
Last Updated: July 13, 2026
Introduction to SCID
Severe combined immunodeficiency (SCID) refers to a group of potentially fatal disorders due to a combined loss of function of both T- and B-lymphocytes. There are at least 13 known and characterized genetic causes of SCID. The most common (45%) cause of SCID is the X-linked recessive disorder resulting from loss of function of the common gamma (γ) chain of the T-cell receptor and other interleukin (IL) receptors. The second most common (15%) form of SCID is caused by defects in the activity of the purine nucleotide catabolism enzyme, adenosine deaminase (ADA). This form of SCID is inherited as an autosomal recessive disorder with an incidence rate as high as 1:200,000, although some studies report an incidence of 1:1,000,000. Deficiency in purine nucleotide phosphorylase (PNP), another purine catabolic enzyme, results in 1%-2% of all cases of SCID.
ADA catalyzes the irreversible deamination of adenosine and deoxyadenosine to inosine and 2′-deoxyinosine, respectively.
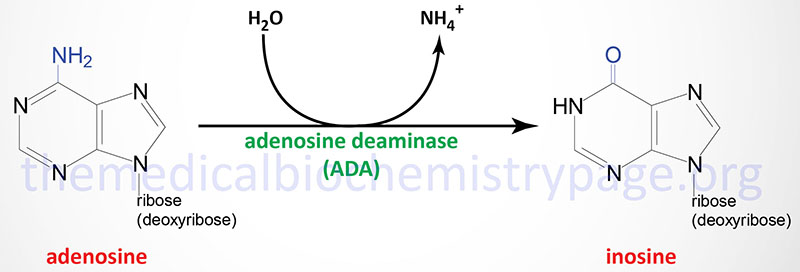
Although ADA activity is found in all tissues of the body, the highest concentrations are found in the thymus and other lymphoid tissues. In addition to the differences in levels of ADA in various tissues, the enzyme is also developmentally regulated such that immature thymocytes express higher levels than do mature thymocytes. The level of expression of ADA also decreases as B cells mature.
Molecular Biology of ADA Deficient SCID
Adenosine deaminase is encoded by the ADA gene. The ADA gene is located on chromosome 20q13.12 and is composed of 12 exons that generate three alternatively spliced mRNAs. These mRNAs encode proteins of 363 amino acids (isoform 1), 228 amino acids (isoform 2), and 339 amino acids (isoform 3).
Over 60 different pathogenic variants have been identified in the ADA gene resulting in immunodeficiency. Nearly 70% of all ADA pathogenic variants result in single amino acid substitutions with the rest being deletions and splicing variants.
Biochemical Consequences of ADA Deficiency
In ADA deficiency there is an elevation in the level of adenosine and 2′-deoxyadenosine in the blood and 2′-deoxyadenosine levels in the urine are also elevated. The consequences of the elevations in these two ADA substrates are impaired lymphocyte differentiation, function, and viability which results in lymphopenia and severe immunodeficiency.
Increases in 2′-deoxyadenosine, through the action of ubiquitous nucleoside phosphorylases, results in dramatic increases in cellular dATP pools. The consequence of increased dATP pools is an inhibition of ribonucleotide reductase (RR), the enzyme responsible for generating deoxyribonucleotides (necessary for DNA replication) from ribonucleotides. The inhibition of RR leads to a block to DNA replication which is critical for the expansion of lymphocytes in response to an antigenic challenge.
Deoxy-ATP also induces DNA strand breakage in non-dividing lymphocytes via a direct activation of a major protease (caspase 9) involved in apoptosis (programmed cell death).
In addition, S-adenosylhomocysteine hydrolase activity is markedly inhibited by 2′-deoxyadenosine resulting in accumulation of S-adenosylhomocysteine which in turn results in reduced synthesis of S-adenosylmethionine (AdoMet), a critical substrate in transmethylation reactions.
Clinical Features of ADA Deficient SCID
Not all forms of SCID are caused by defects in ADA. For example, a clinically related immunodeficiency occurs as a consequence of defects in the purine nucleotide salvage enzyme, purine nucleoside phosphorylase (PNP). However, greater than 85% of all ADA-deficient patients are infants with SCID.
Infants with ADA deficient SCID usually present within the first month of life with failure to thrive and life-threatening interstitial pneumonitis (inflammation of the walls of the airs sacs of the lungs). Pneumonia is often caused by Pneumocystis carinii or a viral infection. The gastrointestinal tract as well as the skin are also frequently involved in initial infections. Candidiasis is first observed as “diaper rash” but then progresses to an extensive infection involving the skin, oral and esophageal mucosa, and the vagina in female patients. Persistent infections prompt most parents to have their baby examined and SCID is thus often diagnosed by 1 to 2 months of age from a determination of immune function.
Most SCID patients lack both cell-mediated (T cell) and humoral (B cell) immunity and thus, the derivation of the disease name: severe combined immunodeficiency. Because this disease is so life-threatening most patients do not survive for more than a few months. If immunological function is not restored, SCID is always fatal by 1 to 2 years of age due to complications from infection with DNA or RNA viruses such as cytomegalovirus, varicella, and parainfluenza.
Treatment of ADA Deficient SCID
The most effective treatment for SCID is bone marrow transplantation. However, many patients do not have a histocompatible sibling and thus other means of treatment were needed. Enzyme replacement therapy (ERT) has proved effective in treating some (but not all) ADA deficient SCID patients. The most common treatment is to use intramuscular injection of highly purified bovine ADA coupled to the inert polymer, monomethoxypolyethylene glycol (PEG-ADA). The use of PEG-ADA therapy is sometimes warranted in SCID patients who are too ill to undergo bone marrow transplantation.
Of clinical significance is the fact that ADA deficient SCID was the first disease for which the use of somatic cell gene therapy was approved in the US. In the original NIH trials carried out in 1990, blood mononuclear cells were collected and transfected with a retroviral vector expressing the human ADA cDNA. After culture of the cells for several days in the presence of a T-cell mitogen the cells were re-infused. Because the efficiency of transfection of the ADA-expressing DNA in these trials was less than 1%, the level of ADA activity attained in the first patients was only 1-20% of normal. The use of PEG-ADA therapy is still superior to the use of the current gene therapy protocols in the treatment of ADA-deficient SCID patients.