Last Updated: May 22, 2026
Introduction to Mechanisms of Signal Transduction
When discussing the process of signal transduction it can be defined at the cellular level as the movement of signals from outside the cell to inside. This page is designed to provide an overview of signal transduction process with many of the specific processes detailed in separate pages, as indicated below.
The movement of signals can be simple, like that associated with receptor molecules of the acetylcholine class: receptors that constitute channels which, upon ligand interaction, allow signals to be passed in the form of small ion movement, either into or out of the cell. These ion movements result in changes in the electrical potential of the cells that, in turn, propagates the signal along the cell.
Signal transduction processes can lead to changes in the growth, proliferation, and/or differentiation state of cells. Changes in cellular growth rates can include alterations in gene expression at the level of transcription, changes in the rates of protein synthesis and/or stability, or changes in various metabolic processes.
More complex signal transduction involves the coupling of ligand-receptor interactions to many intracellular events. These events include phosphorylation by tyrosine kinases and/or serine/threonine kinases. The human genome contains 90 genes that encode tyrosine kinase enzymes including both receptor type and non-receptor type enzymes. Protein phosphorylation results in changes to protein conformations and activities, most often enzymatic activities. The eventual outcome is an alteration in cellular activity and changes in the program of genes expressed within the responding cells.
For detailed discussion of various signal transduction processes that are not covered in this page go to:
Signal Transduction Pathways: Cyclic Nucleotides and Kinases
Signal Transduction Pathways: G-Proteins and GPCR
Signal Transduction Pathways: MAK Kinases
Signal Transduction Pathways: Nucleotides
Signal Transduction Pathways: Phosphatases
Signal Transduction Pathways: Phospholipids
Signal Transduction Pathways: PKC Family
Please refer to the Growth Factors and Other Cellular Regulators page for descriptions of the growth factors described in this page and the explanation of their abbreviations.
Kinases in Signal Transduction
The major mediators of signal transduction processes are enzymes that possess kinase activity. Kinases can be divided into two broad categories, those that phosphorylate tyrosine (Tyr, Y) residues and those that phosphorylate serine (Ser, S) and/or threonine (Thr, T) residues. Humans express a large family of over 500 genes encoding the various kinases that encompass these two broad categories.
The Tyr kinase family includes receptor tyrosine kinases and non-receptor tyrosine kinases.
The Ser/Thr kinases are the largest family of kinases encompassing around 350 of the classified kinases and includes the receptor Ser/Thr kinases and all the remaining Ser/Thr kinases that are divided into at least five broad subfamilies as detailed below and in other Signal Transduction Pathways pages.
Classifications of Signal Transducing Receptors
Signal transducing receptors are of three general classes:
1. Receptors that penetrate the plasma membrane and have intrinsic enzymatic activity. Receptors that have intrinsic enzymatic activities include those that are tyrosine kinases (e.g. PDGF, insulin, EGF and FGF receptors), tyrosine phosphatases (e.g. CD45 [cluster determinant-45] protein of T cells and macrophages), guanylate cyclases (e.g. natriuretic peptide receptors) and serine/threonine kinases (e.g. activin and TGF-β receptors). Receptors with intrinsic tyrosine kinase activity are capable of autophosphorylation (specifically ligand-induced transphosphorylation) as well as phosphorylation of other substrates. Additionally, several families of receptors lack intrinsic enzyme activity, yet are coupled to intracellular non-receptor tyrosine kinases by direct protein-protein interactions.
2. Receptors that are coupled, inside the cell, to GTP-binding and hydrolyzing proteins (termed G-proteins). Receptors of the class that interact with G-proteins all have a structure that is characterized by seven transmembrane spanning domains and as such are sometimes referred to as serpentine receptors. These receptors all belong the superfamily of G-protein coupled receptors, GPCR. Examples of this class are the adrenergic receptors, odorant receptors, and certain hormone receptors (e.g. glucagon, angiotensin, vasopressin and bradykinin).
3. Receptors that are found intracellularly and upon ligand binding migrate to the nucleus where the ligand-receptor complex directly affects gene transcription. Because this class of receptors is intracellular and functions in the nucleus as transcription factors they are commonly referred to as the nuclear receptors. Receptors of this class include the large family of steroid and thyroid hormone receptors. Receptors in this class have a ligand-binding domain, a DNA-binding domain and a transcriptional activator domain.
Receptor Tyrosine Kinases (RTK)
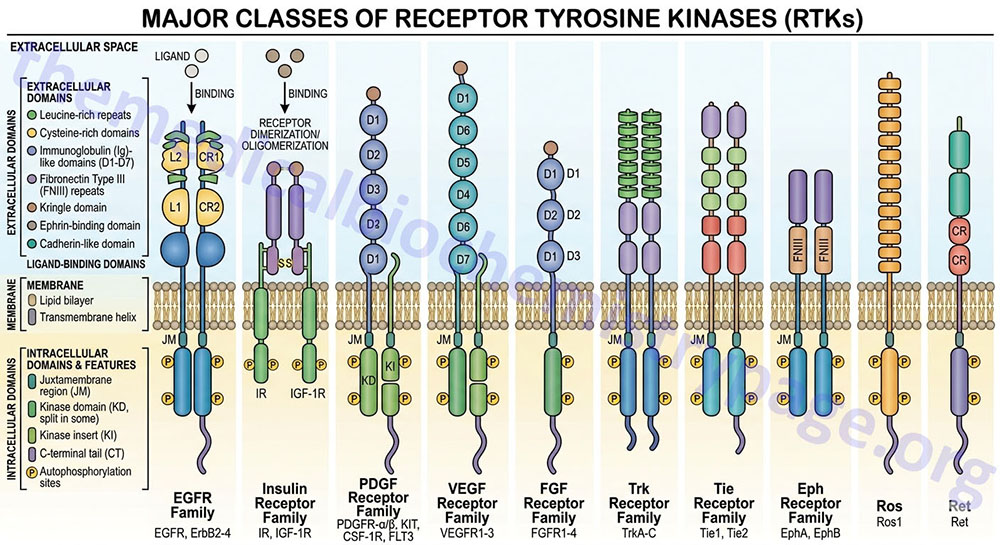
The receptor tyrosine kinase (RTK) family of transmembrane ligand-binding proteins is comprised of 58 members in the human genome. Each of the RTK exhibit similar structural and functional characteristics. Most RTK are monomers, and their domain structure includes an extracellular ligand-binding domain, a transmembrane domain, and an intracellular domain possessing the tyrosine kinase activity. The insulin and insulin-like growth factor receptors are the most complex in the RTK family being disulfide linked heterotetramers.
The amino acid sequences of the tyrosine kinase domains of RTK are highly conserved with those of cAMP-dependent protein kinase (PKA) within the ATP binding and substrate binding regions. Some RTK have an insertion of non-kinase domain amino acids into the kinase domain termed the kinase insert. RTK proteins are classified into families based upon structural features in their extracellular portions (as well as the presence or absence of a kinase insert) which include the cysteine rich domains, immunoglobulin-like domains, leucine-rich domains, Kringle domains, cadherin domains, fibronectin type III repeats, discoidin I-like domains, acidic domains, and EGF-like domains.
Based upon the presence of these various extracellular domains, the 58 proteins of the RTK family have been sub-divided into at least 20 different subfamilies.
Table of the Characteristics of the Common Classes of RTK
| Class | Family Name | Examples | Structural Features of Class |
| I | ErbB Receptor Family | EGF receptor (ERBB1); NEU/HER2 (ERBB2); HER3 (ERBB3); HER4 (ERBB4) | cysteine-rich sequences; receptors dimerize in response to ligand binding; numerous ligands bind and activate the EGFR (ERBB1) including EGF, TGF-α, and amphiregulin |
| II | Insulin Receptor Family | insulin receptor (InsR); IGF-1 receptor (IGF1R); insulin receptor-related receptor (IRR) | cysteine-rich sequences; characterized by disulfide-linked heterotetramers |
| III | PDGF Receptor Family | PDGF receptors (PDGFα and PDGFβ); c-Kit, CSF-1 receptor (CSFR); fms-related tyrosine kinase 3 (FLT3) | contain 5 immunoglobulin-like domains; contain the kinase insert |
| IV | VEGF Receptor Family | vascular endothelial cell growth factor (VEGF) receptor-1 (VEGFR-1; also known as fms-related tyrosine kinase-1 (FLT1); kinase insert domain receptor (VEGFR-2); fms-related tyrosine kinase 4 (VEGFR-3) | contain 7 immunoglobulin-like domains as well as the kinase insert domain |
| V | FGF Receptor Family | fibroblast growth factor receptor 1 (FGFR1); FGFR2; FGFR3; FGFR4 | contain 3 immunoglobulin-like domains as well as the kinase insert; acidic domain |
| VII | Neurotrophin Receptor Family | neurotrophic tyrosine kinase receptor type 1 (TrkA); TrkB; TrkC | contains several closely spaced leucine-rich regions (LRRs); one or two cysteine-rich domains; two immunoglobulin-like domains; no kinase insert |
| VIII | ROR Family | receptor tyrosine kinase-like orphan receptor type 1 (ROR1); ROR2 | |
| X | HGF Receptor Family | met proto-oncogene (Met); macrophage stimulating receptor 1 (MST1R, also Ron) | heterodimeric like the class II receptors except that one of the two protein subunits is completely extracellular. The HGF receptor is a proto-oncogene that was originally identified as the MET oncogene |
Many receptors that have intrinsic tyrosine kinase activity, as well as the tyrosine kinases that are associated with cell surface receptors, contain tyrosine residues, that upon phosphorylation, interact with other proteins of the signaling cascade. These other proteins contain a domain of amino acid sequences that are homologous to a domain first identified in the SRC proto-oncogene. These domains are termed SH2 domains (SRC homology domain 2). The typical SH2 domain is approximately 100 amino acids in length. Different SH2 domains recognize different tyrosine phosphorylated residues based upon the presence of the tyrosine phosphate as well as the amino acid sequences surrounding the tyrosine residue. These variable domains are, therefore, what determine the specificity of SH2 domain-containing protein binding. At least 110 different proteins are expressed in humans that contain SH2 domains.
Another conserved protein-protein interaction domain identified in many signal transduction proteins is related to a third domain in SRC identified as the SH3 domain. Typical SH3 domains are composed of approximately 60 amino acid residues.
The interactions of SH2 domain-containing proteins with RTK or receptor associated tyrosine kinases leads to tyrosine phosphorylation of the SH2 containing proteins. The result of the phosphorylation of SH2 containing proteins. that have enzymatic activity, is an alteration (either positively or negatively) in that activity. Several SH2 containing proteins that have intrinsic enzymatic activity include phospholipase Cγ (PLCγ, PLC-gamma), the proto-oncogene RAS associated GTPase activating protein (rasGAP), phosphatidylinositol-3-kinase (PI3K), protein phosphatase-1C (PTP1C; encoded by the PTPN6 gene), as well as members of the SRC family of protein tyrosine kinases (PTK).
Non-Receptor Protein Tyrosine Kinases (PTK)
Humans express 32 genes that encode intracellular (cytosolic) PTK that are responsible for phosphorylating a variety of intracellular proteins on tyrosine residues following activation of cellular growth and proliferation signals. The proteins in the PTK superfamily can be divided into ten distinct subgroups (ABL, CSK, FES, JAK, PTK2, PTK6, SRC, SYK, TEC, and TNK) where the archetypal PTK subgroup is related to the SRC protein that is a tyrosine kinase first identified as the transforming protein in Rous sarcoma virus and identified as v-src. Subsequently, a cellular homolog was identified and originally designated c-src. Numerous proto-oncogenes were identified as the transforming proteins carried by retroviruses as a result of transduction of host DNA into the viral genome.
The ABL family of tyrosine kinases includes the proteins encoded by the ABL1 (Abelson murine leukemia proto-oncogene 1) and ABL2 (Abelson murine leukemia proto-oncogene 1) genes.
The CSK family of tyrosine kinases includes the proteins encoded by the CSK (C-terminal Src kinase) and MATK (megakaryocyte-associated tyrosine kinase) genes.
The FES family of tyrosine kinases includes the proteins encoded by the FER (feline encephalitis virus-related tyrosine kinase) and FES (feline sarcoma proto-oncogene, tyrosine kinase) genes.
The JAK family of tyrosine kinases includes the proteins encoded by the JAK1 (Janus kinase 1), JAK2 (Janus kinase 2), JAK3 (Janus kinase 3), and TYK2 (tyrosine kinase 2) genes.
The PTK2 family of tyrosine kinases includes the proteins encoded by the PTK2 (protein tyrosine kinase 2) and PTK2B (protein tyrosine kinase 2 beta) genes.
The PTK6 family of tyrosine kinases includes the proteins encoded by the PTK6 (protein tyrosine kinase 6) and SRMS (src-related kinase lacking C-terminal regulatory tyrosine and N-terminal myristylation sites) genes.
The SRC family of tyrosine kinases includes the proteins encoded by the SRC, BLK (B lymphocyte kinase), FGR (Gardner-Rasheed feline sarcoma homolog), FRK (FYN-related kinase), FYN (FGR and YES-related novel tyrosine kinase), HCK (hematopoietic cell kinase), LCK (ymphocyte-specific protein tyrosine kinase), LYN (LCK/YES related novel protein tyrosine kinase), and YES1 (Yamaguchi sarcoma virus homolog) genes.
The SYK family of tyrosine kinases includes the proteins encoded by the SYK (spleen associated tyrosine kinase) and ZAP70 (zeta chain of T cell receptor associated protein kinase 70) genes.
The TEC family of tyrosine kinases is named for the founding member TEC (tyrosine kinase expressed in hepatocellular carcinoma) and includes the proteins encoded by the BMK [BMX (bone marrow kinase in chromosome X) non-receptor tyrosine kinase], BTK (Bruton tyrosine kinase), ITK (IL2 inducible T cell kinase), and TXK (TXK tyrosine kinase) genes.
The TNK family of tyrosine kinases includes the proteins encoded by the TNK1 (tyrosine kinase non-receptor 1) and TNK2 (tyrosine kinase non-receptor 2) genes.
Most of the proteins of the various families of non-receptor PTK couple to cellular receptors that lack enzymatic activity themselves. This class of receptor includes all of the cytokine receptors (e.g. the interleukin-2 receptor, IL2R) as well as the CD4 and CD8 cell surface glycoproteins of T cells, the T cell antigen receptor (TCR), the B cell receptor (BCR), immunoglobulin receptors (IR), erythropoietin receptor (EPOR), and the prolactin receptors.
An example of an alteration in receptor activity in response to association with an intracellular PTK is the nicotinic acetylcholine receptor (AChR). These receptors comprise an ion channel consisting of five distinct subunits (alpha: α, beta: β, gamma: γ, delta: δ, and epsilon: ε). The β, γ, and δ subunits are tyrosine phosphorylated in response to acetylcholine binding which leads to an increase in the rate of desensitization to acetylcholine.
Receptor Serine/Threonine Kinases (RSK)
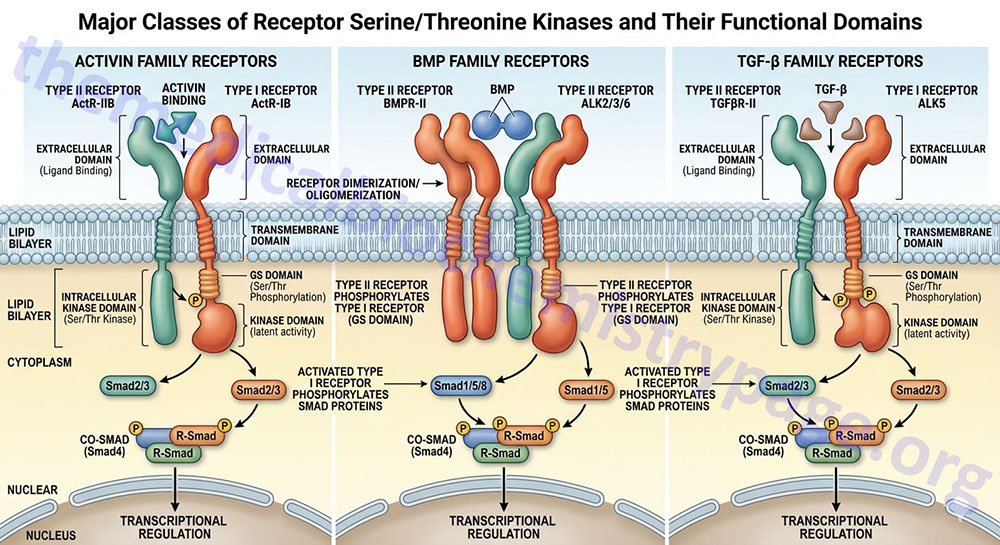
The receptors for the TGF-β (TGF-beta) superfamily of ligands have intrinsic serine/threonine kinase activity. A more complete description of the TGF-β signaling cascade can be found in the Signaling by Wnts and the TGFs-β/BMP Families page.
There are more than 30 multifunctional proteins of the TGF-β superfamily which also includes the activins, inhibins and the bone morphogenetic proteins (BMP). This superfamily of proteins can induce and/or inhibit cellular proliferation or differentiation and regulate migration and adhesion of various cell types. The signaling pathways utilized by the TGF-β, activin and BMP receptors are different than those for receptors with intrinsic tyrosine kinase activity or that associate with intracellular tyrosine kinases.
At least 12 RSK have been characterized as being expressed in humans. These receptors can be divided into two subfamilies identified as the type I and type II receptors. The type I RSKs are also known as activin receptors or activin receptor-like kinases, ALKs. Ligands first bind to the type II receptors which then leads to interaction with the type I receptors. The type II protein phosphorylates the kinase domain of the type I partner leading to displacement of proteins called subunit (or protein) partners. The displacement of these protein partners allows for the binding and phosphorylation of particular members of the Smad family. Once phosphorylated, the particular Smad will migrate to the nucleus and act as complexes that regulate the expression of specific target genes. One predominant effect of TGF-β is regulation of progression through the cell cycle. One nuclear protein involved in the responses of cells to TGF-β is the proto-oncogene, MYC which directly affects the expression of genes harboring MYC-binding elements.
Table of Human Receptor Serine Threonine Kinases (RSK)
| Class | Receptor Name | Common Abbreviation | Gene Symbol |
| I | activin A receptor, type I | ALK2 | ACVR1 |
| I | activin A receptor, type IB | ALK4 | ACVR1B |
| I | activin A receptor, type IC | ALK7 | ACVR1C |
| I | activin A receptor type II-like 1 | ALK1 | ACVRL1 |
| I | bone morphogenetic protein (BMP) receptor, type IA | BMPR1A | BMPR1A |
| I | bone morphogenetic protein (BMP) receptor, type IB | BMPR1B | BMPR1B |
| I | transforming growth factor beta (TGF-β) receptor 1 | TGFBR1 | TGFBR1 |
| II | activin A receptor, type IIA | ActR2 | ACVR2A |
| II | activin A receptor, type IIB | ActR2B | ACVR2B |
| II | anti-Mullerian hormone receptor, type II | MISR2 | AMHR2 |
| II | bone morphogenetic protein (BMP) receptor, type II | BMPR2 | BMPR2 |
| II | transforming growth factor beta (TGF-β) receptor II | TGFBR2 | TGFBR2 |
Serine and Threonine Kinases: STK
Humans express a large superfamily of kinases that phosphorylate serine and/or threonine residues referred to as the STK family. The STK family of kinases includes the RSK discussed in the Receptor Serine/Threonine Kinases (RSK) section above.
The STK regulate a broad array of processes the include control of cellular metabolic rates, cellular proliferation, and cellular differentiation. Humans express more than 350 genes that encode the various STK which are divided into multiple related subfamilies.
Many of the kinases included in the various families outlined below, are covered in detail is the various Signal Transduction Pathways pages.
One major subfamily of the STK superfamily of kinases is identified as the CMGC family. This acronym is derived from the fact that members of the cyclin-dependent kinase (CDK), mitogen-activated protein kinase (MAPK), glycogen synthase kinase 3 (GSK3), and CDC like kinase (CLK) families comprise the CMGC subfamily kinases. The CMGC family is comprised of at least 63 identified members. Three subfamilies of the CMGC family, CDK, MAPK, and GSK3, are introduced in sections below but are also described in greater detail in other linked pages.
PRKAA Family
AMP-regulated protein kinase (AMPK) is a master metabolic regulatory kinase, whose activities are discussed in detail in the AMP-Activated Protein Kinase (AMPK): Master Metabolic Regulator page. AMPK is a trimeric enzyme composed of a catalytic α-subunit and the non-catalytic β- and γ-subunits. The α-subunit can be encoded by one of two genes, PRKAA1 or PRKAA2.
In addition to the catalytic subunit encoding genes of AMPK, there are five additional PRKAA-related subfamilies of kinases that constitute the PRKAA related kinase family. These five PRKAA related kinase subfamilies are the BR kinases, the MELK kinases, the microtubule affinity regulating kinases (MARK), the NUAK kinases, and the SIK kinases.
The BR kinase family is so-called because they were first identified as BRain specific kinases. This family includes two genes, BRSK1 and BRSK2.
The MELK kinase family includes a single gene identified as encoding Maternal Embryonic Leucine zipper Kinase.
The microtubule affinity regulating kinase family includes four genes, MARK1-MARK4.
The NUAK kinase family is so-called from the words “nua”, meaning novel and kinase. The NUAK kinase family is composed of two genes, NUAK1 and NUAK2. The NUAK1 encoded enzyme is also known as AMPK-related protein kinase 5 (ARK5). The NUAK2 encoded enzyme is also known as SNF1/AMP kinase-related kinase (SNARK).
The SIK kinase family is composed of the Salt-Induced Kinases encoded by the SIK1, SIK2, and SIK3 genes.
Mitogen-Activated Protein Kinase Family: MAPK
The mitogen-activated protein (MAP) kinase (MAPK) family constitutes a large family of 13 genes that encode serine/threonine kinases that are involved in a wide range of signal transduction cascades. This family of kinases has been organized into four distinct MAPK cascades named according to the MAPK component that is the central enzyme of each of the cascades. These four MAPK cascades are the extracellular signal-regulated kinase 1/2 (ERK1/2), the c-Jun N-terminal kinase (JNK), the p38, and the ERK5 cascades.
The details of the MAPK family enzymes are covered in the Signal Transduction Pathways: MAP Kinases page.
AGC Kinase Family
The AGC kinase family name is derived from the three major kinases, PKA, PKG, and PKC. The AGC kinase family encompasses at least 63 proteins and can be further divided into 21 subfamilies that includes the PKA family (three genes), the PKG family (two genes), and the PKC family (9 genes).
The details of the PKA and PKG family kinases are covered in the Signal Transduction Pathways: Cyclic Nucleotides and Kinases page.
The details of the PKC family enzymes are covered in the Signal Transduction Pathways: PKC Family page.
Another AGC kinase family of enzymes are the AKT/PKB kinases of which there are three genes, AKT1 (PKBα), AKT2 (PKBβ), and AKT3 (PKBγ).
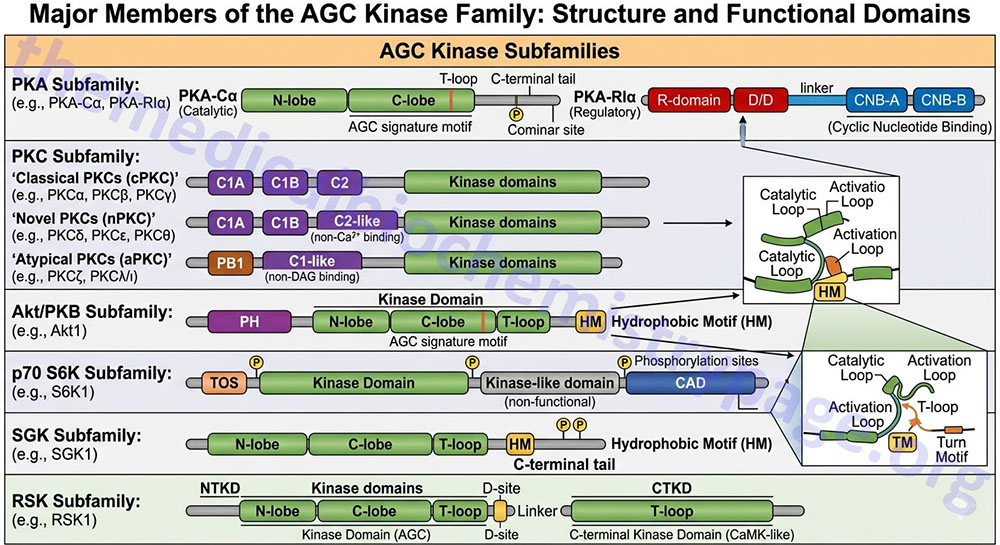
Nomenclature for many of the rest of the subfamilies of AGC kinase family enzymes can be confusing due to historical naming of the enzymes, newer standardized naming conventions, and the identities of the genes encoding these enzymes. For example the enzyme 3-phosphoinositide-dependent protein kinase 1 (encoded by the PDPK1 gene) is also identified as PDK1 in many publications. This can be confusing since the PDK1 gene encodes a pyruvate dehydrogenase (PDH) kinase.
The other subfamilies of the AGC kinase superfamily include the RSK, S6K, RSKL, SGK, MSK, PKN, GRK, NDR/LATS, YANK, ROCK, DMPK, MRCK, CRIK, MAST, SGK494, and PRK subfamilies.
Two additional families, the Aurora kinases and the Polo-like kinases (PLK), are mitotic kinases that are not specifically members of the AGC kinase superfamily but they do share sequences and mechanisms of regulation with other AGC kinase family enzymes. The Aurora family is composed of three enzymes encoded by the AURKA (also known as the Aurora 2 enzyme), AURKB (also known as the Aurora 1 enzyme), and AURKC genes. The PLK family is composed of five enzymes encoded by the PLK1, PLK2, PLK3, PLK4, and PLK5 genes.
The RSK (ribosomal protein S6 kinase A enzymes), S6K (p70 ribosomal protein S6 kinase B enzymes), and RSKL (ribosomal protein S6 kinase C enzymes) subfamily enzymes are all encoded by genes identified as RPS6K (ribosomal protein S6 kinase).
The RSK family of enzymes consists of four members that are encoded by the RPS6KA1 (enzyme is RSK1), RPS6KA2 (enzyme is RSK3), RPS6KA3 (enzyme is RSK2), and RPS6KA6 (enzyme is RSK4) genes.
The S6K family of enzymes consists of two enzymes that are encoded by the RPS6KB1 (enzyme is S6K as well as S6Kβ1) and RPS6KB2 (enzyme is S6Kβ2) genes.
The RSKL family of enzymes consist of two enzymes that are encoded by the RPS6KC1 (enzyme identified as RSKL1) and RPS6KL1 (enzyme identified as RSKL2) genes.
The MSK (mitogen- and stress-activated protein kinases) family of enzymes consists of two enzymes that are also members of the ribosomal protein S6 kinase group and as such are encoded by the RPS6KA4 (enzyme is MSK2) and RPS6KA5 (enzyme is MSK1) genes.
The SGK (serum- and glucocorticoid-induced protein kinases) family of enzymes consists of three enzymes that are encoded by the SGK1, SGK2, and SGK3 genes.
The PKN (PKC-related protein kinases) family of enzymes consists of three enzymes that are encoded by the PKN1, PKN2, and PKN3 genes.
The GRK (G protein-coupled receptor kinases) family of enzymes consists of seven enzymes that are encoded by the GRK1, GRK2, GRK3, GRK4, GRK5, GRK6, and GRK7 genes.
The NDR (nuclear DbF2-related kinases) and LATS (large tumor suppressor kinases) family of enzymes are often listed as separate AGC kinase subfamilies but they are all related due to the presence of an internal extended segment that may serve as an autoinhibitory domain. The NDR/LATS family consists of four enzymes that are encoded by the serine/threonine kinase 38 [STK38 (enzyme is NDR1)], STK38L (enzyme is NDR2), LATS1 and LATS2 genes.
The YANK (yet another novel kinases) family enzymes belong to the serine/threonine kinase 32 family composed of three enzymes that are encoded by the STK32A, STK32B, and STK32C genes.
The ROCK (Rho-activated protein kinases) family of enzymes consists of two enzymes that are encoded by the ROCK1 and ROCK2 genes.
The DMPK (myotonin protein kinase) family consists of a single gene, DMPK, that encodes the kinase identified as DM1 protein kinase. The DMPK gene is a member of the trinucleotide repeat disorder gene family and as such expansion of a repeat in this gene is associated with myotonic dystrophy type 1.
The MRCK (myotonic dystrophy kinase-related CDC42-binding kinase)family of enzymes consists of two member, MRCKα and MRCKβ, that are encoded by the CDC42BPA (CDC42 binding protein kinase alpha) and CDC42BPB (CDC42 binding protein kinase beta) genes, respectively.
The CRIK (citron Rho-interacting kinase) family consists of a single member that is encoded by the CIT gene.
The MAST (microtubule associated serine/threonine kinases) family of enzymes consists of five enzymes that are encoded by the MAST1, MAST2, MAST3, MAST4, and MASTL genes.
The PDK1 (3-phosphoinositide-dependent protein kinase 1) family consists of a single enzyme encoded by the PDPK1 gene. The common nomenclature of PDK1 for this enzyme can be confusing due to the fact that the PDK1 gene encoded a pyruvate dehydrogenase (PDH) kinase.
The SGK494 (Sugen kinase 494) family is composed of a single enzyme encoded by the RSKR (ribosomal protein S6 kinase related) gene.
The PRK enzymes are encoded by X-linked and Y-linked genes PRKX (protein kinase cAMP-dependent X-linked catalytic subunit) and PRKY (protein kinase Y-linked), respectively. The human PRKY encoded mRNA is likely a target for nonsense mediated decay (NMD).
Ca2+/Calmodulin-Dependent Kinase Family: CaMK
There are two broad classes of kinase families that are regulated by interaction with complexes of Ca2+ and the regulatory protein, calmodulin (CaM), defined as Ca2+/CaM complexes.
One family of Ca2+/CaM regulated kinases have restricted substrate specificity such as that of phosphorylase kinase (PHK). There are two additional restricted Ca2+/CaM regulated kinases, elongation factor 2 kinase (eEF2K), and myosin light chain kinase (MLCK).
The other family is the multifunctional Ca2+/CaM regulated kinase family. This family contains four subfamilies identified as CaMKK, CaMKI, CaMKII, and CaMKIV.
More information on the CaMK family kinases can be found in the Glycogen Metabolism page.
STE Kinase Family
The name of STE kinase family name is derived from the identification of mammalian homologs of the yeast, Sacharomyces cerevisiae genes identified as STErile 7, sterile 11, and sterile 20. The STE kinases play important role in MAP kinase signaling pathways. The STE kinases are members of the MAP2K, MAP3K, and MAP4K families of MAP kinase signaling kinases.
Casein Kinase Family: CK
The casein kinases (CK) are divided into two main subfamilies identified as casein kinase 1 (CK1) and casein kinase 2 (CK2). The CK1 family contains nine genes while the CK2 family contains three genes.
Two of the CK1 family enzymes were originally identified as vaccinia-related kinases and as such are identified as VRK1 and VRK2.
The CK2 enzyme is a heterotetrameric enzyme composed of two α-subunits and two β-subunits. The α-subunits are derived from one of two genes (CSNK2A1 or CSNK2A2) while the β-subunit is derived from the CSNK2B gene.
Cyclin-Dependent Kinase Family: CDK
All dividing cells require cell cycle control mechanisms to exert their influences at specific times during each transit through a cell cycle. The heart of this timing control is the responsibility of a family of protein kinases that are called cyclin-dependent kinases, CDKs. Details of the CDK family are covered in the Eukaryotic Cell Cycles: Mitosis and Meiosis page.
The human genome contains a total of 26 genes that encode proteins of the CDK family. The kinase activity of these enzymes rises and falls as the cell progresses through a cell cycle. These oscillating changes are controlled by a complex series of proteins, the most important of which are the cyclins, hence the name of the enzymes as cyclin-dependent kinases.
Glycogen Synthase Kinase 3 Family: GSK3
Humans express two genes encoding glycogen synthase kinase 3 enzymes, GSK3A (encoding GSK3α) and GSK3B (encoding GSK3β). Details of the GSK3 enzymes are covered in the Glycogen Metabolism page.
Intracellular Hormone Receptors (Nuclear Receptors)
The steroid/thyroid hormone receptor superfamily, which includes but is not limited to, the glucocorticoid (GR), vitamin D (VDR), retinoic acid (RAR) and thyroid hormone (TR) receptors, is a class of proteins that reside in the cytoplasm, or the nucleus, and bind their lipophilic hormone ligands in these locations since the hormones are capable of freely penetrating the hydrophobic plasma membrane. Because these receptors bind ligand intracellularly and then interact with DNA directly they are more commonly called the nuclear receptors (NR). In addition to binding hormone, all receptors of this class are capable of directly activating gene transcription.
All of the steroid hormones (e.g. progesterone, aldosterone, estradiol, cortisol) bind their receptors in the cytosol, whereas the non-steroidal hormones that bind nuclear receptors (e.g. thyroid hormones, calcitriol, retinoic acid), do so within the nucleus.
The cytosol localized nuclear hormones are “trapped” in this location through interaction with proteins of the heat shock family. When steroid hormones bind their receptors in the cytosol the ligand-receptor complex is dissociated from the heat shock proteins and the complex migrates to nucleus where it binds to specific DNA sequences termed hormone response elements (HRE). The binding of the complex to an HRE results in altered transcription rates of the associated gene.
The non-steroidal hormones that activate nuclear receptors are constitutively present in the nucleus bound to their target genes in the absence of their cognate hormones. These receptors exhibit potent transcriptional repression function in the absence of hormones and the repressor function is mapped to the domain that is responsible for binding ligand.
Analysis of the human genome has revealed 48 nuclear receptor genes that can be classified into seven defined subfamilies. Many of these genes are capable of yielding more than one receptor isoform. The nuclear receptors all contain a ligand-binding domain (LBD), a DNA-binding domain (DBD) and, in most cases, two activation function domains (identified as AF-1 and AF-2). The activity of the AF-1 domain is independent of the presence of ligand bound to the LBD, whereas the activity of the AF-2 domain is dependent upon ligand being bound to the LBD. Based upon the sequences of these two domains the nuclear receptor family is divided into six sub-families.
Some members of the family bind to DNA as homodimers such as is the case for subfamily III receptors which comprises the steroid receptors such as the estrogen receptor (ER), mineralocorticoid receptor (MR), progesterone receptor (PR), androgen receptor (AR), and the glucocorticoid receptor (GR). Other family members (such as all subfamily I members) bind to DNA as heterodimers through interactions with the retinoid X receptors (RXR). In addition to the steroid hormone and thyroid hormone receptors there are numerous additional family members that bind lipophilic ligands. These include the retinoid X receptors (RXR), the liver X receptors (LXR), the farnesoid X receptors (FXR) and the peroxisome proliferator-activated receptors (PPAR).
Table of the Nuclear Receptor Families
| Receptor Nomenclature | Receptor Common Name | Human Gene Name |
| Type 1A: Thyroid Hormone Receptors | ||
| NR1A1 | thyroid hormone receptor-α | THRA |
| NR1A2 | thyroid hormone receptor-β | THRB |
| Type 1B: Retinoic Acid Receptors (RAR) | ||
| NR1B1 | retinoic acid receptor-α (RARα) | RARA |
| NR1B2 | retinoic acid receptor-β (RARβ) | RARB |
| NR1B3 | retinoic acid receptor-γ (RARγ) | RARG |
| Type 1C: Peroxisome Proliferator-Activated Receptors (PPAR) | ||
| NR1C1 | peroxisome proliferator-activated receptor-α (PPARα) | PPARA |
| NR1C2 | peroxisome proliferator-activated receptor-β/δ (PPARβ/δ) | PPARD |
| NR1C3 | peroxisome proliferator-activated receptor-γ (PPARγ) | PPARG |
| Type 1D: Reverse ERBA Receptors | ||
| NR1D1 | Thyroid hormone receptor, α-1-like (Rev-erbα) | THRAL |
| NR1D2 | Rev-erbα-related receptor (Rev-erbγ) | RVR |
| Type 1F: RAR-Related Orphan Receptors | ||
| NR1F1 | RAR-related orphan receptor-α | RORA |
| NR1F2 | RAR-related orphan receptor-β | RORB |
| NR1F3 | RAR-related orphan receptor-γ | RORC |
| Type 1H: Liver X Receptor-Like Receptors | ||
| NR1H2 | liver X receptor-β (LXRβ) | NR1H2 |
| NR1H3 | liver X receptor-α (LXRα) | NR1H3 |
| NR1H4 | farnesoid X receptor (FXR) | NR1H4 |
| NR1H5 | farnesoid X receptor-β (FXRβ) | NR1H5P |
| Type 1I: Vitamin D Receptor-Like Receptors | ||
| NR1I1 | vitamin D receptor (VDR) | VDR |
| NR1I2 | pregnane X receptor (PXR) | NR1I2 |
| NR1I3 | constitutive androstane receptor (CAR) | NR1I3 |
| Type 2A: Hepatocyte Nuclear Factor-4 (HNF4) Receptors | ||
| NR2A1 | hepatocyte nuclear factor-4-α (HNF-4α) | HNF4A |
| NR2A2 | hepatocyte nuclear factor-4-γ (HNF-4γ) | HNF4G |
| Type 2B: Retinoid X Receptors (RXR) | ||
| NR2B1 | retinoid X receptor-α (RXRα) | RXRA |
| NR2B2 | retinoid X receptor-β (RXRβ) | RXRB |
| NR2B3 | retinoid X receptor-γ (RXRγ) | RXRG |
| Type 2C: Testis Receptors | ||
| NR2C1 | testes receptor 2 | TR2 |
| NR2C2 | testicular nuclear receptor 4 | TR4, TAK1 |
| Type 2E: Orphan Ligand Receptors | ||
| NR2E1 | homolog of Drosophila tailless | TLX |
| NR2E3 | photoreceptor-specific nuclear receptor | PNR |
| Type 2F: Chicken Ovalbumin Upstream Promoter Transcription Factor-Related Receptors | ||
| NR2F1 | chicken ovalbumin upstream promoter transcription factor 1 | TFCOUPI |
| NR2F2 | chicken ovalbumin upstream promoter transcription factor 2 | TFCOUPII |
| NR2F6 | ERBA-related 2 | EAR2 |
| Type 3A: Estrogen Receptors | ||
| NR3A1 | estrogen receptor-α | ESR1 |
| NR3A2 | estrogen receptor-β | ESR2 |
| Type 3B: Estrogen Receptor-Related Receptors | ||
| NR3B1 | estrogen receptor-relatedα | ESRRA |
| NR3B2 | estrogen receptor-relatedβ | ESRRB |
| NR3B3 | estrogen receptor-relatedγ | ESRG |
| Type 3C: Steroid Receptors | ||
| NR3C1 | glucocorticoid receptor | GCCR |
| NR3C2 | mineralocorticoid receptor | MR |
| NR3C3 | progesterone receptor | PGR |
| NR3C4 | androgen receptor | AR |
| Type 4A: Orphan Ligand Receptors | ||
| NR4A1 | nerve growth factor (NGF)-induced factor B | NGFI-B |
| NR4A2 | nuclear receptor-related 1 | NURR1 |
| NR4A3 | neuron-derived orphan receptor 1 | NOR1 |
| Type 5A: Orphan Ligand Receptors | ||
| NR5A1 | steroidogenic factor 1 | SF1 |
| NR4A2 | liver receptor homolog 1 | LRH-1 |
| Type 6A: Orphan Ligand Receptors | ||
| NR6A1 | germ cell nuclear factor | GCNF |
| Type 0B: DAX-Like Receptors | ||
| NR0B1 | DAX1 | NR0B1 |
| NR0B2 | small heterodimer partner, SHP | NR0B2 |
Although all members of the NR family possess activation function domains that are responsible for the regulation of transcription of target genes, the regulation of transcription is much more complex due to the association of numerous coregulatory proteins. These coregulatory proteins are of two distinct classes: those that function to co-activate the NR and those that function to co-repress the receptor complex. Co-activators are found to be associated with ligand-bound NR and, thereby, induce gene expression. Co-repressors selectively repress gene expression through interaction with NR that are ligand free or bound to antagonists.
In addition, coregulators can be classified into two main groups: one that modifies histones (e.g, by acetylation/deacetylation or methylation/demethylation) and the other that includes ATP-dependent chromatin remodeling factors. These remodeling factors modulate promoter accessibility to other transcription factors as well as to the basal transcriptional machinery. The properties of several members of each class of coregulator are discussed below.
In addition to the nuclear receptors discussed below, additional members are being identified all the time such as the estrogen related receptors (ERRβ and ERRγ), the retinoid-related orphan receptor (RORα), and the constitutive androstane receptor (CAR).
Retinoid X Receptors: RXR
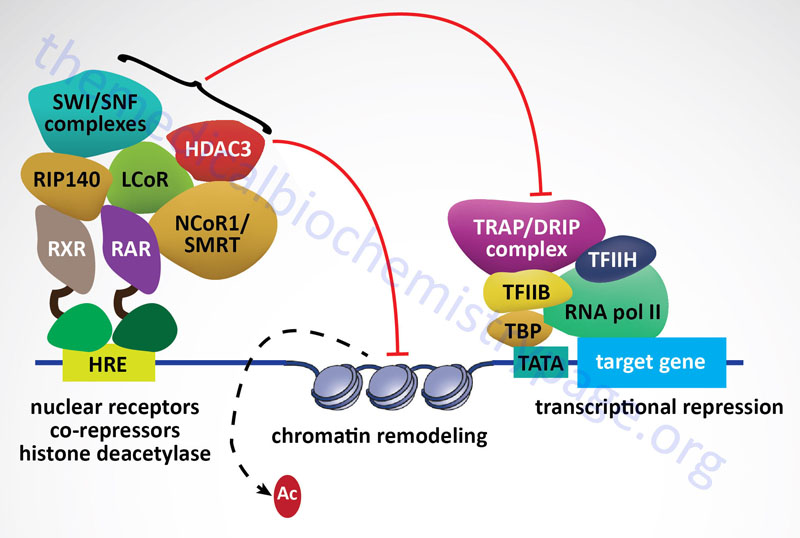
The RXR represent a class of receptors that bind the retinoid 9-cis-retinoic acid. There are three isotypes of the RXR: RXRα, RXRβ, and RXRγ and each isotype is composed of several isoforms. The RXR serve as obligatory heterodimeric partners for numerous members of the nuclear receptor family including those discussed below (PPAR, LXR, and FXR). In the absence of a heterodimeric binding partner the RXR are bound to hormone response elements (HRE) in DNA and are complexed with co-repressor proteins that include histone deacetylases (HDACs) and nuclear receptor corepressor 1 (NCoR1), or silencing mediator of retinoid and thyroid hormone receptor (SMRT; also called NCoR2).
RXRα is widely expressed with highest levels liver, kidney, spleen, placenta, and skin. The critical role for RXRα in development is demonstrated by the fact that null mice are embryonic lethals. RXRβ is important for spermatogenesis and RXRγ has a restricted expression in the brain and muscle.
Peroxisome Proliferator-Activated Receptors: PPAR
The PPAR family is composed of three family members: PPARα, PPARβ/δ, and PPARγ. Each of these receptors forms a heterodimer with the RXR. For more detailed information on the PPAR visit the PPAR page.
The first family member identified was PPARα and it was found by virtue of it binding to the fibrate class of anti-hyperlipidemic drugs and resulting in the proliferation of peroxisomes in hepatocytes, hence the derivation of the name of the protein. Although PPARγ and PPARδ are related to PPARα they do not stimulate peroxisome proliferation. Subsequently it was shown that PPARα is the endogenous receptor for polyunsaturated fatty acids. PPARα is highly expressed in the liver, skeletal muscle, heart, and kidney. Its function in the liver is to induce hepatic peroxisomal fatty acid oxidation during periods of fasting. Expression of PPARα is also seen in macrophage foam cells and vascular endothelium. Its role in these cells is thought to be the activation of anti-inflammatory and anti-atherogenic effects.
PPARγ is a master regulator of adipogenesis and is most abundantly expressed in adipose tissue. Low levels of expression are also observed in liver and skeletal muscle. PPARγ was identified as the target of the thiazolidinedione (TZD) class of insulin-sensitizing drugs. The mechanism of action of the TZDs is a function of the activation of PPARγ activity and the consequent activation of adipocytes leading to increased fat storage and secretion of insulin-sensitizing adipocytokines such as adiponectin.
PPARδ is expressed in most tissues and is involved in the promotion of mitochondrial fatty acid oxidation, energy consumption, and thermogenesis. PPARδ serves as the receptor for polyunsaturated fatty acids and VLDL. Current pharmacologic targeting of PPARδ is aimed at increasing HDL levels in humans since experiments in animals have shown that increased PPARδ levels result in increased HDL and reduced levels of serum triglycerides.
Liver X Receptors: LXR
There are two forms of the LXR: LXRα and LXRβ. The LXRs form heterodimers with the RXR and as such can regulate gene expression either upon binding oxysterols (e.g. 22R-hydroxycholesterol) or 9-cis-retinoic acid. Because the LXRs bind oxysterols they are important in the regulation of whole body cholesterol levels. The function of LXR in the liver is to mediate cholesterol metabolism by inducing the expression of SREBP-1c. SREBP-1c is a transcription factor involved in the control of the expression of numerous genes including several involved in cholesterol synthesis. For more detailed information on the LXR visit the LXR page.
Farnesoid X Receptors: FXR
There are two genes encoding FXR identified as FXRα and FXRβ. In humans at least four FXR isoforms have been identified as being derived from the FXRα gene as a result of activation from different promoters and the use of alternative splicing; FXRα1, FXRα2, FXRα3, and FXRα4. The FXR gene is also known as the NR1H4 gene (for nuclear receptor subfamily 1, group H, member 4). The FXR genes are expressed at highest levels in the intestine and liver.
FXR forms a heterodimer with members of the RXR family. Following heterodimer formation the complex binds to specific sequences in target genes resulting in regulated expression. One major target of FXR is the small heterodimer partner (SHP) gene. Activation of SHP expression by FXR results in inhibition of transcription of SHP target genes. Of significance to bile acid synthesis, SHP represses the expression of the cholesterol 7-hydroxylase gene (CYP7A1). CYP7A1 is the rate-limiting enzyme in the synthesis of bile acids from cholesterol.
The FXR were originally identified by their ability to bind farensol (an isoprene derivative) metabolites. However, subsequent research has demonstrated that FXR are receptors for bile acids which is the primary mechanism by which bile acids negatively regulate their own expression. In addition to binding bile acids, FXR have been shown to bind polyunsaturated fatty acids (PUFA) such as the omega-3 PUFA docosahexaenoic acid (DHA) and α-linolenic acid (ALA). Most recently, FXR has been shown to bind the androgen hormone, androsterone, derived via testosterone metabolism. For more detailed information on the FXR visit the FXR page.
Pregnane X Receptors: PXR
A particular receptor of this family that has been shown to bind numerous structurally unrelated chemicals was originally identified as the pregnane X receptor (PXR). PXR is highly expressed in the liver and is involved in mediating drug-induced multi-drug clearance. For this reason PXR is important in protecting the body from harmful metabolites. Its ability to bind various steroids and xenobiotics has led to PXR also being identified as steroid and xenobiotic sensing nuclear receptor, SXR.
An additional physiologically significant function of PXR is in the regulation of bile acid synthesis. PXR is a recognized receptor for lithocholic acid and other bile acid precursors. PXR activation leads to repression of bile acid synthesis due to its physical association with hepatocyte nuclear factor 4α (HNF-4α) causing this transcription factor to no longer be able to associate with the transcriptional co-activator PGC-1α (PPARγ co-activator 1α) which ultimately leads to loss of transcription factor activation of the rate-limiting enzyme of bile acid synthesis CYP7A1 which, as described above, is also the target of FXR action. In addition to regulation of bile acid metabolism, PXR represses the expression of the gluconeogenic enzyme PEPCK.
PXR is encoded by the nuclear receptor subfamily 1, group I, member 2 (NR1I2) gene.
Nuclear Receptor Coactivators
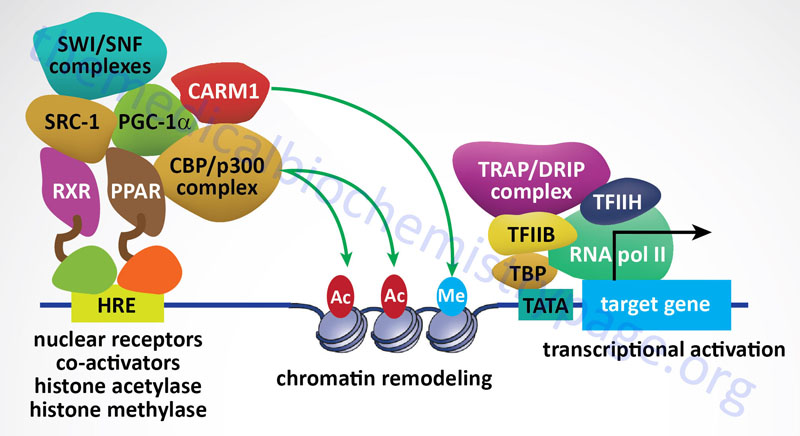
The first nuclear receptor coactivator to be identified was steroid receptor coactivator-1 (SRC-1). To date, more than 400 coregulators (both coactivators and corepressors) have been identified. There are now known to exist three SRC gene families. SRC-1 (encoded by the NCOA1 gene), SRC-2 (also known as GRIP1 for glucocorticoid receptor-interacting protein 1 and TIF2 for transcriptional intermediary factor 2) encoded by the NCOA2 gene, and SRC-3 (also known as AIB1 for amplified in breast cancer 1 and TRAM-1 for thyroid hormone receptor activator molecule 1) encoded by the NCOA3 gene. The three members of the SRC family contain homologous domains and share between 50% and 54% amino acid sequence similarity. There is also a diverse family of enzymes that interact with and modify SRCs which includes histone acetyltransferases (HAT), histone methyltransferases (HMT), kinases, phosphatases, ubiquitin ligases, and small ubiquitin-related modifier (SUMO) ligases.
Peroxisome proliferator-activated receptor gamma, coactivator 1 alpha (PGC-1α) is another critical NR coregulator. PGC-1α has been shown be involved in the regulation of metabolism and energy homeostasis. Indeed, expression levels of PGC-1α have been associated with genetic diseases associated with impaired mitochondrial function, including type 2 diabetes and obesity. Another important coactivator is cAMP response-element binding protein (CREB)-binding protein, CBP. CBP is closely related to another protein identified as p300 forming a family identified as p300-CBP.
Humans express seven genes in the CREB subfamily of the basic leucine zipper (bZIP) family of transcription factors. The seven CREB gene are identified as CREB1, CREB3, CREB5, and CREB3-like 1, 2, 3, and 4 (CREB3L1, CREB3L2, CREB3L3, and CREB3L4). The CREB3L3 encoded protein is commonly identified as CREBH. The CREB1 encoded proteins are most closely related in structure and function to two additional transcription factors called cAMP response element modulator (CREM) and activating transcription factor 1 (ATF-1). Another member of the activating transcription factor (ATF) family, ATF-4, was originally identified as CREB2.
CBP and p300 possess intrinsic histone acetyltransferase (HAT) activity that leads to relaxation of the chromatin structure near a NR target gene. Other chromatin remodeling complexes, such as coactivator-associated arginine methyltransferase 1 (CARM1), can also stimulate gene transcription by NRs as well as other transcription factors in combination with the SRC family of coactivators.
In addition to acting a coactivators for NRs, the SRC family proteins also interact with many different types of transcription factors and potentiate their transcriptional activity. These include p53, signal transducers and activators of transcription (STATs), nuclear factor-κB (NF-κB), hypoxia-inducible factor 1 (HIF1), and hepatocyte nuclear factor-4 (HNF4) to name just a few. Several extracellular stimuli, such as growth factors and cytokines, that activate membrane-spanning signal transducing receptors, generating phosphorylation codes on SRCs that lead to increased coactivator affinity for the androgen receptor (AR), estrogen receptor-alpha (ERα), and progesterone receptor (PR).
Nuclear Receptor Corepressors
As a general rule it has been established that when nuclear receptors are free of activating ligand they preferentially interact with corepressor complexes to mediate transcriptional repression. Nuclear receptor corepressor 1 (NCoR1) and silencing mediator of retinoic and thyroid receptors (SMRT) are the most well-characterized NR corepressor complexes. The core NCoR/SMRT protein complex consists of NCoR/SMRT, transducin β-like 1/related 1 (TBL1/TBLR1), histone deacetylase 3 (HDAC3), and G-protein pathway suppressor 2 (GPS2). NCoR and SMRT serve as the docking sites for corepressor complex assembly. NCoR/SMRT bind NR and associate with each of the other complex subunits.
As discussed above, when the NR interacts with ligand, transcriptional activation results due to the ability of the NR-ligand complex to recruit coactivator proteins and displace corepressor proteins. Nuclear receptor corepressors can inhibit the transcriptional activity of most members of the NR superfamily. As always in biology, there are a few exceptions to the general rule of unliganded NR binding corepressors. These exceptions include LCoR (ligand-dependent nuclear-receptor corepressor), RIP140 (receptor-interacting protein-140) and REA (repressor of estrogen-receptor activity). These repressors bind to NR in a ligand-dependent manner and compete with coactivators by displacing them. In addition, there are several coregulatory factors, such as the ATP-dependent chromatin remodeling complexes SWI/SNF (switching of mating type/sucrose non-fermenting, chromatin remodeling complex), which have been shown to be involved in the regulation of both transcriptional activation and repression.