceramide synthase (UDP-glucose ceramide glucosyltransferase, UGCG) inhibitors results in improved glucose tolerance and increased insulin sensitivity in muscle and liver. Collectively, these studies strongly implicate a role for glycosylated ceramides in increased adipose tissue inflammation, peripheral insulin resistance, and hepatic steatosis.
The most potent reagent used to study the effects of the manipulation of enzymes involved in sphingolipid biosynthesis is the compound myriocin [2-amino-3,4-dihydroxy-2-(hydroxymethyl)-14-oxoicos-6-enoic acid]. Myriocin is a highly specific inhibitor of serine palmitoyltransferase (SPT), which is the first and rate-limiting enzyme in the de novo pathway of ceramide synthesis (see the Figure above showing sphingosine and ceramide synthesis). Myriocin (also known as antibiotic ISP-1 and thermozymocidin) was isolated from thermophilic fungi such as Mycelia sterilia and Isaria sinclairii. Extracts from these fungi have been used in traditional Chinese medicine as a treatment for numerous conditions including diabetes. Myriocin can be administered chronically to rodents and it appears to be well tolerated. Addition of myriocin to animals, that are models of obesity, prevents insulin resistance and the development of diabetes, atherosclerosis, and cardiomyopathy. In addition, myriocin improved glucose tolerance, insulin sensitivity, and ameliorates hypertension when administered to rodents.
Genetic manipulation of several enzymes in ceramide metabolism has also been shown to insulin sensitizing. In mice heterozygous for the SPT subunit SPTLC2 there is a reduction in peripheral ceramide levels and improved insulin sensitivity when these animals are fed a high-fat diet. Similar results are seen in mice heterozygous for dihydroceramide desaturase 1 (DES1). Both SPT and DES1 are required for ceramide biosynthesis. As described above, a large family of ceramide synthases (CerS) have been identified in mammals. CerS1 is the most abundant isoform expressed in skeletal muscle and is involved primarily in the synthesis of C18:0 ceramides. The level of expression of CerS1 was shown to be significantly elevated in mice fed a high-fat diet. This increase in CerS1 expression was associated with alterations in ceramide levels and reduced glucose tolerance.
Collectively these data demonstrate a complex interrelationship between sphingosine and ceramide metabolism and insulin resistance. As pointed out ceramides, can be deacylated by ceramidases to form sphingosine. As discussed below, sphingosine can be phosphorylated to sphingosine-1-phosphate (S1P) which is an important biologically active lipid.
As pointed out earlier, obesity is associated with a low-grade systemic inflammatory state. One of the mechanisms involved in this inflammatory status is the activation of toll-like receptors (TLRs). TLR activation leads to enhanced transcription of pro-inflammatory cytokines such as TNFα and interleukin-6 (IL6). Saturated fatty acids are known to activate TLR4 and this activation is requisite for lipid induction of TNFα and other cytokines. When TLRs are knocked-out in mice the animals are protected from lipid-induced insulin resistance. The signal transduction cascade initiated by TLR activation involves the downstream effectors IKKβ (inhibitor of nuclear factor kappa-B kinase subunit beta) and NFκB (nuclear factor kappa-B). TLR4 activation has been shown to selectively and strongly increase the levels of sphingolipids within cells. Several studies have shown that ceramide is indeed an obligate intermediate linking TLR4 activation to the induction of insulin resistance.
Ceramides can also act as substrates for the sphingomyelin synthases yielding sphingomyelins or they can be phosphorylated (by ceramide kinase) to yield ceramide-1-phosphate. Thus, it is clear that multiple products of the actions of SPT, CerS, and DES1 could all potentially contribute to the development of insulin resistance and diabetes.
Sphingomyelin Synthesis and Metabolism
Sphingomyelins are sphingolipids that are also phospholipids. Sphingomyelins are important structural lipid components of nerve cell membranes. The predominant sphingomyelins contain palmitic or stearic acid N-acylated at carbon 2 of sphingosine.
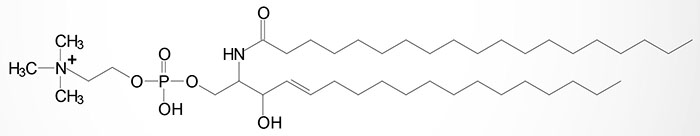
Sphingomyelin Synthases
The sphingomyelins are synthesized by the transfer of phosphorylcholine from phosphatidylcholine to a ceramide in a reaction catalyzed by sphingomyelin synthases (SMS). There are two SMS genes in humans identified as SMS1 and SMS2. The SMS1 enzyme is found in the trans-Golgi apparatus while the SMS2 enzyme is predominantly associated with the plasma membrane.
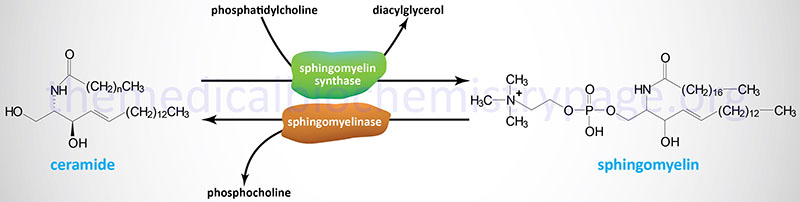
The SMS1 enzyme is encoded by the SGMS1 gene. The SGMS1 gene is located on chromosome 10q11.23 and is composed of 20 exons that encode a 413 amino acid protein with five transmembrane-spanning domains. Expression of the SGMS1 gene predominates in the brain.
The SMS2 enzyme is encoded by the SGMS2 gene. The SGMS2 gene is located on chromosome 4q25 and is composed of 11 exons that generate nine alternatively spliced mRNAs, seven of which encode the same 365 amino acid protein.
In addition to the phosphocholine head group of phosphatidylcholines (PC), that is transferred to a ceramide to generate the sphingomyelins by the SMS1 and SMS2 enzymes, the phosphoethanolamine head group of phosphatidylethanolamines (PE) can also be transferred to ceramides. The transfer of phosphoethanolamine from PE is catalyzed by the enzyme encoded by the SAMD8 (sterile alpha motif domain containing 8) gene. The SAMD8 encoded enzyme is localized to the endoplasmic reticulum (ER) and is commonly referred to as ceramide phosphoethanolamine synthase since the products of the SAMD8 catalyzed reaction are referred to as ceramide phosphoethanolamines (CPE). Although CPE are abundant sphingolipids in invertebrates, they are present only in trace amounts in human tissues.
Sphingomyelinases
As shown in the Figure above, sphingomyelins are substrates for the action of sphingomyelinases resulting in release of ceramides and phosphocholine. This reaction represents a significant pathway for the synthesis of ceramides. Sphingomyelinases are classified based upon their requirement for Mg2+ as well as pH optimum such that there are acidic, alkaline, and neutral sphingomyelinases. These enzymes belong to a family of enzymes that are identified as sphingomyelin phosphodiesterases.
The sphingomyelinase that functions at acidic pH is commonly referred to as acid sphingomyelinase (ASMase or aSMase). The human ASMase is encoded by the sphingomyelin phosphodiesterase-1 (SMPD1) gene. The SMPD1 gene is located on chromosome 11p15.4 and is composed of 6 exons that generate five alternatively spliced mRNAs, each of which encode a distinct protein isoform. The major protein isoform (isoform 1) is a 631 amino acid preproprotein. Following processing, and due to differential modification, there are two forms of aSMase, one is extracellular and one is localized to the endoplasmic reticulum and lysosomal membrane compartment.
Defects in the SMPD1 gene result in the lysosomal storage disease known as Niemann-Pick disease. There are in fact two major forms of Niemann-Pick (NP) disease. NP disease caused by acid sphingomyelinase deficiencies comprises types A and B, referred to as NPA and NPB. The other form of NP disease comprises types C1 and C2, the former due to defects in the NPC1 gene and the latter due to defects in a gene identified as NPC2.
Humans express four genes in the sphingomyelin phosphodiesterase family that encode Mg2+-dependent neutral sphingomyelinases. These four genes are the SMPD2, SMPD3, SMPD4, and SMPD5 genes. The activity of the SMPD2 encoded enzyme is a lysophospholipase and not a sphingomyelinase. The SMPD3 encoded enzyme is identified as neutral sphingomyelinase 2 (nSMase2) and the SMPD4 encoded enzyme is identified as nSMase3. The SMPD5 gene is designated a pseudogene in humans. However, an open reading frame in the SMPD5 gene encodes a protein with a mitochondrial localization signal and evidence suggests that this enzyme functions as a mitochondria-localized member of the nSMase family. There is also a Mg2+-independent neutral sphingomyelinase activity that has been characterized in mammalian tissues, but its encoding mRNA is yet to be determined.
The alkaline sphingomyelinase is encoded by the ENPP7 (ectonucleotide pyrophosphatase/phosphodiesterase 7) gene.
Role of SMPD3 Encoded Enzyme in Liver Disease
Recent work (2025) has demonstrated a role for the SMPD3 encoded sphingomyelinase in the progression of hepatic lipotoxicity leading to the development of metabolic-dysfunction-associated steatohepatitis, MASH. MASH was previously referred to as non-alcoholic steatohepatitis, NASH. MASH represents the state of liver disease in the progression from the early stages of lipotoxicity defined as metabolic dysfunction-associated fatty liver disease (MAFLD), also known as metabolic dysfunction-associated steatotic liver disease (MASLD). MAFLD was formerly referred to as non-alcoholic fatty liver disease, NAFLD.
As indicated above, elevated levels of serum ceramides have been found to be contributory to the development of cardiovascular disease and the insulin resistance associated with type 2 diabetes. The major enzyme contributing to increased levels of ceramides in the serum has been shown to be the SMPD3 encoded enzyme, which is identified as neutral sphingomyelinase 2 (nSMase2).
The normal level of expression of the SMPD3 gene is very low in the livers of healthy individuals. However, in the hepatocytes of individuals with the hepatic lipotoxicity associated with MAFLD and MASH the level of expression of the SMPD3 gene is upregulated. This upregulation of SMPD3 expression has been found to be the result of the lipotoxicity-mediated repression of the activity of the deacetylase encodes by the SIRT1 gene. The activity of SIRT1 normally leads to repression of SMPD3 expression thus, loss of SIRT1 activity results in increased SMPD3 expression.
In mice, the hepatocyte-specific knock-out of the SMPD3 gene results in reduced lipid uptake resulting in amelioration of the development of MASH. This effect is the result of a block to the role of the SMPD3 encoded enzyme in endocytosis of extracellular fatty acids. The release of ceramides from plasma membrane-associated sphingomyelins, via the action of SMPD3, activates caveolin-1 mediated endocytosis.
The mechanism of lipotoxicity-induced increases in SMPD3 expression involves DNA damage. It is known that hepatic lipotoxicity increases the level of DNA damage, which in turn results in increased activity of NAD+-dependent PARP enzymes. This leads to a depletion in overall levels of NAD+ resulting in decreased activity of SIRT1 and a consequent de-repression of SMPD3 gene expression.
The Glycosphingolipids
Glycosphingolipids, or glycolipids, are composed of a ceramide backbone with a wide variety of carbohydrate groups (mono- or oligosaccharides) attached to carbon 1 of sphingosine. The four principal classes of glycosphingolipids are the cerebrosides, sulfatides, globosides, and gangliosides.
With the exception of galactocerebrosides and the GM4 gangliosides, all complex glycosphingolipids begin via the attachment of glucose to a ceramide. Attachment of glucose to ceramide occurs in the Golgi following the transfer of ceramides from the ER via the lipid transfer protein, FAPP2. The enzyme that catalyzes glucose attachment utilizes UDP-glucose and is commonly called glucosylceramide synthase, GCS. GCS is encoded by the UGCG (UDP-glucose:N-acylsphingosine D-glucosyltransferase) gene.
Cerebrosides
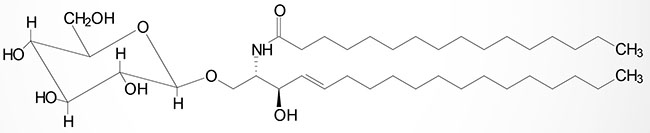
Cerebrosides have a single sugar group linked to ceramide. The most common of these is galactose generating the galactocerebrosides (also termed galactosylcerebrosides or galactosylceramides). Glucose attachment yields the glucocerebrosides (glucosylcerebrosides or glucosylceramides) although these are, in humans, primarily intermediates in the synthesis of more complex glycosphingolipids.
The attachment of galactose to a ceramide is catalyzed by the UGT8 (UDP glycosyltransferase 8) encoded enzyme. UDP glycosyltransferase 8 is commonly termed galactosylceramide synthase. The UGT8 encoded enzyme is a member of the large family of UDP glucuronosyltransferases (UGT). The UGT8 encoded enzyme utilizes UDP-galactose as the substrate for galactose attachment to a ceramide. As discussed below, GM4 gangliosides have galactose attached to the ceramide backbone and thus, begin as galactocerebrosides (galactosylceramides).
Galactocerebrosides are found predominantly in neuronal cell membranes. By contrast glucocerebrosides are not normally found in neuronal membranes; instead, they primarily represent intermediates in the synthesis or degradation of more complex glycosphingolipids.
Excess lysosomal accumulation of glucocerebrosides is observed in Gaucher disease. Glucocerebrosides are only intermediates in the synthesis of complex gangliosides or are found at elevated levels only in disease states such as Gaucher disease, where there is a defect in the catabolism of the complex gangliosides. Thus, the presence of high concentrations of glucocerebrosides in cells such as monocytes and macrophages is indicative of a metabolic defect.
Sulfatides
The sulfuric acid esters of galactocerebrosides (galactosylceramides) are the sulfatides. Sulfatides are synthesized from galactocerebrosides and the activated sulfate, 3′-phosphoadenosine 5′-phosphosulfate (PAPS), the synthesis of which is shown in the Figure below. There is one major class of sulfatide in humans identified as the SM4 sulfatides.
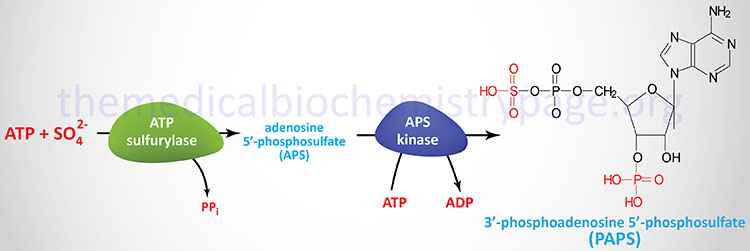
The sulfation reaction is catalyzed via the action of enzymes of the sulfotransferase family. Humans express 50 different sulfotransferase genes, 37 of which encode membrane-bound enzymes and 13 of which encode cytosolic enzymes. These enzymes are involved in the synthesis of sulfate esters and sulfo-amines such as those found in the glycosaminoglycans. Excess accumulation of sulfatides is observed in metachromatic leukodystrophy (sulfatide lipidosis).
As described in the section above, galactocerbrosides are synthesized from a ceramide via the actions of UGT8 encoded enzyme and a UDP-galactose molecule. Following synthesis of galactocerebrosides in the endoplasmic reticulum (ER) they are transferred to the Golgi apparatus where the SM4 sulfatides are then generated. Synthesis of SM4 sulfatides is catalyzed by the homodimeric enzyme commonly called cerebroside sulfotransferase (CST). CST is also called galactosylceramide sulfotransferase. CST is encoded by the GAL3ST1 (galactose-3-O-sulfotransferase 1) gene.
Globosides
Globosides represent cerebrosides that contain additional carbohydrates, predominantly galactose, glucose, or GalNAc. Globosoides are generated by sequential addition of various sugars via the action of glycosyltransferases. Human cells contain primarily three distinct globosides; lactosylceramide, Gb3, and Gb4.
Lactosylceramide, which is the precursor for the synthesis of the majority of the gangliosides, is a globoside found in erythrocyte plasma membranes.
The Gb3 globoside (also referred to as globotriaosylceramide and also as ceramide trihexoside) is synthesized from a lactosylceramide and UDP-galactose via the A4GALT (alpha 1,4-galactosyltransferase) encoded enzyme. The A4GALT encoded enzyme is also known as Gb3 synthase. The Gb3 globoside accumulates, primarily in the kidneys, of patients suffering from Fabry disease. Gb3 is the P(k) antigen of the P blood group system.
The Gb4 globoside is catalyzed from Gb3 by the addition of N-acetylgalactosamine (GalNAc) which is catalyzed by the B3GALNT1 (beta-1,3-N-acetylgalactosaminyltransferase 1) encoded enzyme.
Gangliosides
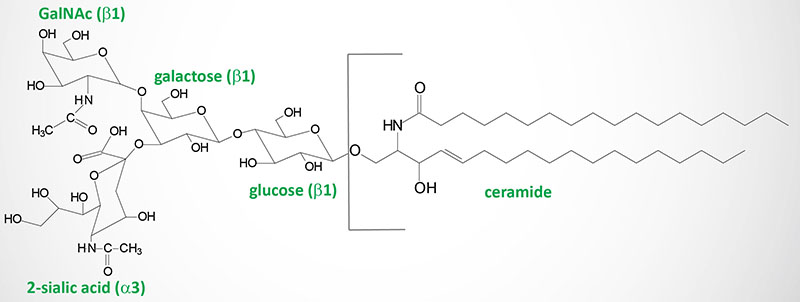
Gangliosides are very similar to globosides except that they also contain N-acetylneuraminic acid (NANA; also known as sialic acid) in varying amounts. The specific names for gangliosides are a key to their structure. The first letter on the nomenclature, G, refers to ganglioside. The following letters, A, M, D, T, Q, and P, indicate that the molecule contains none (absent or asialo), mono-, di-, tri, quatra(tetra), or poly(more than 4)-sialic acid residues. The numerical designations, 1, 2 and 3, refer to the carbohydrate sequence that is attached to ceramide:
1 stands for Galβ1-3GalNAcβ1-4Galβ1-4Glcβ1-1′-ceramide
2 stands for GalNAcβ1-4Galβ1-4Glcβ1-1′-ceramide
3 stands for Galβ1-4Glcβ1-1′-ceramide.
Finally the presence of lower case letters a, b, and c refer to one, two, or three sialic acid residues attached to the innermost galactose residue within the gangliosides that also have sialic acid residues attached to the terminal galactose residue.
Within the nervous system, gangliosides serve numerous important functions that include being the main carriers of sialic acid, an amino sugar that plays important roles in cell-cell and pathogen-cell interactions. Gangliosides are most abundant within the plasma membrane where their hydrophobic ceramide tails are inserted into the outer phospholipid leaflet, while the glycan headgroup extends out into the extracellular space where it can interact with molecules on other cells and those in the extracellular space. These ganglioside-mediated interactions lead to the modulation of cell signaling and cell-to-cell communication.
Simple gangliosides, like GM3, predominate in most peripheral tissues. Within the brain approximately 90% of the gangliosides consist of four complex gangliosides (GM1, GD1a, GD1b, and GT1b). Within the brain, gangliosides are most abundant in myelin, followed by neuronal cells. Myelin contains large amounts of GM1 and GM4. Oligodendrocyte membranes contain GM1, GD3, and GM4. Astrocyte membranes possess mostly simple gangliosides, including GM3, GD3 and, to a lesser extent, GM4
The GM4 series gangliosides (referred as the gala-series) contain the single sugar, galactose, attached to the ceramide backbone. These gangliosides also contain a single sialic acid residue attached to the galactose.
Synthesis of Gangliosides
Ganglioside biosynthesis begins with a ceramide and will involve the sequential attachment of one or more monosaccharides as well as one or more sialic acid residues. Gangliosides are synthesized in the Golgi apparatus and then transported, primarily, to the plasma membrane.
As a result of the heterogeneity in the sugar composition of the glycan headgroup of the gangliosides there are more than 200 different ganglioside structures that have been identified. Additional molecular diversity is conferred by the ceramide itself as a result of different fatty acids generating the sphingosine base of ceramides as well as the various N-acylated fatty acids. These fatty N-acyl groups include fatty acyl chains of different lengths and various degrees of saturation.
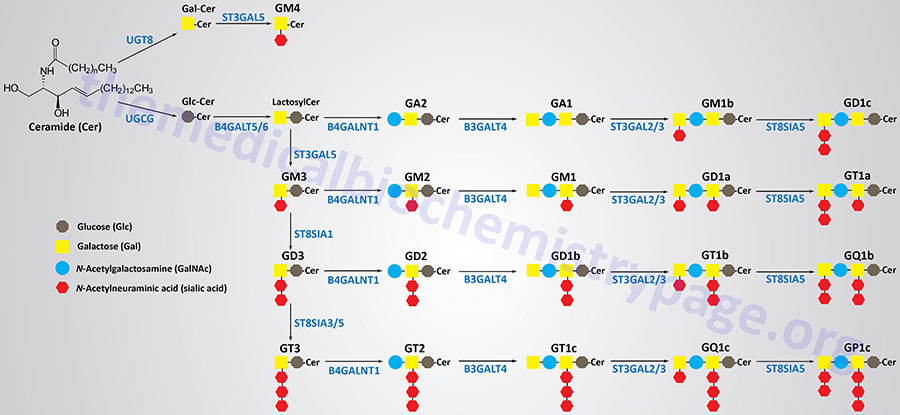
Synthesis of the largest number of gangliosides begins with the attachment of glucose to the ceramide backbone. This reaction involves UDP-glucose and a ceramide and is catalyzed by the enzyme commonly called glucosylceramide synthase, GCS. GCS is also commonly referred to as ceramide glucosyltransferase 1. GCS is encoded by the UGCG (UDP-glucose:N-acylsphingosine D-glucosyltransferase) gene.
Following the addition of the glucose residue, a galactose residue is added forming a family of lactosylceramides. The lactosylceramides serve as the building blocks for all the other gangliosides, excluding the GM4 series. The addition of the galactose residue can be catalyzed by one of two related enzymes, beta-1,4-galactosyltransferase 5 or 6, which are encoded by the B4GALT5 and B4GALT6 genes, respectively.
The sequential addition of sialic acid residues to a lactosylceramide will generate a GM3, then a GD3, and finally a GT3 ganglioside. The first sialic acid residue is added by the enzyme, ST3 beta-galactoside alpha-2,3-sialyltransferase 5, which is encoded by the ST3GAL5 gene. The ST3GAL5 encoded enzyme is also referred to as GM3 synthase and as lactosylceramide alpha-2,3-sialyltransferase.
The second sialic acid residue is added by the enzyme, ST8 alpha-N-acetyl-neuraminide alpha-2,8-sialyltransferase 1, encoded by the ST8SIA1 gene.
The third sialic acid residue is added by either one of two related enzymes, ST8 alpha-N-acetyl-neuraminide alpha-2,8-sialyltransferase 3 or 5, encoded by the ST8SIA3 and ST8SIA5 genes, respectively.
The addition of N-acetylgalactosamine (GalNAc) to a lactosylceramide generates the GA2 gangliosides. Addition of GalNAc to a GM3 ganglioside generates a GM2 ganglioside, to a GD3 ganglioside generates a GD2 ganglioside, and to a GT3 ganglioside generates a GT2 ganglioside. All of the GalNAc additions are catalyzed by the enzyme, beta-1,4-N-acetyl-galactosaminyltransferase 1, encoded by the B4GALNT1 gene.
The enzyme, beta-1,3-galactosyltransferase 4, which is encoded by the B3GALT4 gene, adds a galactose residue to GA2, GM2, GD2, and GT2 gangliosides generating GA1, GM1, GD1b, and GT1c gangliosides, respectively.
One of two related enzymes, ST3 beta-galactoside alpha-2,3-sialyltransferase 2 or 3, encoded by the ST3GAL2 and ST3GAL3 genes, respectively, adds a sialic acid residue to the terminal galactose of GA1, GM1, GD1b, and GT1c gangliosides which generates the GM1b, GD1a, GT1b, and GQ1c gangliosides, respectively.
Finally, the ST8SIA5 encoded enzyme, ST8 alpha-N-acetyl-neuraminide alpha-2,8-sialyltransferase 5, adds a second sialic acid residue to the terminal galactose of the GM1b, GD1b, GT1b, and GQ1c gangliosides to generate the GD1c, GT1a, GQ1b, and GP1c gangliosides, respectively
The GM4 series of gangliosides have galactose attached to the ceramide backbone and this addition is catalyzed by the UDP glycosyltransferase 8 enzyme encoded by the UGT8 gene. Sialic acid is added to the galactose residue by the enzyme, ST3 beta-galactoside alpha-2,3-sialyltransferase 5, which is encoded by the ST3GAL5 gene.
Clinical Significance of Ganglioside Metabolism
Deficiencies in several of the enzymes responsible for the synthesis of the gangliosides have been identified and shown to be associated with disease. Loss of function mutations in the ST3GAL5 gene are associated with an infantile epileptic seizure syndrome. This disorder is often referred to as salt and pepper developmental regression syndrome (SPDRS). SPDRS is a rare autosomal recessive disorder characterized by infantile onset of severe, recurrent and refractory seizures, failure to thrive, psychomotor delay, developmental stagnation, and cortical blindness. Mutations in the B4GALNT1 gene are associated with hereditary spastic paraplegia, also known as autosomal recessive spastic paraplegia type 26.
Deficiencies in lysosomal enzymes, which normally are responsible for the degradation of the carbohydrate portions of various gangliosides, underlie the symptoms observed in rare autosomal recessive diseases of the lysosomal storage disease family, (also called the sphingolipidoses or lipid storage diseases) many of which are listed below.
Sphingosine-1-Phosphate (S1P) and Signal Transduction
Lysophospholipids (LP) are minor lipid components compared to the major membrane phospholipids such as phosphatidylcholline (PC), phosphatidylethanolamine (PE), and sphingomyelin. The LP were originally presumed to be simple metabolic intermediates in the de novo biosynthesis of phospholipids. However, subsequent studies demonstrated that the LP exhibited biological properties resembling those of extracellular growth factors or signaling molecules.
The most biologically significant LP are sphingosine-1-phosphate (S1P), lysophosphatidic acid (LPA), lysophosphatidylcholine (LPC), and sphingosylphosphorylcholine (SPC). More information on the biological activities of the lysophospholipids can be found in the Signal Transduction Pathways: Phospholipids page.
Synthesis of S1P occurs exclusively from sphingosine via the action of sphingosine kinases. Sphingosine is phosphorylated in humans through the action of two related sphingosine kinases encoded by the SPHK1 and SPHK2 genes. The intermediate in sphingosine synthesis, dihydrosphingosine, is also a substrate for sphingosine kinases. The importance of the action of the sphingosine kinases can be shown by the fact that when both are knocked out in transgenic mice embryos are not viable due to the incomplete development of the brain and the vascular system.
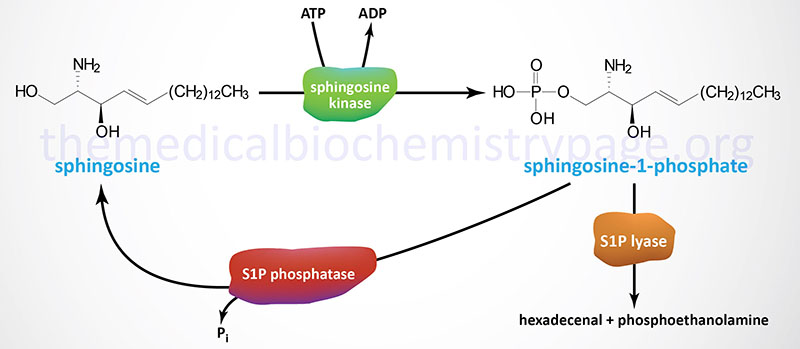
The SPHK1 gene is located on chromosome 17q25.1 and is composed of 7 exons that generate five alternatively spliced mRNAs that collectively encode three distinct protein isoforms.
The SPHK2 gene is located on chromosome 19q13.33 and is composed of 8 exons that generate five alternatively spliced mRNAs that collectively encode four distinct protein isoforms.
In vertebrates, S1P is secreted into the extracellular space by specific transporters, one of which (sphingolipid transporter 2) was originally identified as spinster 2 homolog-2. The sphingolipid transporter 2 protein is encoded by the SPNS2 gene. Plasma levels of S1P are high, whereas interstitial fluids contain very low levels. This results in an S1P gradient in different compartments. Hematopoietic cells and vascular endothelial cells are the major sources of the high plasma S1P concentrations. Lymphatic endothelial cells are also thought to secrete S1P into the lymphatic circulation. The majority of plasma S1P is bound to HDL (65%) with another 30% bound by albumin.
Recent work has demonstrated that the ability of HDL to induce vasodilation and migration of endothelial cells, as well as to serve a cardioprotective role in the vasculature is dependent on S1P. These studies suggest that the beneficial property of HDL to reduce the risk of cardiovascular disease may be due, in part, on its role as an S1P chaperone.
Nuclear Sphingosine-1-Phosphate
Recent studies have shown that SPHK2 (which contains a nuclear localization signal), and its product S1P, are found in the nucleus associated with transcriptional co-repressor complexes that contain histone deacetylase 1 (HDAC1) and HDAC2. The S1P-containing HDAC complexes are prevented from deacetylating lysine residues in the histone tails, thereby altering gene expression patterns.
One gene whose expression is upregulated in cells with S1P-HDAC complexes is the cell cycle regulating protein p21 which is an inhibitor of cyclin-dependent kinases (CDK) and is involved in p53-mediated apoptosis. This observation suggests that enhancing SPHK2-S1P-associated complexes in the nucleus could be of clinical benefit in p53-deficient types of cancer. Given that mutations in p53 are some of the most commonly occurring genetic abnormalities in cancer, this could prove highly significant.
Sphingosine-1-Phosphate Receptors
Each of the lysophospholipids (LP), including S1P, function via interaction with specific G-protein coupled receptors (GPCR) leading to autocrine or paracrine effects. The first GPCR shown to bind S1P was called S1P1. Currently there are five characterized S1P receptors. Because several of the LP receptors were independently identified in unrelated assays, there are several different names for some members of this receptor family. In particular, there is a group of genes that were originally identified as GPCR and called endothelial differentiation genes (EDG) that were later found to be the same as several of the LP receptors. Thus S1P1 is also known as EDG1, S1P2 as EDG5, S1P3 as EDG3, S1P4 as EDG6, and S1P5 as EDG8.
The biological activities attributed to S1P interaction with any of the five identified receptors are broad. These activities include involvement in vascular system and central nervous system development, viability and reproduction, immune cell trafficking, cell adhesion, cell survival and mitogenesis, stress responses, tissue homeostasis, angiogenesis, and metabolic regulation.
Table of the Sphingosine-1-phosphate (S1P) Receptors
| S1P Receptor | Alternative Name | Gene Symbol | G-Proteins | Comments |
| S1P1 | EDG1 | S1PR1 | Gi/o | Expressed in brain, heart, spleen, liver, kidney, skeletal muscle, thymus, pancreatic β-cells, and numerous white blood cells. Within the immune system, activation of S1P1 has been shown to block B cell and T cell chemotaxis and infiltration into tissues. In addition S1P1 activation results in inhibition of late-stage maturation processes associated with T cells. Within the central nervous system S1P1 is involved in astrocyte migration and increased migration of neural stem cells. Within the vasculature S1P1 is involved in early vascular system development and endothelial cell functions such as adherens junction assembly and vascular smooth muscle cell development. Within the pancreas S1P1 functions in islet cell survival and insulin secretion. |
| S1P2 | EDG5 | S1PR2 | Gi/o, Gq, G12/13 | Expressed in the brain, heart, spleen, liver, lung, kidney, skeletal muscle, and thymus. S1P2 is involved in the development of epithelial cells, enhancing the survival of cardiac myocytes to ischemic-reperfusion injury, and hepatocyte proliferation and matrix remodeling. Within the vasculature S1P2 promotes mast cell degranulation and decreases vascular smooth muscle cell responses to PDGF-induced migration. In the eye S1P2 activation can result in pathologic angiogenesis and disruption in adherens junction formation. |
| S1P3 | EDG3 | S1PR3 | Gi/o, Gq, G12/13 | Expressed in the brain, heart, spleen, liver, lung, kidney, skeletal muscle, testis, and thymus. S1P3 activation is associated with a worsening of sepsis, increased inflammation and coagulation. However, with respect to cardiac tissues S1P3 promotes survival in response to ischemic-reperfusion injury.t |
| S1P4 | EDG6 | S1PR4 | Gi/o, G12/13 | Expressed in lymphoid tissues (leukocytes) and within a restricted subset of cells in the lung that includes airway smooth muscle cells. The primary responses to S1P4 activation are increased T cell migration and secretion of cytokines. |
| S1P5 | EDG8 | S1PR5 | Gq, G12/13 | Expressed in the brain, spleen, and the skin. Within the brain S1P5 activation is associated with inhibition of migration of oligodendrocyte progenitors while increasing the survival of oligodendrocytes. S1P5 also stimulates natural killer (NK) cell trafficking. |
Sphingosine-1-Phosphate Catabolism
Degradation of S1P occurs through dephosphorylation and cleavage. The dephosphorylation occurs via the action of enzymes of the lipid phosphatase family, specifically the two enzymes in the sphingosine-1-phosphate phosphatase subfamily (S1P phosphatase-1 and -2) as well as lipid phosphate phosphatase 3 (LPP3). LPP3 is a member of the phospholipid phosphatase (PLPP) family. Cleavage of S1P is catalyzed by sphingosine-1-phosphate lyase 1 (S1P lyase).
S1P lyase is encoded by the SGPL1 gene. The SGPL1 gene is located on chromosome 10q22.1 and is composed of 16 exons that encode a 568 amino acid protein.
S1P phosphatase 1 is encoded by the SGPP1 gene. The SGPP1 gene is located on chromosome 14q23.2 and is composed of 3 exons that encode a 441 amino acid protein.
S1P phosphatase 2 is encoded by the SGPP2 gene. The SGPP2 gene is located on chromosome 2q36.1 and is composed of 7 exons that generate three alternatively spliced mRNAs encoding two distinct protein isoforms.
Lipid phosphate phosphatase 3 (LPP3) is encoded by the PLPP3 gene. The PLPP3 gene is located on chromosome 1p32.2 and is composed of 6 exons that encode a 311 amino acid protein.
The different S1P phosphatases remove the phosphate, thus, regenerating sphingosine which can re-enter the sphingolipid metabolic pathway. When used as a substrate for phospholipid synthesis, S1P is degraded by S1P lyase to yield hexadecenal and phosphoethanolamine. Phosphoethanolamine is the direct precursor for the synthesis of the phospholipid phosphatidylethanolamine (PE). The hexadecenal is converted into hexadecenoic acid by hexadecenal dehydrogenase and then into palmitoyl-CoA. The degradation of S1P by the S1P lyase pathway serves as an important pathway for the conversion of sphingolipids into glycerolipids.
Ceramide-1-Phosphate (C1P)
Ceramide-1-phosphate (C1P) is a bioactive lipid that has been shown to play important roles the regulation of cell survival, apoptosis, and autophagy, primarily as an intracellular second messenger. C1P also inhibits the activity of serine palmitoyltransferase (SPT) the rate-limiting enzyme of sphingosine and ceramide synthesis. C1P also inhibits acid sphingomyelinase.
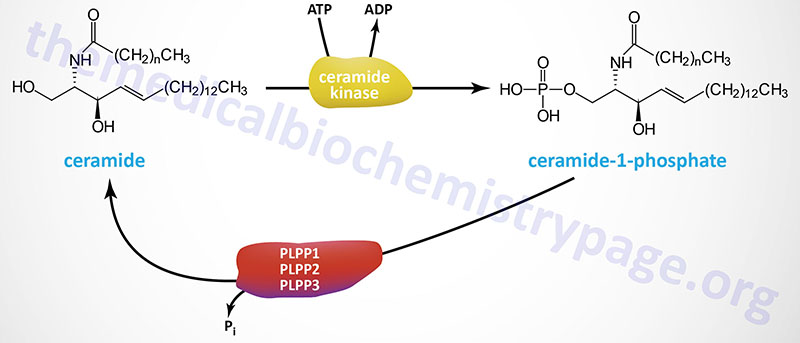
The synthesis of C1P is catalyzed by a single enzyme in humans identified as ceramide kinase. Ceramide kinase is encoded by the CERK gene. The CERK gene is located on chromosome 22q13.31 and is composed of 13 exons that encode a protein of 537 amino acids.
Ceramide-1-phosphate is hydrolyzed back to ceramide via the actions of several phosphatases of the phospholipid phosphatase (PLPP) family. The most likely phosphatases are the three enzymes of the lipid phosphate phosphatase (LPP) subfamily of PLPP enzymes. These three enzymes are encoded by the PLPP1, PLPP2, and PLPP3 genes.
Following synthesis C1P is transported to the plasma membrane where it exerts effects as an intracellular second messenger. The transport of C1P is the function of ceramide-1-phosphate transfer protein, CPTP.
As an intracellular second messenger C1P exerts effects that includes the regulation cell growth by stimulating cell
cycle progression or cellular proliferation pathways. These pathways include the the kinases, phosphatidylinositol-3 kinase (PI3K), AKT (also called PKB), JNK, PKCα, and ERK1/2 kinase.
The activation of AKT by C1P results in the subsequent activation of glycogen synthase kinase-3β, GSK-3β. Activation of GSK-3β leads to phosphorylation and activation of the transcription factor, MYC, and the cell cycle regulatory protein, cyclin D1. AKT activation by C1P also results in the activation of the potent immune system modulating transcription factor, NFκB, the effect of which is inhibition of apoptosis and activation of cellular proliferation. Activated AKT also phosphorylates and activates the mTORC1 complex which leads to activation of cellular proliferation and migration.
CPTP can also facilitate C1P transport to the extracellular compartment where is has been shown to interact with a Gi-type G-protein coupled receptor, GPCR. The activation of this receptor by C1P is associated with mobilization of GLUT3 transporters resulting in increased glucose uptake.
C1P has also been shown to regulate apoptosis through its ability to inhibit the activity of pro-apoptotic ceramide synthesis enzymes. C1P activated AKT also activates the expression of the inducible form of nitric oxide synthase (iNOS) by which it can promote a reduction in inflammation as well as inhibit apoptosis and autophagy.
Although in some contexts C1P inhibits inflammation, in others it acts to promote inflammation. C1P has been shown to promote the translocation and activation of the group IV cytosolic phospholipase A2 (cPLA2α). The activation of cPLA2α results in increased release of arachidonic acid from membrane phospholipids. Arachidonic acid then serves as the substrate for the synthesis of many different pro-inflammatory eicosanoids.
Clinical Significances of Glycosphingolipids
Some of the most devastating inborn errors in metabolism are those associated with defects in the enzymes responsible for the lysosomal degradation of membrane glycosphingolipids which are particularly abundant in the membranes of neural cells. Many of these disorders lead to severe psychomotor impairment and early lethality. Because the disorders are caused by defective lysosomal enzymes, with the result being lysosomal accumulation of pathway intermediates, these are often referred to as lysosomal storage diseases or lysosomal storage disorders. The mucopolysaccharidoses are another class of disorders that are members of the lysosomal storage diseases.
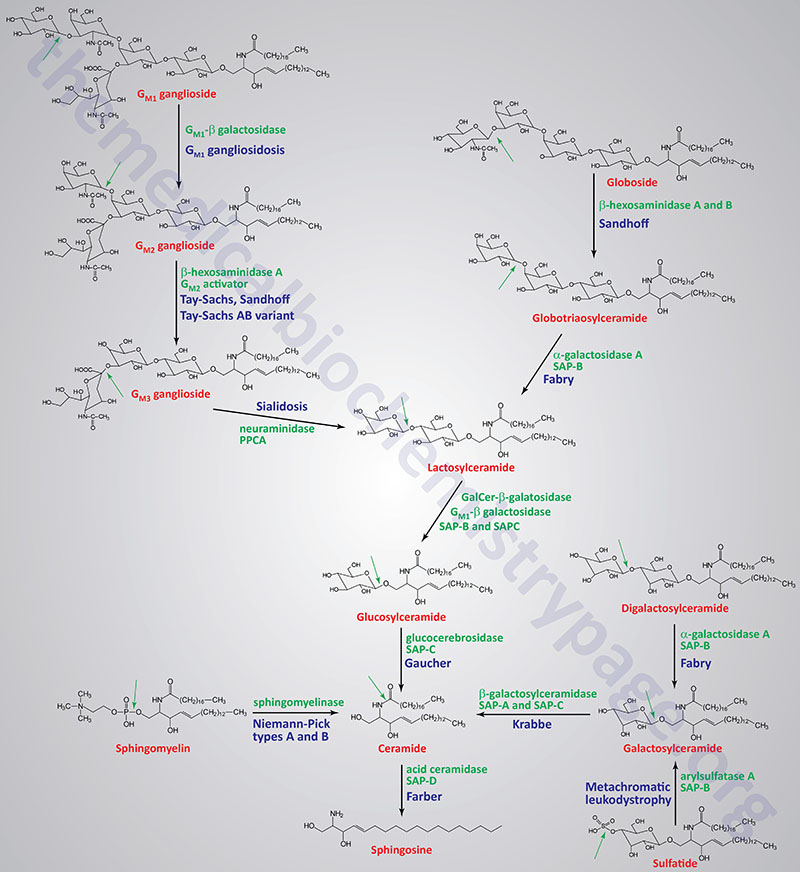
Table of the Disorders Associated with Abnormal Sphingolipid Metabolism
| Disorder | Enzyme Deficiency | Accumulating Substance | Comments/Symptoms |
| Tay-Sachs disease | β-hexosaminidase A | GM2 ganglioside | β-hexosaminidase A is a heterodimer composed of an α-subunit encoded by the HEXA gene and a β-subunit encoded by the HEXB gene; in the infantile form there is rapidly progressing psychomotor impairment, blindness, early mortality |
| Sandhoff disease | β-hexosaminidases A and B | globoside; GM2 ganglioside | β-hexosaminidase A is a heterodimer of composed of an a-subunit encoded by the HEXA gene and a β-subunit encoded by the HEXB gene; β-hexosaminidase B is a homodimer of two β-subunits encoded by the HEXB gene; the infantile form manifests with the same symptoms as Tay-Sachs; disease progresses more rapidly than Tay-Sachs |
| Tay-Sachs AB variant GM2 activator deficiency | GM2 ganglioside activator | GM2 ganglioside | the GM2 ganglioside activator protein is encoded by the GM2A gene; the infantile form manifests with the same symptoms as Tay-Sachs |
| Gaucher disease | glucocerebrosidase (glucosylceramidase beta) | glucocerebrosides | glucocerebrosidase is encoded by the glucosylceramidase beta (GBA) gene; most common form has average age of onset of 30 years; hallmark of disease is accumulation of lipid-engorged cells of the monocyte/macrophage lineage in multiple tissues |
| Fabry disease | α-galactosidase A | globotriaosylceramide; also called ceramide trihexoside (CTH) | kidney failure, skin rashes |
| Niemann-Pick diseases Types A and B Type C | sphingomyelinase NPC1 protein | sphingomyelins LDL-derived cholesterol | the sphingomyelinase that is defective in Niemann-Pick type A (NPA) and NPB is encoded by the SMPD1 (sphingomyelin phosphodiesterase 1) gene; humans express four genes with sphingomyelin phosphodiesterase activity (SMPD1, SMPD2, SMPD3, and SMPD4; type A is severe disorder with hepatosplenomegaly, severe neurological involvement leading to early death, type B is only associated with visceral involvement |
| Krabbe disease; globoid cell leukodystrophy (GLD) | galactocerebrosidase | galactocerebrosides | galactocerebrosidase is encoded by the GALC (galactosylceramidase) gene; intellectual impairment, myelin deficiency |
| GM1 gangliosidosis | β-galactosidase-1 | GM1 gangliosides | β-galacatosidase-1 is encoded by the GLB1 (galactosidase beta 1) gene; intellectual impairment, skeletal abnormalities, hepatomegaly |
| Metachromatic leukodystrophy; sulfatide lipodosis | arylsulfatase A | sulfatides | arylsulfatase A is encoded by the ARSA gene; intellectual impairment, metachromasia of nerves |
| Farber lipogranulomatosis | acid ceramidase | ceramides | acid ceramidase is encoded by the ASAH1 (N-acylsphingosine amidohydrolase 1) gene; hepatosplenomegaly, painful swollen joints |
| Sialidosis: type 1 (adult) and type 2 (infantile) | neuraminidase 1 (sialidase) | sialyloligosaccharides in glycolipids and glycoproteins | enzyme is lysosome-localized hydrolase; type 1 sialidosis is also known as cherry-red spot myoclonus syndrome; type 2 is also identified as mucolipidosis I (ML-I); neuraminidase 1 requires the accessory protein: protective protein/cathepsin A (PPCA) which is encoded by the CTSA gene; type I associated with myoclonus, gait disturbances, reduced visual acuity with cherry-red macula; type II is associated hydrops fetalis, hepatosplenomegaly, dysostosis multiplex, coarse facial features, myoclonus, ataxia, tremor and angiokeratoma; see the Sialidosis page for details |
One of the most clinically important classes of sphingolipids are those that confer antigenic determinants on the surfaces of cells, particularly the erythrocytes. The ABO blood group antigens are the carbohydrate moieties of glycolipids on the surface of cells as well as the carbohydrate portion of serum glycoproteins. When present on the surface of cells the ABO carbohydrates are linked to sphingolipid and are therefore of the glycosphingolipid class. When the ABO carbohydrates are associated with protein in the form of glycoproteins they are found in the serum and are referred to as the secreted forms. Some individuals produce the glycoprotein forms of the ABO antigens while others do not. This property distinguishes secretors from non-secretors, a property that has forensic importance such as in cases of rape.
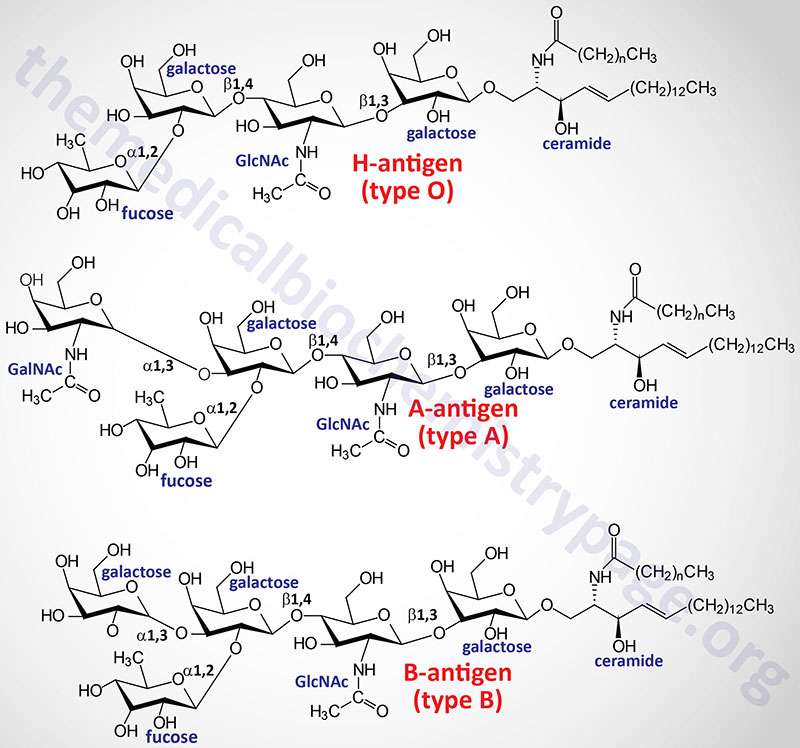
A significant cause of death in premature infants and, on occasion, in full term infants is respiratory distress syndrome (RDS) or hyaline membrane disease. This condition is caused by an insufficient amount of pulmonary surfactant. Under normal conditions the surfactant is synthesized by type II endothelial cells and is secreted into the alveolar spaces to prevent atelectasis following expiration during breathing. Surfactant is comprised primarily of dipalmitoyllecithin; additional lipid components include phosphatidylglycerol and phosphatidylinositol along with proteins of 18 and 36 kDa (termed surfactant proteins).
During the third trimester the fetal lung synthesizes primarily sphingomyelin, and type II endothelial cells convert the majority of their stored glycogen to fatty acids and then to dipalmitoyllecithin. Fetal lung maturity can be determined by measuring the ratio of lecithin to sphingomyelin (L/S ratio) in the amniotic fluid. An L/S ratio less than 2.0 indicates a potential risk of RDS. The risk is nearly 75-80% when the L/S ratio is 1.5.
The carbohydrate portion of the ganglioside, GM1, present on the surface of intestinal epithelial cells, is the site of attachment of cholera toxin, the protein secreted by Vibrio cholerae.
These are just a few examples of how sphingolipids and glycosphingolipids are involved in various recognition functions at the surface of cells. As with the complex glycoproteins, an understanding of all of the functions of the glycolipids is far from complete.