
Last Updated: October 30, 2025
Introduction to SLOS
Smith-Lemli-Opitz syndrome (SLOS) is an autosomal recessive disorder that was first described in 1964 by three doctors whose last names constitute the name of this syndrome. SLOS results in multiple malformations and behavioral problems as a consequence of a defect in cholesterol synthesis. The defect resides in the terminal enzyme of the cholesterol biosynthesis pathway, namely 7-dehydrocholesterol reductase. Defects in this gene result in increased levels of 7-dehydrocholesterol and reduced levels (15% to 27% of normal) of cholesterol in SLOS patients. The highest frequency of SLOS appears in Caucasians of northern European descent with a frequency of around 1 in 20,000 to 1 in 70,000.
Molecular Biology of SLOS
The 7-dehydrocholesterol reductase gene (DHCR7) is found on chromosome 11q13.4 spanning 14 kb and encompassing 9 exons that generate two alternatively spliced mRNAs, both of which encode the same 475 amino acid protein. The highest levels of expression of DHCR7 occur in the liver, adrenal glands, and brain tissue.
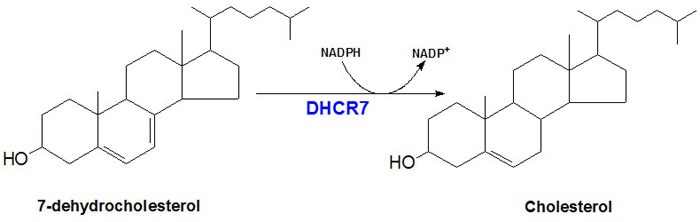
Functional DHCR7 protein is a 55.5 kDa integral membrane protein localized to the microsomal membrane. Activity of DHCR7 requires NADPH as a co-factor and the reaction catalyzed by the enzyme is diagrammed in the Figure below.
Over 130 different mutations in the DHCR7 gene have been identified in SLOS patients including missense, nonsense and splice site mutations. Six mutations have been shown to account for greater than 60% of all SLOS mutations. These mutations include a cysteine for arginine substitution at amino acid 404 (identified as the R404C mutation), a methionine for threonine substitution at amino acid 93 (identified as the T93M mutation), a leucine for valine substitution at amino acid 362 (identified as the V362L mutation), a nonsense mutation at amino acid 151 resulting in termination of translation (identified as the W151X mutation), and a splice site mutation in intron 8 of the DHCR7 gene. The splice site mutation is the most frequently observed mutation in the DHCR7 gene accounting for one third of all mutations in SLOS.
Biochemical Consequences of DHCR7 Mutations
Because cholesterol serves a broad range of functions in the body it is not entirely surprising that a deficiency in its synthesis would lead to such profound developmental anomalies. For a clear understanding of the functions of cholesterol see the Cholesterol Synthesis and the Steroid Hormones pages.
Recent evidence has demonstrated that one of the most important development regulating proteins of early embryos is modified by cholesterol addition. This protein is called sonic hedgehog (abbreviated Shh). Shh is necessary for patterning events that take place in the central nervous system, during limb development, and in the formation of facial structures. The involvement of cholesterol in normal Shh function explains many of the abnormalities that are observed in SLOS patients.
Altered levels of cholesterol in SLOS affect the normal physiochemical processes of cell membranes as well due to the role of cholesterol in overall membrane function. One major defect that results from reduced membrane cholesterol is an inability to form ordered lipid domains that are necessary for normal signal transduction events to take place. These defects then alter normal cellular functions resulting in many of the symptoms of SLOS.
Clinical Features of SLOS
The clinical spectrum of SLOS is very broad, ranging from the most severe form manifesting as a lethal malformation syndrome, to a relatively mild disorder that encompasses behavioral and learning disabilities. Frequent observations in SLOS infants is poor feeding and postnatal growth failure. The growth failure may necessitate the insertion of a gastronomy tube to ensure adequate nutritional support. There are distinct craniofacial anomalies associated with SLOS. These include microcephaly (head size smaller than normal), micrognathia (abnormally small lower jaw), ptosis (drooping eyelids), a small upturned nose, and cleft palate or bifid uvula.
Male infants with SLOS exhibit genital abnormalities that range from a small penis to ambiguous genitalia or gender reversal. Abnormalities in limb development are common in SLOS patients and include short thumbs, postaxial polydactyly, and single palmar creases. In addition, the most common clinical finding in SLOS patients is syndactyly (fusion of digits) of the second and third toes. This latter limb deformity is found in over 95% of SLOS patients.